Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(6):1053-1061. doi:10.7150/jca.16584 This issue Cite
Research Paper
Effects of prolonged exposure to low dose metformin in thyroid cancer cell lines
Safar Kheder ![]() , Karen Sisley, Sirwan Hadad, Sabapathy P Balasubramanian
, Karen Sisley, Sirwan Hadad, Sabapathy P Balasubramanian
Department of Oncology & Metabolism, University of Sheffield, S10 2RX.
Received 2016-6-24; Accepted 2016-12-16; Published 2017-4-9
Abstract
Background: Thyroid cancer is generally associated with an excellent prognosis, but there is significant long-term morbidity with standard treatment. Some sub-types however have a poor prognosis. Metformin, an oral anti-diabetic drug is shown to have anti-cancer effects in several types of cancer (breast, lung and ovarian cancer). The proposed mechanisms include activation of the Adenosine Mono-phosphate-activated Protein Kinase (AMPK) pathway and inhibition of the mTOR pathway (which promotes growth and proliferation). By inhibiting hepatic gluconeogenesis and increasing glucose uptake by muscles, metformin decreases blood glucose and circulating Insulin levels.
Aims: Explore the effect of metformin on the growth and proliferation of thyroid cancer cell lines.
Methods: The effects of metformin on thyroid cancer cell lines (FTC-133, K1E7, RO82-W-1, 8305C and TT) and normal thyroid follicular cells (Nthy-ori 3-1) were investigated using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay for cell proliferation; clonogenic assays; FACS analysis for apoptosis and cell cycle, H2A.X phosphorylation (γH2AX) assay for DNA repair and scratch assay for cell migration.
Results: Metformin inhibited cell proliferation and colony formation at 0.03 mM and above and inhibited cell migration at 0.3 mM. At concentrations of 0.1 mM and above metformin increased the percentage of apoptotic cells and induced cell cycle arrest in G0/G1 phase at minimum concentration of 0.3 mM. Unlike previous reports, no effect on DNA repair response was demonstrated.
Conclusion: Metformin suppressed growth of all thyroid cancer cell lines, at concentrations considered to be within in the therapeutic range for diabetic patients on metformin (<0.3 mM).
Keywords: Metformin, thyroid cancer, human, in vitro
Introduction
Thyroid cancer is a relatively uncommon malignancy (less than 1% of all cancers registered in the UK); but it is the most common endocrine cancer, accounting for more than 90% of these tumours [1, 2]. The majority of thyroid cancer types belong to the category of differentiated thyroid cancer (DTC); the prognosis of which is good to excellent with >80% surviving 10 years [3]. However, there are concerns over long-term morbidity associated with standard treatment regimes. For example, thyroid stimulating hormone (TSH) suppression in these patients is associated with side effects due to high dose of Thyroxine used [4].
Metformin (N',N' -dimethybiguanide) is an oral antidiabetic drug of the Biguanide class and one of the most commonly prescribed drugs worldwide; accounting for nearly 120 million prescriptions a year [5]. It is the drug of choice in most patients with type 2 diabetes and has been licensed for use in in UK since 1958 [6-8]. Several population based studies have demonstrated the potential of metformin as a chemo-preventative agent in cancer [9-11]. Evans et al [9] reported the first such association, demonstrating metformin use to be associated with low cancer incidence among type 2 diabetic patients. Other data suggests that metformin can reduce the risk of breast cancer [12-14], prostatic cancer [13, 15], gastric cancer [13, 16], lung cancer [12, 13, 16], pancreatic cancer [13, 16], and colorectal cancer [13]. There is also observational evidence that cancer related mortality is reduced in patients on metformin [17-20]. Furthermore, investigations of the anti-cancer effects of metformin in vitro, using a number of human cancer cell lines, including thyroid cancer [21, 22], have confirmed these effects, indicating the response is in a time and concentration-dependent manner. The concentrations however of metformin used in these studies were much higher than normal human plasma concentrations (0.01-0.3 mM) [23, 24], limiting the extrapolation of these findings to clinical practice.
The possible anti-cancer effects of metformin are thought to arise through activating the Adenosine Mono-phosphate-activated Protein Kinase (AMPK) pathway in several types of cancer [25]. More recently, the anti-tumour effect of metformin in breast cancer patients correlated with reduced Ki67 expression and altered expression of numerous genes [26, 27]. In mapping these genes into biological pathways, several pathways seem to be involved; in particular the TNFR1 and G1/S checkpoint regulation signaling pathways were most significantly affected [26].
The anti-proliferative action of metformin on thyroid cancer [28, 29] may be particularly pertinent, as studies suggest metformin decreases TSH level without changes in free thyroxine (FT4) [28, 29]. The TSH lowering effect of metformin was observed in a retrospective case study of 4 hypothyroid patients [28], however once metformin use was discontinued the level of TSH increased again in all patients. Furthermore, in a pilot study, 11 patients with type 2 diabetes and hypothyroidism on stable thyroxine doses, were treated with metformin for 6 months [29]. A significantly decreased TSH level was observed after 6 months of treatment. The morbidity associated with the long term use of high dose thyroxine to achieve TSH suppression in patients with differentiated thyroid cancer may be countered by the use of metformin which is relatively free of side effects. This may facilitate a move from TSH suppression to TSH replacement therapy.
The prognosis in certain types of thyroid cancer such as medullary thyroid cancer (MTC) and anaplastic thyroid cancer (ATC) is intermediate to poor with 14% surviving 10 years [3]. New adjuvant treatment options are invaluable for these types of thyroid cancer. Given recent information on the role of metformin as an anti-cancer agent, there is potential for metformin to be used as adjuvant treatment in these cancer subtypes. To facilitate the development of metformin as a potential clinically useful anti-cancer drug for thyroid cancer, a detailed understanding of its effects on different types of thyroid cancer is required. The aim of this study is to determine its effects on the growth and proliferation of several thyroid cancer and normal cell lines at concentrations relevant to clinical settings.
Material and methods
Cell culture
Nthy-ori 3-1 (human thyroid follicular epithelium immortalised by transfection with a plasmid containing an origin-defective SV40 genome (SV-ori)), K1E7 (a subclone of K1 cell line from primary papillary thyroid cancer) and FTC-133 (derived from primary follicular thyroid cancer) were available within the department and were authenticated by STR profiling. RO82-W-1 (derived from metastases follicular thyroid cancer), 8305C (derived from primary anaplastic thyroid cancer) and TT (derived from primary medullary thyroid cancer) were purchased as authenticated cell lines from the European Collection of Cell Culture (ECACC). One additional breast cancer cell line (MCF7) on which the effect of metformin has been reported [30, 31] has been included for comparison. This was available in the department. All cell lines were cultured in DMEM containing 2 g/L glucose supplemented with 10% FBS, 1% fungizone and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 95% and 5% CO2.
Cell proliferation assay
Cell proliferation was determined using the MTT colorimetric metabolic activity assay (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide ((M5655-1G) purchased from Sigma) [32]. For each cell line, cells were seeded in 96 well plates (5000 cells per well) and treated in triplicate with different concentrations of metformin (Control, 0.03 mM, 0.1 mM, 0.15 mM, 0.3 m, 1 mM, 5 mM, 10 mM and 20 mM) (metformin (1,1-Dimethylbiguanide Hydrochloride, 5g stored at 4°C purchased from Enzo). A broad range of metformin concentrations was examined to establish the response to sub therapeutic and toxic levels of metformin. Proliferation was determined at specific time points (1, 2, 3, 4, 5 and 6 days) and the assay was repeated for each cell line on 3 separate occasions. For logistic reasons, subsequent investigations used a more focused range of metformin concentrations.
Clonal formation assay
A standard protocol to assess the ability of single cells to form a colony was followed as previously described [33]. Briefly, each cell line was cultured in 10cm petri dishes (500 cells per dish). At 24 hours, fresh media containing a more focused range of metformin was added (0.03 mM, 0.1 mM, 0.3 mM, 1 mM, and 5 mM) and cells were incubated for 14 days. The assay was repeated for each cell line on 3 separate occasions.
Scratch assay or cell migration assay
Cells were cultured in silicone inserts with a defined cell free gap in each chamber. After 24 hours, the silicone insert was removed and the medium were changed with medium containing metformin (control, 0.3 mM and 10 mM) and incubated for 24 hours. The cells were washed with PBS and stained with methylene blue. For each cell line the experiment was repeated on 3 occasions.
H2AX assay
Metformin has been previously shown to induce DNA damage in mammalian cells [34]. To investigate this, the H2AX assay (H2A.X protein gamma-H2A.X) was used to measure DNA Double Strand Breaks (DNA-DSBs), as phosphorylation of H2A.X is an effective measure of DSBs [35], and was performed as previously reported [36]. For each cell line, cells were seeded (2x 104 cells per slide) into 6 well plates and after 24 hours, the wells corresponding to each cell line were divided in groups and subject to different combinations of treatments:
a) Metformin (20 mM) + radiation (2Gy IR).
b) Metformin (0.1 mM) + radiation (2Gy IR).
c) Radiation (2Gy IR).
d) Metformin (20 mM).
e) Metformin (0.1 mM).
f) Control (not treated with either metformin or IR)
Cells labelled with metformin were treated with medium containing metformin (0.1 mM and 20 mM) for 6 days. After 6 days of incubation with metformin, cells in the IR group were irradiated with 2Gy IR using a Cesium137 irradiator (CIS Bio International). Post IR 3 times points were assessed (0.5 hour, 1 hour and 2 hours), allowing recovery of cells after DNA damage. The same time points were also examined in the control and metformin only conditions. Cells were fixed by adding 1.5 ml of 4% PFA then 2ml of 0.2% Triton X-100 in PBS and blocked by adding 1.5ml of blocking buffer (10% normal goat serum in PBS). 100µl of Phospho-histone rabbit anti-у-H2A.X (purchased from Cell signaling technology®) was added on each coverslip and incubated for 24 hours at 4°C in a humidity chamber. Cells were washed 3 times with PBS (5 minutes wash on the rotate shaker). They were incubated with 100µl of Cy3 goat anti-rabbit IgG (purchased from Invitrogen™, Molecular Probes®) in the dark for 1 hour at room temperature. The numbers of γH2AX foci were counted by using Cytovision software (Newcastle upon Tyne, UK) in 100 cells per slide via fluorescent microscope with CCD camera at 100x magnification. For each cell line, the experiment was repeated in triplicate wells and on 3 occasions.
Flow cytometric analysis for apoptosis assay
Cells were cultured in 6 well plates (4 x 105 cells per well) with 2 ml of medium. Fresh medium containing various metformin concentrations (control, 0.1 mM, 0.3 mM, 1 mM and 10 mM) was added at 24 hours and incubated for 6 days (37°C, 95% humidity, 5% CO2). Cells were stained with 5µl of FITC anexin V and 5µl of propidium iodide (PI) (FITC Annexin V Apoptosis Detection Kit (Cat: 556547) purchased from BD Biosciences), and analysed with flow cytometer (FACS) (BD Biosciences) using Cell Quest Pro software. For each cell line, the experiment was repeated on 3 occasions.
Flow cytometric analysis for cell cycle assay
Cells were cultured in 6 well plates (4 x 105 cells per well) and incubated for 24 hours. Cell lines were treated with different concentrations of metformin (control, 0.3 mM, 1 mM and 10 mM) and incubated for 6 days, fixed with 70% ethanol and left overnight at 4°C. Cells were treated with 50 µl of ribonuclease (100µg/mL solution) for 15 minutes at room temperature, followed by 200µl of propidium iodide (PI) (50 µg/mL stock solution) (Propidium iodide 1mg/ml (ab14083) was purchased from Abcam) and left for 2 hours at 4°C. The samples were analysed with flow cytometer (FACS) (BD LSRII) using FACSDiva 7.0 software. For each cell line the experiment was repeated on 3 occasions.
Statistical analyses
All the data was non-parametric and the statistical analyses were performed using Graphpad Prism software v. 6 for Mac. Kruskal-Wallis test was used to compare the results of the experiments for the treated (cells treated with different concentrations of metformin) and untreated (cells not treated with metformin) groups.
Results
Metformin inhibits cell proliferation, colony formation and migration of thyroid cancer cell lines
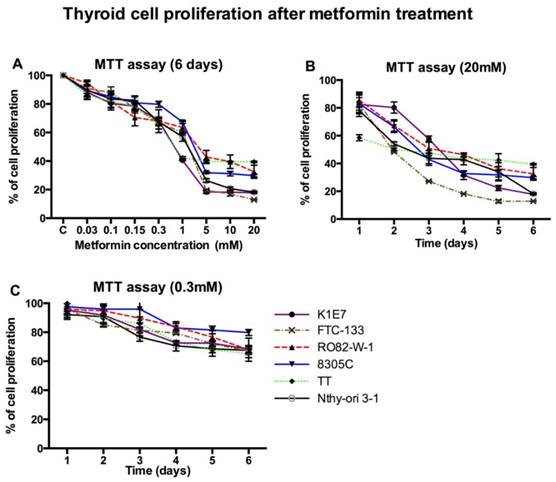
Metformin inhibited cell proliferation of all thyroid cancer cell lines and Nthy-ori 3-1 at concentrations of 0.03 mM and above in a time dependent manner (figure 1). The minimum statistically significant concentration of metformin that inhibited cell proliferation was 5 mM, at day 3 for K1E7 (p=0.01), FTC (p=0.04) and TT (p=0.01); day 4 for 8305C (p=0.02) and Nthy-ori 3-1 (p=0.02); day 5 for RO82-W-1 (p=0.02); and day 1 for MCF7 (p=0.03) (data not shown). The anti-proliferative effects on all cell lines were significantly enhanced at higher concentrations (figure 1b). Even at low physiological doses (0.3 mM) longer exposure reduced proliferation, although not significantly (figure 1c).
The effect of increasing concentrations of metformin on thyroid cancer cell lines over 6 days was accessed by the MTT assay. Each data point represents the mean of three experiments performed in triplicate. Error bars show the standard error of mean (SEM) for the 3 experiments. Panel A shows proliferation after six days with various concentrations of metformin. The supra-physiological and physiological concentrations of metformin decreased thyroid cell proliferation in time dependent manner as shown in panel B and C respectively.
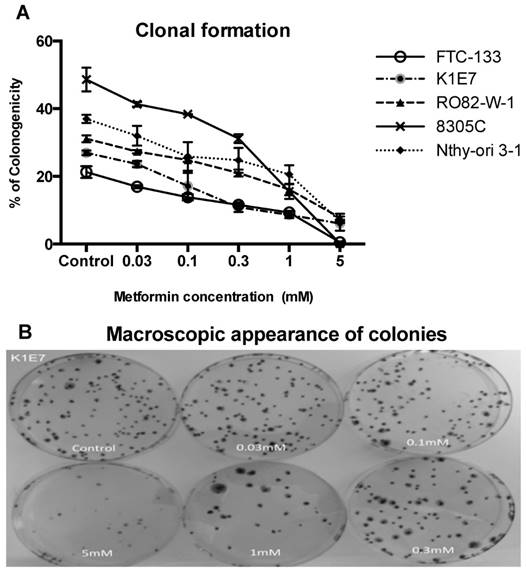
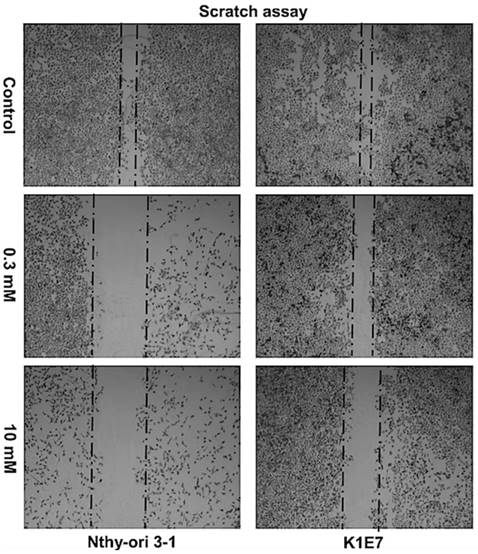
Inhibition of colony formation, although not significant was evident at metformin concentrations of 0.03 mM. The minimum statistically significant concentration of metformin that was demonstrated to inhibit colony formation was 1 mM for K1E7 (p=0.02), FTC-133 (p=0.02), RO82-W-1 (p=0.03) and 8305C (P=0.02) and 5 mM for Nthy-ori 3-1 cells (p=0.005) (figure 2). Colony formation was further decreased by increasing the concentration of metformin (figure 2a & 2b). The clonogenic assay was also studied on the TT cell line but the TT cells did not form colonies. The inhibitory effect of metformin can be seen even earlier (although not significant) when considering the results of cell migrations assays. Metformin inhibited cell migration at 24 hours (figure 3), and the effect was seen at low physiological concentrations with 0.3 mM metformin showing clear inhibition of migration and 10mM providing evidence of cell stress. Lower concentrations of metformin were not investigated.
Metformin inhibition of colony formation. Panel A demonstrates percentage of cells that formed colonies (y-axis) in various thyroid cancer cell lines at 14 days over a range of concentrations of metformin (x-axis). Each data point represents the mean of 3 experiments done in triplicate. Error bars show the standard error of mean (SEM) for the 3 experiments. Panel B shows macroscopic morphology of colony formation of K1E7 cells. After 14 days, pictures were taken of colonies of untreated cells and cells treated with various concentrations of metformin.
Metformin inhibited cell migration of all thyroid cancer cell lines (only data for K1E7 and Nthy-ori 3-1 is presented). After 24 hours, pictures of cells were taken of untreated cells and cells treated with metformin (0.3 mM and 10 mM) using the Nikon glipse TS100 microscope at 4x magnification. This experiment repeated 3 times independently for each cell lines.
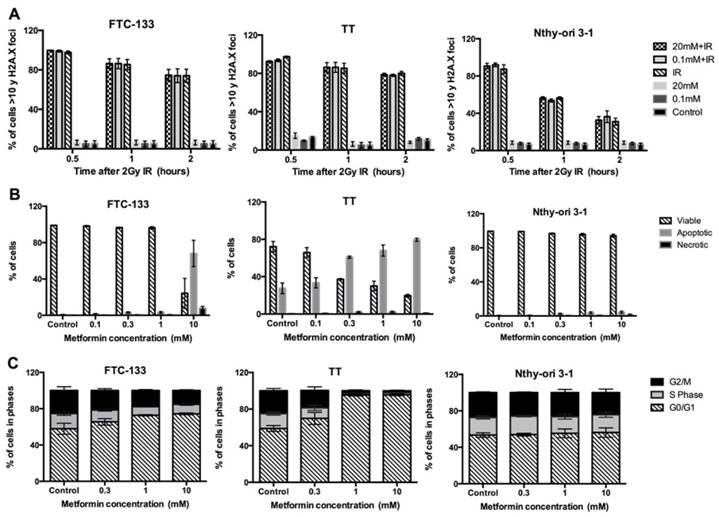
Metformin had no effect on the DNA repair in thyroid cancer cell lines
In untreated cells (control), and cells treated with metformin (0.1 mM and 20 mM) very low levels of phosphorylation of γH2AX foci were detected at the comparable post irradiation time points (figure 4a). Following radiation for the K1E7 cell line, the percentage of cells with more than 10 γH2AX foci was 99.6% at 30 minutes, 83% at 1 hour and 76% at 2 hours. Similar results were observed in FTC-133, RO82-W-1, 8305C, TT and Nthy-ori 3-1 cells.
Effects of DSBs-DNA and DNA repair, apoptosis and cell cycle arrest. Panel A demonstrated as percentage of cells > 10 γH2AX foci (y-axis) over time in hours (x-axis) in thyroid cell lines treated either with metformin (0.1 mM or 20 mM) for 6 days; metformin followed by radiation (2Gy); or with 2Gy radiation only as shown in different colours. Panel B shows the percentage of apoptotic, necrotic and viable cells (y-axis) after treatment with different concentrations of metformin for 6 days (x-axis). Panel C shows the percentage of viable cells in different phases (y-axis) after treatment with metformin (x-axis) for 6 days. In all panels each value represents the mean of 3 experiments. Error bars show SEM reported for the 3 experiments and only 3 cell lines presented.
Metformin induces cell cycle arrest and apoptosis in thyroid cancer cell lines
Apoptosis at concentrations of 0.1 mM and above was observed for all cell lines and increased in a dose dependent manner (figure 4b). Similar results were observed in K1E7, FTC-133, RO82-W-1, 8305C, Nthy-ori 3-1 and MCF7 but the TT cells already had high levels of apoptosis and were more sensitive to metformin compared to other cells. The minimum statistically significant concentration of metformin that induced apoptosis was 10 mM for K1E7 (P=0.01), FTC-133 (p=0.007), 8305C (P=0.01), TT (P=0.01), Nthy-ori 3-1 (p=0.04) and MCF7 (P=0.02). For the RO82-W-1 cell line, statistical significance could not be demonstrated at 10 mM of metformin (P>0.9). It is also of interest that the Nthy-ori 3-1 normal thyroid cells appeared less responsive, with apoptosis only marginally increased (p=0.04), since cell cycle arrest appeared unaltered in Nthy-ori 3-1 following metformin treatment (figure 4c). Thyroid cancer cell lines however, in particular TT, did demonstrate cell cycle arrest with increased number of cells in the G0/G1 phase from a concentration of 0.3 mM metformin (figure 4c).
Discussion
The current study evaluated a spectrum of thyroid cancer cell lines representing the different types seen in clinical practice. Initially, proliferation of all cell lines (normal thyroid, thyroid cancers and breast cancer cell line MCF-7) was studied following exposure to a broad range of metformin concentrations, accounting for supra-physiological to sub-physiological concentrations (figure 1). Our findings showing high dose metformin reducing proliferation, support earlier investigations on thyroid cancer [21, 37] and the control breast cancer cell line MCF-7 [30, 38]. In previous studies however, cells were treated with high concentrations of metformin for relatively short periods of up to 3 days [22]. In this study, metformin was also tested at therapeutic doses over a longer time period (1 -14 days). These results suggest that at lower physiological doses (0.01-0.3 mM) [23, 24], metformin although not significantly reducing proliferation, does dampen cell proliferation in cancer and normal (Nthy-ori 3-1) cell lines (figure 1c).
When combining these observations with those of the clonogenic assays (figure 2), the dampening effect appears enhanced over time, with the lowest significant dose (affecting proliferation) dropping from 5 mM at day 3 to 1 mM metformin at day 14 significantly inhibiting colony formation (figures 1 and 2). The effects of metformin on clonogenicity have been intensively investigated in other cancers and also thyroid cancer cell lines. As with these earlier studies [22, 30, 39] we found that supra-physiological concentrations of metformin (1 mM and 5 mM) virtually obliterated colony formation for some cell lines (8305C and FTC -133). Furthermore we found that at concentrations above 5 mM no colonies were formed and cells at these concentrations at 14 days were dead (data not shown). The action of metformin appears wide ranging, and whilst only high doses clearly affect proliferation at 24hrs (figure 1b) its effect on other processes is evident even at lower doses, with 0.3 mM inhibiting migration in the scratch assay (figure 3), suggesting that the effects of metformin are wide ranging and target different pathways to varying degrees.
How metformin exerts its action on thyroid cancer cells is unclear, but unlike previous investigations [34] we did not find that exposure to metformin induced DNA damage; even treatment with 20mM Metformin for 6 days (prior to measuring DNA damage) although significantly reducing proliferation did not increase DNA DSB's in the absence of IR (figure 4a). It is thought that the growth inhibitory effect of metformin on cancer cell proliferation may possibly be through the activation of AMPK via activation of LKB1 [40], which causes inactivation of mammalian target of rapamycin (mTOR) [41]. In numerous human cancers, mTOR increased cell growth and proliferation [42]. In addition, the growth inhibitory effect of metformin on tumour growth may be by indirect effects by decreasing circulatory tumour growth factors such as glucose [6, 43], insulin [43] and TSH [28, 29]. Insulin and glucose play a critical role in tumour growth and malignant transformation through stimulation of insulin and IGF1 receptors [44-47]. As TSH specifically promotes thyroid cancer growth in rat [48] and mouse models [49] it may suggest that metformin would be specifically effective against thyroid cancers. Indeed evidence from this study suggests this may be the case, when the observations on apoptosis and cell cycle are considered.
Our findings demonstrated that metformin induced apoptosis and cell cycle arrest in thyroid cancer cells, increasing both the percentage of apoptosis cells and accumulating cells in G0/G1 phase (figures 4b, 4c). Similar findings were reported in other studies on different cancers, including thyroid [22, 50, 51]. It is however of interest that the normal thyroid cell line Nthy-ori 3-1 seemed to be relatively unaffected by metformin even at doses of 10 mM at 6 days, with only low levels of apoptotic cells, negligible necrosis and no change to the number of cells in GO/G1 (figures 4 b and c). Furthermore, colony formation for all thyroid cancer cell lines was inhibited at 1mM, but for Nthy-ori 3-1 significance was at 5mM. In contrast, TT (medullary thyroid cancer cell line) had naturally high levels of apoptosis, which increased dramatically on exposure to metformin, and at 10 mM virtually obliterated cell division and stalled the majority of the cells in GO/G1 (figures 4b and 4c). The observations of apoptosis and cell cycle assist in explaining the varying proliferative responses of the thyroid cancers to metformin. For example at high does (20mM), proliferation of TT is blocked even on day 1 (figure 1b), presumably because metformin at 10mM induced higher apoptosis in this already high turnover cell line, but more importantly stalls cells at G0/G1. The high level of apoptosis helps to explain the acute sensitivity of the TT line to metformin, since low doses of 0.3mM (figure 1c) induce the most inhibition for TT (although not reaching statistical significance) of all lines at day 6. Apparently in contrast to this observation high doses of 20 mM (figure 1b) have the least effect on TT at 6 days. It seems that the high natural rate of apoptosis in TT means that it is the most sensitive to low doses (figure 1a and c) but that high does will have a catastrophic effect even at day 1 (figure 1b) that tips the balance in favour of apoptosis and stalls the remainder of the cells in GO/G1 (figure 1b and 4 b and c). As only one medullary thyroid cancer cell line was studied, it is not possible to say if this subtype is particularly sensitive, or whether the naturally high level of apoptosis affects the response. Recently however it has been found that the RET induced medullary thyroid cancers increase apoptosis in response to therapy targeting the RET oncogene [52, 53] with inhibition of the MAPK and mTOR pathways, supporting other studies suggesting metformin targets these pathways [54]. These observations in combination with our findings suggest that different subtypes of thyroid cancer may be more sensitive to metformin than others, although it is too early to say whether is clinically applicable. Variation in subtypes may explain why a recent study has not found any decreased risk of developing thyroid cancer for metformin users [55].
Although this study did not include surgical samples and only immortalized lines were available; our current investigation is the first in vitro study that looks at longer term exposure to low dose metformin. Practically it is not possible to increase the exposure due to technical constrains, but our investigation has observed that the cumulative effects of metformin increase over time for all types of thyroid cancer, and suggests that even low sub-physiological doses alter behavior and can act to dampen down the proliferative response. In combination, our results possibly explain why prolonged exposure to therapeutic doses, as given to diabetic patients, may account for the observed lower level of cancers in these populations and are associated with a favorable outcome for thyroid cancer [56].
Conclusions
In summary, metformin inhibits all thyroid cancer cell lines, and its action is wide ranging impacting on cell proliferation, apoptosis and cell cycle arrest, as well as colony formation and migration. Our findings suggest that even at low does metformin has the capability to exert an effect, which over time will be cumulative. For some forms of thyroid cancers, such as the medullary thyroid cancer, these effects may be enhanced. Further work is aimed at understanding the molecular mechanisms of the above effects of metformin and its interactions with TSH and glucose in the environment.
Acknowledgements
The authors thank Rare Tumor Research Group (RTRG), Department of Oncology and Metabolism, University of Sheffield for their support for this project.
Funding
This work is funded by the Human Capacity Development Program (HCDP) in higher education in Kurdistan Regional government-Iraq (KRG).
Conflict of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
1. Vanderpump M. The epidemiology of thyroid disease. British medical bulletin. 2011;99:39
2. Middendorp M, Grünwald F. Update on recent developments in the therapy of differentiated thyroid cancer. Seminars in nuclear medicine: Elsevier. 2010:145-52
3. Sipos J, Mazzaferri E. Thyroid cancer epidemiology and prognostic variables. Clinical Oncology. 2010;22:395-404
4. Reverter JL, Colome E. [Potential risks of the adverse effects of thyrotropin suppression in differentiated thyroid carcinoma]. Endocrinol Nutr. 2011;58:75-83
5. Sahra IB, Le Marchand-Brustel Y, Tanti J-F, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Molecular Cancer Therapeutics. 2010;9:1092-9
6. Nicholson G, Hall G. Diabetes mellitus: new drugs for a new epidemic. British journal of anaesthesia. 2011;107:65-73
7. Micic D, Cvijovic G, Trajkovic V, Duntas LH, Polovina S. Metformin: its emerging role in oncology. Hormones (Athens). 2011;10:5-15
8. Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC medicine. 2011;9:33
9. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Research Pointers: Metformin and reduced risk of cancer in diabetic patients. BMJ: British Medical Journal. 2005;330:1304
10. Currie C, Poole C, Gale E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52:1766-77
11. Monami M, Lamanna C, Balzi D, Marchionni N, Mannucci E. Sulphonylureas and cancer: a case-control study. Acta diabetologica. 2009;46:279-84
12. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New Users of Metformin Are at Low Risk of Incident Cancer A cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620-5
13. Ruiter R, Visser LE, van Herk-Sukel MP, Coebergh J-WW, Haak HR, Geelhoed-Duijvestijn PH. et al. Lower Risk of Cancer in Patients on Metformin in Comparison With Those on Sulfonylurea Derivatives Results from a large population-based follow-up study. Diabetes Care. 2012;35:119-24
14. Bosco JLF, Antonsen S, Sørensen HT, Pedersen L, Lash TL. Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiology Biomarkers & Prevention. 2011;20:101-11
15. Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes & Control. 2009;20:1617-22
16. Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, Melani C. et al. Metformin and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care. 2011;34:129-31
17. Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes impact of metformin on survival. Diabetes Care. 2012;35:299-304
18. Romero IL, McCormick A, McEwen KA, Park S, Karrison T, Yamada SD. et al. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstetrics and gynecology. 2012;119:61-7
19. Tan BX, Yao WX, Ge J, Peng XC, Du XB, Zhang R. et al. Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer. 2011;117:5103-11
20. Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin Associated With Lower Cancer Mortality in Type 2 Diabetes ZODIAC-16. Diabetes Care. 2010;33:322-6
21. Klubo-Gwiezdzinska J, Jensen K, Costello J, Patel A, Hoperia V, Bauer A. et al. Metformin inhibits growth and decreases resistance to anoikis in medullary thyroid cancer cells. Endocrine-related cancer. 2012;19:447-56
22. Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. The Journal of clinical endocrinology and metabolism. 2012;97:E510-20
23. Schulz M, Schmoldt A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Die Pharmazie-An International Journal of Pharmaceutical Sciences. 2003;58:447-74
24. Lalau JD, Lemaire-Hurtel AS, Lacroix C. Establishment of a database of metformin plasma concentrations and erythrocyte levels in normal and emergency situations. Clinical drug investigation. 2011;31:435-8
25. Hawley S, Boudeau J, Reid J, Mustard K, Udd L, Mäkelä T. et al. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. Journal of Biology. 2003;2:28
26. Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L. et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast cancer research and treatment. 2011;128:783-94
27. Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N. et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast cancer research and treatment. 2012;135:821-30
28. Vigersky RA, Filmore-Nassar A, Glass AR. Thyrotropin suppression by metformin. Journal of Clinical Endocrinology & Metabolism. 2006;91:225-7
29. Cappelli C, Rotondi M, Pirola I, Agosti B, Gandossi E, Valentini U. et al. TSH-Lowering Effect of Metformin in Type 2 Diabetic Patients Differences between euthyroid, untreated hypothyroid, and euthyroid on L-T4 therapy patients. Diabetes Care. 2009;32:1589-90
30. Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE. et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909-15
31. Hadad SM, Hardie DG, Appleyard V, Thompson AM. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2014;16:746-52
32. Sylvester PW. Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods in molecular biology (Clifton, NJ). 2011;716:157-68
33. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature protocols. 2006;1:2315-9
34. Amador RR, Longo JP, Lacava ZG, Dorea JG, Almeida Santos Mde F. Metformin (dimethyl-biguanide) induced DNA damage in mammalian cells. Genetics and molecular biology. 2012;35:153-8
35. Foster ER, Downs JA. Histone H2A phosphorylation in DNA double-strand break repair. Febs J. 2005;272:3231-40
36. Hoh L, Gravells P, Canovas D, Ul-Hassan A, Rennie IG, Bryant H. et al. Atypically low spontaneous sister chromatid exchange formation in uveal melanoma. Genes, Chromosomes and Cancer. 2011;50:34-42
37. Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. Journal of Clinical Endocrinology & Metabolism. 2012;97:E510-E20
38. Rocha GZ, Dias MM, Ropelle ER, Osório-Costa F, Rossato FA, Vercesi AE. et al. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clinical Cancer Research. 2011;17:3993-4005
39. Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10:2959-66
40. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA. et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642-6
41. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer res. 2007;67:10804-12
42. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-84
43. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. Journal of molecular endocrinology. 2012;48:R31-R43
44. Hardie DG. AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25:1895-908
45. Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. International Journal of Cancer. 2012;131:752-9
46. Bo S, Ciccone G, Rosato R, Villois P, Appendino G, Ghigo E. et al. Cancer mortality reduction and metformin: a retrospective cohort study in type 2 diabetic patients. Diabetes, Obesity and Metabolism. 2012;14:23-9
47. Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A. et al. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. 2008;114:23-37
48. Goldberg RC, Lindsay S, Nichols CW Jr, Chaikoff IL. Induction of Neoplasms in Thyroid Glands of Rats by Subtotal Thyroidectomy and by the Injection of One Microcurie of I-131. Cancer res. 1964;24:35-43
49. Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer res. 2007;67:8002-6
50. Malki A, Youssef A. Antidiabetic drug metformin induces apoptosis in human MCF breast cancer via targeting ERK signaling. Oncology research. 2011;19:275-85
51. Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ. et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9:e98207
52. Gild ML, Landa I, Ryder M, Ghossein RA, Knauf JA, Fagin JA. Targeting mTOR in RET mutant medullary and differentiated thyroid cancer cells. Endocr Relat Cancer. 2013;20:659-67
53. Gild ML, Bullock M, Pon CK, Robinson BG, Clifton-Bligh RJ. Destabilizing RET in targeted treatment of thyroid cancers. Endocrine connections. 2016;5:10-9
54. Han B, Cui H, Kang L, Zhang X, Jin Z, Lu L. et al. Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:6295-304
55. Becker C, Jick SS, Meier CR, Bodmer M. No evidence for a decreased risk of thyroid cancer in association with use of metformin or other antidiabetic drugs: a case-control study. BMC Cancer. 2015;15:719
56. Jang EK, Kim WG, Kwon H, Choi YM, Jeon MJ, Kim TY. et al. Metformin Is Associated with a Favorable Outcome in Diabetic Patients with Cervical Lymph Node Metastasis of Differentiated Thyroid Cancer. European thyroid journal. 2015;4:181-8
Author contact
![]() Corresponding author: Safar Kheder, Department of Oncology and Metabolism, University of Sheffield, S10 2RX. E-mail address: mdp12shkac.uk or safardeloyicom
Corresponding author: Safar Kheder, Department of Oncology and Metabolism, University of Sheffield, S10 2RX. E-mail address: mdp12shkac.uk or safardeloyicom