Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(7):1205-1213. doi:10.7150/jca.18255 This issue Cite
Review
E2F8 is a Potential Therapeutic Target for Hepatocellular Carcinoma
Yi Lv1, 2, Jia Xiao 1, Jing Liu3 ![]() , Feiyue Xing1, 2
, Feiyue Xing1, 2 ![]()
1. Department of Immunobiology, Institute of Tissue Transplantation and Immunology, Jinan University, Guangzhou, China;
2. Key Laboratory of Functional Protein Research of Guangdong, Higher Education Institutes, Jinan University, Guangzhou, China;
3. Department of Stomatology, Jinan University, Guangzhou, China.
Received 2016-11-8; Accepted 2017-1-11; Published 2017-4-9
Abstract
E2F transcriptional factors are widely expressed in a number of tissues and organs, possessing many regulatory functions related to cellular proliferation, differentiation, DNA repair, cell-cycle and cell apoptosis. E2F8 is a recently identified member of the E2F family with a duplicated DNA-binding domain feature discriminated from E2F1-6, controlling gene expression in a dimerization partner-independent manner. It is indispensable for angiogenesis, lymphangiogenesis and embryonic development. Although E2F8 and E2F7 perform complementary and overlapping functions in many cell metabolisms, E2F8, but not E2F7, overexpresses remarkably in hepatocellular carcinoma (HCC) to facilitate the HCC occurrence and development via activating a E2F1/ Cyclin D1 signaling pathway to regulate the G1- to S-phase transition of cell cycle progression or transcriptionally suppressing CDK1 to induce hepatocyte polyploidization. It also involves closely a variety of cellular physiological functions and pathological processes, which may bring a new breakthrough for the treatment of certain diseases, especially the HCC. Here, we summarize the latest progress of E2F8 on its relevant functions and mechanisms as well as potential application.
Keywords: E2F8, Hepatocellular carcinoma, angiogenesis, lymphangiogenesis, polyploidization, DNA damage.
Introduction
E2F family of transcriptional factors has many regulatory functions implicating cellular proliferation, differentiation, DNA repair, cell cycle and cell apoptosis [1, 2]. The study of the E2F family began in 1980s, of which most extensive researches involve E2F1 [3]. As yet, E2F family has been found to have eight members in mammals, including from E2F1 to E2F8 [4]. The alternative splicing of some E2F family members and generation of different isoforms result in that the regulation of E2F functions is more complicated than other members [5]. E2F3 and E2F7 have two highly relevant isoforms, respectively [6]. Indeed, studies have shown that E2F evolution in plants and animals is very conservative but not in yeast [1]. According to their structure and function, E2F family can be divided into two groups (canonical E2F1-6 and atypical E2F7-8) and four subgroups (E2F1-3a, E2F3b-5, E2F6 and E2F7-8). Nowadays we get the difference between canonical and atypical E2F molecular structures, but it is still undefined for its evolutionary mechanism [3].
The canonical E2Fs possess a leucine-zipper domain in the N terminus, which is necessary for dimerization with a partner protein DP (dimerization partner). Except E2F6, there are also a transactivation domain and a retinoblastoma-binding domain for E2F1 to E2F5 in the C terminus [5, 7]. Based on functional division, E2F1-3a are usually considered to be a transcriptional activator. When overexpressed, it can drive G0-phase cells into S-phase, while E2F3b-6 are considered to be a transcriptional repressor [6]. The overexpression of E2F1-3a can drive G0-phase cells into S-phase, while the cells subjected to E2F1-3 triple knock-out were demonstrated to have defect for S-phase entry [6, 8]. In contrast, E2F3b-6 attach to the E2Fs-related regulatory promoters at the G0/G1 phase [9, 10]. The classic E2F-dependent transcriptional activation is executed by a RB/E2F/DP approach. E2F1-5 are associated with one of two DP (DP1/DP2) proteins and one member of pRB (pRB/p107/p130) family from a tripolymer [11, 12]. In the G1 to S period of the cell cycle, RB is phosphorylated by the cyclin-dependent kinases (CDKs), resulting in the dissociation of RB from the RB-E2F complex and dysfunction of E2F inhibition [9, 13]. Due to the lack of the retinoblastoma-binding domain, E2F6 is considered to regulate E2F-targeted genes that are independent on pRB family proteins [14]. E2F6 does not be regulated by pRB family. It can repress transcription by recruitment of chromatin remodeling complexes that include polycomb group proteins to E2F-target promoters [12, 15, 16]. It is further clarified that E2F1, E2F2 and E2F3 predominantly connect with pRB, and E2F4 is associated with all of the pRB/p107/p130 proteins, whereas E2F5 tends to bind with p130 [3, 14].
The atypical E2F consists of E2F7 and E2F8. They have two distinct DNA-binding domains (DBD), but lack DP-dimerization, retinoblastoma-binding, and transcriptional activation domains contained in the canonical E2F. Thus, the atypical E2F binding to DNA is different from other E2F members, which does not rely on DP [7, 17]. E2F7 and E2F8 control cell cycle genes through homodimers or heterodimers from DBD, functioning predominantly as transcriptional repressors [5, 18] (Fig.1). E2F8 is a recently discovered member of the E2F family. Christensen et al. noticed that there were several murine and human expressed sequence tags showing high similarity to E2F7, using the E2F7 sequence as query searching in the GenBank. Thus, they have designated the cloned transcripts as E2F8 [19]. E2F8 shows high similarity to E2F7 in structure and has two special domains exhibiting a high degree of resemblance to the E2F family in DNA-binding domain [8]. E2F8 shares the unique structure of E2F7 and performs overlapping functions in many cell metabolisms [16].
Structures of the E2F family members: E2F8 has the duplicated DNA-binding domain (DBD), but lacks the dimerization partner (DP) and retinoblastoma-binding (RB) domains.
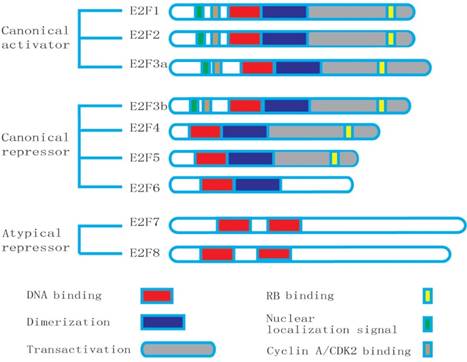
In spite of that the DNA binding domain of E2Fs is conservative, there is a lot of accumulated evidence that they also bind and regulate disparate sets of target genes through interactions with specific cofactors [20]. In fact, E2F family works more sophisticatedly than it is simply divided into an activator and repressor. Sometimes, they can play as either an activator or a repressor of transcription that depends on target genes, cofactors and cellular context. More importantly, E2Fs usually work as a balanced network [3].
Structural properties of E2F8
Accessions NM 024680 (human) and XM149937 (mouse) were early identified as E2F8. They encode the FLJ23311 protein of Homo sapiens and the RIKEN cDNA 4432406C08 gene of Mus musculus, respectively [19]. NM 024680 and XM149937 include highly homologous ORFs that have the ability to encode proteins consisting of 867 and 860 amino acids, the molecular weights of which are 94272 and 93382 Da, respectively [2].
E2F8 shows a high resemblance to E2F7. E2F8 is also found to be the high conservation and unique repeat DNA-binding domains. The result of alignment test shows that the human E2F8 has a 31.9% homology with the E2F7 in overall and the identity area mainly focuses on the DNA binding domain [19]. The conserved RRXYD motif is necessary for DNA binding in the E2F family. E2F8 presents the conserved motif in domain 1 (RRIYD) and domain 2 (RRLYD), respectively. The latest data demonstrate that they are located in the residues 113-182 and 261-347 of the E2F8's N-terminal. These two domains of E2F8 are highly similar to the DNA-binding domains of E2F1 to E2F7 [2]. In fact, the similarity of DNA binding domains in E2F8 and E2F7 is far more than the conserved RRXYD motif, which extends to about 120 amino acid residues. The sequence between the domain 1 and domain 2 in E2F8 and E2F7 contains about 160 residues, appearing highly similar [2]. Like E2F7, E2F8 lacks a marked box, DP-dimerization, pRb-binding and transcriptional activation domains required for other E2F family members [19]. E2F8 has duplicated DNA-binding domains and three putative nuclear localization signal sequence (NLS) domains. The integrity of DNA-binding domains is essential for combination with consensus E2F-binding DNA elements. The amino acid mutation of E2F8 in the conserved RRXYD motif will result in failure to bind to DNA [2]. Similar to E2F7, the increasing expression of E2F8 negatively influences E2F-target genes, and represses cellular proliferation [19]. E2F7 and E2F8 can form homodimers or heterodimers. They usually present in the same adult tissues. These results suggest that the both may have partial overlapping and perhaps synergistic functions [2]. The similarity of genomic structure and function of the E2F8 and E2F7 strongly supports that they probably originate from a same ancestral gene.
Both of human and murine E2f8 genes are made up of 13 exons and 12 introns. The human and murine E2f8 genes are located in the short arm of chromosome11p15.1 and chromosome 7, respectively [8]. Their initiation codon ATGs are located in the second exons, and the termination codon TGAs present in the exon 13 [19]. The human E2f8 mRNA consists of 323bp long 5'-UTR, 2604bp long ORF, and 610bp long 3'-UTR, appearing in a variety of tissues. The mouse E2f8 mRNA comprises 490 bp long 5'-UTR, 2583 bp long ORF and a 632 bp long 3'-UTR. They have a consensus polyadenylation signals ATTAAA that is located in upstream of the poly (A) tail, respectively [8]. The similarity of sequence and genomic structure of the human and murine E2F8 genes strongly supports that they are true orthologues. The entire E2F8 protein is necessary for fully functional activity, but the first 200 residues are more important in the majority of the signaling activity. Molecular weight of E2F8 has slight differences reported in literatures. Predicted molecular mass of the human and mouse E2F8 proteins is about 105KD and 95KD, respectively [8]. E2F8 is predominantly expressed in liver, thymus, skin and testis, but not in the brain, stomach, and muscle [8]. Studies have shown that the E2F8 is very prone to degradation so that the level of the endogenous protein E2F8 is relatively low [19].
E2F8 regulates the cell cycle and cellular proliferation
According to the current knowledge of E2F family, we know that E2Fs play a key role in the regulation of cell cycle [13]. The E2Fs control transcription of the cell cycle-related genes in the different phases of the cell cycle [21, 22]. E2F3b-5 bind to E2F-regulated promoters to recruit chromatin remodeling factors in the G0/G1 phase of the cell cycle by dissociating with the pRB family proteins [23]. When cells reenter into late G1 phases, the CDKs phosphorylate RB and the free activators E2F1-3a replace the repressor E2F complexes [10]. The activators E2F1-3 reach the peak in this G1/S phases. Then, they are attenuated in G2 [9]. E2F1-3 also possess the ability to overcome arrest by the cyclin-dependent kinase inhibitor and induce quiescent cells to enter an S-phase [23, 24]. On the other hand, the group of atypical repressors play a vital role in the cell cycle exit, promoting the process of cell differentiation [7].
E2F8 is not only similar to E2F7 in structure, but also has a lot of overlap in many metabolic functions [8, 20]. The synchronization treatment of HeLaS3 cells and qPCR analysis show that the E2F8 reaches the peak at early S phase, then drops after through S phase ,which is consistent with E2F7 [19, 25]. Ectopic expression of E2F7 and E2F8 causes the delayed cell cycle progression and the increased proportion of G1 cells [2]. E2F7 and E2F8 have an overlapping mechanism in the control of cell cycle. They reciprocally compensate to repress a network of fluctuant cell cycle genes to modulate S-phase progression [2, 25]. E2F7 and E2F8 can compete with E2F1 to combine with the promoters of E2Fs-targeted genes, inhibiting E2F1-activated gene transcription. On the other hand, E2F7 and E2F8 can directly bind to E2F1 to repress the expression of E2F1 [26]. E2F8 is also one of the E2F-targeted genes, and several family members (E2F1, E2F3, E2F4 and E2F7) have been found to combine to the E2F8 promoter, suggesting that there is a feedback-loop in E2Fs [19, 27]. E2F8 plays as a repressive arm component in monitoring the E2F network, coordinating the cell cycle progression [2].
E2Fs are to be critical players in orchestrating the control of cellular proliferation [28, 29]. The roles of each family member in controlling cellular proliferation, cell cycle transition and apoptosis are different. The expressions of atypical E2Fs during the cell cycle are different from canonical E2Fs, and the former peak levels are found in the S/G2 phase. Interestingly, the ectopic expression of E2F8 leads markedly to the inhibition of E2F-targeted gene expression and reduces the proliferation capacity of mouse embryonic fibroblasts, whereas over-expression of E2F8 in liver cancer cells could promote cell proliferation and tumor generation [8, 29]. The specific mechanism for the opposite phenomenon of cellular proliferation is still not clear, needing to be further studied. These results also suggest that E2f8 may be an oncogene in the progression of HCC.
E2F8 with E2F7 is essential for the placenta architecture and embryonic development
A large number of studies suggest that E2F functions are involved in regulating a variety of cellular metabolism, and playing a key role in cellular homeostasis [15]. In fact, E2F function is closely related to the cellular context, whereas in vivo environment is more complicated than in vitro one. In order to further explore E2F7 and E2F8 functions in vivo, scientists utilized a homologous recombination technique and a conditional knockout technology to interfere with E2F7 and E2F8 functions in mice [16]. Indeed, genetic ablation of E2F7 and E2F8 demonstrates that at least one allele of E2f7 or E2f8 is necessary for embryonic development and viability. The lack of E2f7 or E2f8 in mice showed no significant effect on embryo development. However, their combined ablation resulted in massive apoptosis, dilation of blood vessels and hemorrhaging so as to finally induce embryonic death at day E11.5 [16]. Interestingly, E2F7 and E2F8 did not offer an equal contribution towards mice postnatal development. Studies have shown that E2f7+/-E2f8-/- mice are normal in postnatal development, but most of E2f7-/-E2f8+/- ones usually appear to be runts, died within the first three months of life [16]. In fact, E2F7 and E2F8 could form homodimers or heterodimers to bind to target DNA, which is not equivalent. E2F7:E2F7 is the first priority, and E2F7:E2F8 precedes E2F8:E2F8 [1,3]. These data may explain that E2F7 and E2F8 offer an unequal contribution towards mouse postnatal development, while their molecular basis is not yet clear [16].
It was confirmed that there were a large number of cell apoptosis in the E2f7 and E2f8 double knockout (DKO) embryos by immunohistochemistry with BrdU-specific antibodies and TUNEL at embryonic day 9.5 (E9.5) [16]. The sequential ChIP assays in HEK 293 cells showed that E2F7 and E2F8 could directly bind to E2f1. The PCR genotyping of genomic DNA assays in DKO mouse embryonic fibroblast (MEFs) demonstrated that ablated E2F7 and E2F8 would significantly up-regulate E2F1 with a concomitant increase of p53 protein [15]. E2f7+/-E2f8-/- mice were mated with either E2f1-/- or p53-/- mice in order to generate cohorts of triple knockout (TKO) embryos. The result of TUNEL assays of the TKO mice showed that the massive apoptosis caused by a deficiency in E2f7 and E2f8 could be rescued on E9.5 TKO embryos by the loss of either E2f1 or p53 [16, 30]. These results reveal that E2F7 and E2F8 can go through the E2F1-p53 axis to control cell apoptosis what is indispensable for the E2F network. Intriguingly, although cell apoptosis was rescued in both E2f7/8/1 and E2f7/8/p53 triple knockout mice, the extent of dilated vessels and hemorrhaging was still similar to DKO embryos when embryos were harvested at E10.5. In fact, because of the vascular defect of TKO, no embryos could exceed E12.5. These results suggest that TKO-rescued apoptosis is independent on vascular defects and embryonic lethality [16, 31]. DKO-resulted E2F7 and E2F8 target gene deregulation is likely to be involved in angiogenesis. At the same time, deletion of E2F7 and E2F8 resulted in a spectrum of embryonic defects impacting the vasculature and cell survival [31]. Their synergistic function may be considered as a special arm of the E2F network implicated in repression of transcription during S-G2, and the E2F1/p53 axis represents a particularly critical target that if not appropriately repressed can cause widespread apoptosis in developing embryos. These findings show that E2F7 and E2F8 are indispensable for embryonic development. They represent a critical role in cell survival and embryonic development by acting as a unique repressive arm [16, 30, 31].
Further researches reveal that the deficiency of E2F7 and E2F8 in trophoblast progenitor cells of placental is sufficient to kill embryos in mid-gestation. Indeed, a wild type placenta possesses the ability to support the E2f7-/- and E2f8-/- fetuses to birth and beyond. These support that the functions of atypical E2Fs in the extro-embryos are necessary and sufficient for mice embryonic development. Studies showed the TKO of E2f3a with E2f7/8 at the same time rescued placental defects and lethality of fetuses, suggesting that E2F3a resists E2F7 and E2F8 to mediate repression by activating the relevant transcriptional program, which is important for placental development [30, 32].
E2F8 mediates the DNA damage-correlated cell-cycle
The structure of atypical E2Fs is similar to a plant E2F-like (E2L) protein, which has two separate DNA-binding domains [33]. The function of the plant E2L has been involved in regulation of endo-replication so that atypical E2Fs may also have similar functions [34]. With a DNA-damaging agent etoposide or bleomycin to treat different mammal cell lines, including U2OS, T98G, MCF7, HeLa, HCT15, HCT116 and MEF, there were results similar to fluctuation of E2F7 and E2F8 which are concomitant with E2F1 [4]. The latter can regulate not only cell cycle, but also cell apoptosis [35-37]. Co-immunoprecipitation (Co-IP) and chromatin immunoprecipitation (ChIP) showed that E2F7 and E2F8 can interact to form dimmer to locate at the promoter of E2f1. The DD is independent on DBD, and mutant residues within the DBD do not influence the interaction [4]. Besides, the integrity of DD and DBD is necessary for forming dimers and performing biological functions [38, 39].
Either E2F7- or E2F8-specific siRNA into U2OS cells caused a significant acceleration of cell death, and their combined knockout led to worse results [4]. Consistently, the sensitivity of E2f7-/-E2f8-/- cells to DNA-damage-inducing drugs camptothecin and cisplatin showed that these drugs induced a marked increase in apoptosis of DKO MEFs, compared to wild type MEFs [16]. Because E2F1 has ability to control apoptosis, overexpression of E2F1 can lead to the apoptosis in the cells treated with DNA damage-inducing agents [40-42]. E2F1 siRNA transfected into DNA-damaged U2OS cells can partly rescue apoptosis, which is caused by defective E2F7 and E2F8 [4]. In summary, E2F7 and E2F8 mediate the DNA damage-correlated cell-cycle through modulating E2F1 activity [18].
Atypical E2Fs promote angiogenesis and lymphangiogenesis
With a large number of target genes identified, we have a further understanding of their other functions. Weijts et al deleted E2f7/8 at the same time in zebrafish and mice embryonic, respectively. The results showed an interesting phenomenon, i.e., embryonic death without proliferation defects, but displaying massive cell apoptosis and vascular defects [16, 30]. The specific mutant of E2f7/8 in the extra-embryonic trophoblastic cells also can lead to a poor structure of the placental vascular network [30]. Besides, embryos alone or federatively injected with E2f7/8 MOs (morpholino oligomers) and genetic mutant embryos of E2f7/8 showed different degrees of defects in angiogenesis and apoptosis, indicating that the E2f7/8 are necessary for angiogenesis. Intriguingly, deletion of E2f1 or p53 can rescue apoptosis caused by deficiency E2f7/8, but not do the defect of the blood vessels [31]. From these phenomena they reasonably speculate that E2F7/8 can regulate angiogenesis by an alternative mechanism.
As we know that vascular endothelial growth factor A (VEGFA) is an essential factor for angiogenesis, VEGFA acts as a chemoattractant through forming a correct concentration gradient to guide the formation of an appropriate vascular system [43-47]. The hypoxia-induced factor 1 (HIF1) is a major modulator of VEGFA in the background of angiogenesis [48-50]. The scarce Hif1α, Hif2α or Hifβ mice are similar to E2f7/8 knockout mice that also die due to vascular defects around E10.5 [51]. The injection of E2f7/8 mRNA increases the endothelial cell numbers to concomitantly raise the mRNA levels of vascular endothelial growth factor A (VEGFA), suggesting that E2f7/8 excite endothelial cell proliferation through up-regulation of the VEGFA expression. Chromatin immunoprecipitation (ChIP) further confirmed that E2f7/8 could directly bind to stimulate the VEGFA promoter in a way independent on canonical E2F-binding elements [31, 48]. Indeed, E2f7/8 stimulate the VEGFA promoter activity by binding with HIF1 to form a transcriptional complex independent on the canonical E2F-binding site. In sum, E2f7/8 cooperate with HIF1 to form a transcriptional complex to activate the VEGFA promoter, and then to raise the VEGFA level to promote angiogenesis.
After finding that atypical E2Fs (E2F7 and E2F8) can control angiogenesis, Weijts et al. also have a study on the E2F role in lymphangiogenesis [52]. The genome-wide microarray analysis on E2f7/8 deficiency in mouse fetuses at E10.5 revealed that only Ccbe1 and Flt4 were deregulated among the necessary genes for lymphangiogenesis. Ccbe1 and Flt4 contain a canonical E2f binding domain near the promoter, and ChIP showed that E2F7 and E2F8 can bind to their promoter [52]. The vascular endothelial VegfC/Flt4 (vascular endothelial growth factor receptor 3) signaling pathway is one of main ways to modulate the formation of lymphatic vessels [53, 54]. Collagen and calcium-binding EGF domain-containing protein 1 (CCBE1) was considered to enhance lymphangiogenesis by increasing the biological effect of VEGFC [55-57]. Atypical E2Fs can direct targets FLT4 and CCBE1, which was identified by a genome-wide approach. Loss of E2f7/8 resulted in deregulated expression of CCBE1 and FLT4, and impaired venous sprouting and lymphangiogenesis. Intriguingly, E2F7/8 modulated CCBE1 and FLT4 in opposite ways. Inactivation of E2f7/8 down-regulated CCBE1 and up-regulated Flt4. The expressional analysis in different cell lines (mesenchymal cells and endothelial cells) shows that E2f7/8 are almost equally expressed in these cell lines, while productions of CCBE1 and FLT4 are dependent on cell types [52, 58, 59]. These results suggest that E2F7/8 do not function as an ON/OFF switch, but fine-tune the expression levels of CCBE1 and FLT4 to ensure the proper signal strength during lymphangiogenesis [52]. The regulation of CCBE1 and FLT4 expression by E2F7/8 enhances the biological effect of VEGFC partial monitoring of lymphangiogenesis, but the elaborating mechanism of lymphangiogenesis needs to be further elucidated.
E2F8 influences polyploidization
Polyploidization occurs in all mammalian species, being the most significant in well-known genome alterations [60-63]. It often exists in some specific tissues, and is an important feature of hepatocytes following weaning and trophoblast giant cells (TGCs) during gestation [61, 64]. Oxidative damage and surgery regeneration of liver often lead to the enhancement of proportion of polyploid cells in liver, which is also associated with aging [65]. The biological functions of polyploidy possibly include the increasing ability to resist adverse conditions, weaken sensitivity to apoptosis, and buffer defence on detrimental mutations [64, 66].
The E2F transcriptional factors are closely related to cell-cycle progression, with E2F activators and atypical repressors oppositely to adjust a common set of target genes [67, 68]. Using a quantitative PCR assay, E2F expression was determined at RNA levels of TGCs in the placenta and postnatal liver development, respectively. The result shows that the endocycling nuclei in TGCs and hepatocytes are most active when total E2F1-3 activator levels are minimized. E2f1-3 ablation of TGCs in the placenta and hepatocytes in postnatal development results in hyperploidy, accompanying with abnormally large nuclei and increased liver weights. On the contrary, E2f7-/- and E2f8-/- DKO reduce ploidy and increase the proportion of G2/M- and M phase-related events. Indeed, the ploidy of wild-type TGCs could go up to 1,000C, whereas the E2f7-/- and E2f8-/- TGCs with genomes never exceeded 64C [64]. The canonical E2F mRNAs showed relatively high levels in 1 week old livers and then precipitous decrease from 3 weeks after birth, whereas they are relatively low in the livers of E2f7-/- and E2f8-/- mice at p0 (postnatal day 0) and have a markedly increase in 3 to 5 weeks. Atypical E2F expressional levels are consistent with the polyploidization of hepatocytes. Separated or combined inactivation of E2f7 (Alb-7ko) and E2f8 (Alb-8ko) showed that Alb-8ko and Alb-78dko mice had apparently smaller hepatocytes than Alb-7ko, but did not significantly affect hepatic mass. Flow cytometry revealed that Alb-8ko and Alb-78dko prevented hepatocyte endocycles over the whole mouse life, whereas Alb-7ko only had a modest reduction in genome ploidy. Indeed, E2F8 was more enriched on the polyploidy-related gene promoter than E2F7. Compared with the wild type mice, the up-regulated genes in 3-week-old livers were more marked in the Alb-7ko, Alb-8ko and Alb-78dko mice than the down-regulated genes, consistent with their inhibitor properties. Further analysis for these genes raised in both Alb-8ko and Alb-78dko livers revealed that they tended to regulate cytokinetic G2/M progression and mitosis. These data suggest that E2F8 has a vital role for mediating the repression of a set of cell-cycle-related target genes during the process of cell polyploidization [69].
It is worth noting that knock-out of E2f4 or E2f5 in mice seems to be little affection of the endocycling in TGCs and hepatocytes. The atypical E2Fs binding to DNA are in the way independent on DP so that they can mediate repression when CDK activity is high in S/G2 phases without the disturbing of the cyclin-CDK axis. Other studies have showed that through activation of the same target genes, deficiency of E2F1 can partially rescue the defect in hepatocyte polyploidization caused by a deficiency in E2f7 and E2f8 [69]. E2F8 can transcriptionally suppress CDK1 to induce the polyploidization of decidual cells [44]. It is necessary to maintain appropriate ploidy of cells, E2F8 abnormal ploidy of hepatocytes might be one of the important causes of HCC. However, the precise molecular mechanism of how the canonical activators and atypical repressors coordinate to control endocycling remains to be further elucidated.
E2F8 and diseases
The E2F family has long been expected as potential therapeutic targets of cancer. However, the molecular mechanism about E2F in the occurrence and development of tumor is still not clear, especially the new member E2F8 [7, 70, 71]. Recently, literatures show that E2F8 is obviously up-regulated in HCC, speculating that it is implicated in oncogenesis and progression [3, 6, 11]. Ectopic overexpression of E2F8 by transiently transfecting the recombinant pcDNA3.1-E2F8 into Huh-7, Hep3B, YY-8103 and PLC/PRF/5 HCC cells causes the enhanced cell proliferation, the promoted colony formation and tumorigenicity. On the contrary, knock down of endogenous E2F8 by introduction of siRNAs and shRNA in some HCC cell lines inhibits cellular proliferation and colony formation, and reduces tumor burden [29]. Mechanism analysis supports that E2F8 overexpression partially attenuates the E2F1 binding to the oncogenic cyclin D1 promoter in HCC cells in a dominant-negative manner. Cyclin D1 functions as an oncogene involved in many cancers by regulating the G1- to S-phase transition of cell cycle progression. These data support that E2F8 can contribute to cell proliferation of HCC partially via E2F8/E2F1/ Cyclin D1 promoting the entry of S phase in cell cycle [29].These results suggest that E2F8 promotes HCC occurrence partly through cyclin D1, possibly becoming a potential therapeutic target. E2F8 is also shown to overexpress in ovarian cancer, lung cancer, breast cancer and prostate cancer, and up-regulated in the clinical samples from patients with cancer [10, 72-74]. The knockdown of E2F8 significantly inhibits prostate cancer cell growth via inducing G2/M arrest, but its mechanism is not fully understood.
Although most studies indicate that E2F8 functions as an oncogene, recently few reports suggest that it might act as a tumor suppressor. Under the in vivo stress of DMBA/TPA skin carcinogenesis model, inactivation of E2f7/8 gives rise to the acceleration of tumorigenesis and deteriorating progression [75]. Temporal-specific ablation of E2f8 in the liver at 1-2 weeks of age resulted in severe liver cancer, but not in older age, suggesting that E2F8 acts as a tumor suppressor in postnatal liver development [27]. These contradictory results might be due to studies performed in different cell lines and context. No matter how to say, E2F8 expression must be affiliated to the occurrence and development of HCC. It may be a potential therapeutic target of HCC.
E2F8 is not only associated with cancer, but also involved in other diseases, including obesity and hepatic steatosis [76-78]. Loss of RecQ-mediated genome instability 1 (RMI1) inhibits E2F8 expression [78], Knockdown of E2f8 suppresses this glucose inducing up-regulation of RMI1 expression [77]. RMI1 and E2F8 consist of a feedback machinery to regulate energy balance in adipose tissue through modulating preadipocyte proliferation. E2F8 is up-regulated in hepatic steatosis. Ectopic expression of E2F8 significantly promotes the fatty acid binding protein 3 (FABP3) expression to cause hepatic steatosis [76]. As we know, the advanced stage of nonalcoholic hepatic steatosis results in ultimately cirrhosis and liver cancer, named trilogy for liver lesions [79, 80]. These results further support the possibility of E2F8 as a therapeutic target for liver cancer.
Concluding remarks
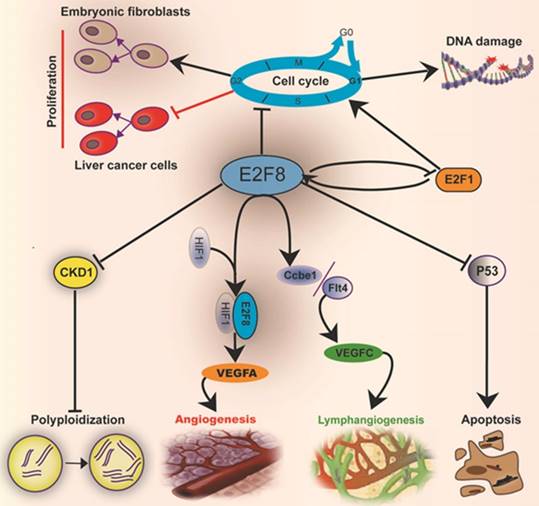
Taken together, the mammalian transcriptional factor E2F8 plays an essential role in embryonic development and regulation of cellular functions, including cell cycle, cell proliferation, cell survival, DNA damage, angiogenesis, lymphangiogenesis and cell polyploidization (Fig.2). Actually, it plays not only a role in regulating cell functions, but also an important one in balancing the E2F network. E2F8 has a remarkable increase in HCC and ovarian cancer, but no enhancement of E2F7 occurs though the both have functional compensation in many respects. E2F8 can transcriptionally suppress CDK1 to induce hepatocyte polyploidization. Moreover, E2F8 mediates the DNA damage-correlated cell-cycle through modulating E2F1 activity. Both E2F8 and E2F7 cooperate with HIF1 to form a transcriptional complex to raise the VEGFA level to promote angiogenesis. The regulation of CCBE1 and FLT4 expressions by both E2F7 and E2F8 can enhance the biological effect of VEGFC monitoring lymphangiogenesis. Besides, E2F8 also acts as a nonreceptor activator of heterotrimeric G proteins [81] that is an important second messenger, involving multiple cell signaling pathways [82, 83]. These provide a strongly theoretical support that E2F8 may become a promising therapeutic target for some tumors, especially HCC, and hepatic steatosis. The detailed mechanism by which E2F8 regulates cell functions still merits to be elucidated in the future.
Mechanisms by which E2F8 mediates the cell metabolic functions and histological development: E2F8 regulates the functions related to cell cycle, proliferation, DNA repair, apoptosis, polyploidization, angiogenesis and lymphangiogenesis.
Main similarities and differences between E2F7 and E2F8
| Object | Similarity | Difference | Reference |
|---|---|---|---|
| Structure | a. With two distinct DBD domains b. Lacking DP-dimerization and retinoblastoma-binding domains c. Absence of transcriptional activation domains | E2F7 has two highly relevant isoforms, but not E2F8. | 2,6,7,17,19 |
| Methods and functions of regulating gene | a. Controlling cell cycle genes through homodimers or heterodimers by DBD b. Functioning predominantly as transcriptional repressors | Efficiency for the both to form dimerization is not equivalent. | 5,16,18 |
| Embryo and postnatal development | Being necessary for embryonic development and viability | E2F7 appears more important than E2F8 in mice postnatal development. | 16 |
| Angiogenesis and lymphangiogenesis | a. Being necessary for angiogenesis b. Being essential for lymphangiogenesis | 16,30,51,51 | |
| DNA demage | Resisting DNA damage | 4,18 | |
| Cell polyploidization | E2F8 plays a vital role in the process of cell polyploidization, but E2F7 only has a modest effect. | 69 | |
| HCC | E2F8 shows overexpression in HCC, but not E2F7. | 3,6,11 |
DBD: DNA-binding domains; DP: dimerization partner; HCC: hepatocellular carcinoma
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant numbers 81172824, 30971465) and the Foundation of Pearl River Science and Technology New Star (grant number 201506010087).
Authors' contribution
All authors contribution to the writing of the review manuscript. F.X and Y.L initiated, conceptualized and wrote the manuscript. F.X, J.L and X.J revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lammens T, Li J, Leone G, De Veylder L. Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 2009;19:111-8
2. Logan N, Graham A, Zhao X, Fisher R, Maiti B, Leone G. et al. E2F-8: an E2F family member with a similar organization of DNA binding domains to E2F-7. Oncogene. 2005;24:5000-4
3. Zhan L, Huang C, Meng XM, Song Y, Wu XQ, Miu CG. et al. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell Signal. 2014;26:1075-81
4. Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS. et al. DNA-damage response control of E2F7 and E2F8. EMBO Rep. 2008;9:252-9
5. L. DS, Jensen MR HK. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003;22(23):6289-98
6. Xanthoulis A, Tiniakos DG. E2F transcription factors and digestive system malignancies: how much do we know? World J Gastroenterol. 2013;19:3189-98
7. Tsantoulis PK, Gorgoulis VG. Involvement of E2F transcription factor family in cancer. Eur J Cancer. 2005;41:2403-14
8. Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R. et al. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem. 2005;280:18211-20
9. Dynlach HCaBD. Emerging roles for E2F:Beyond the G1/S transition and DNA replication. Cancer press. 2003;3(4):311-6
10. Reimer D, Sadr S, Wiedemair A, Goebel G, Concin N, Hofstetter G. et al. Expression of the E2F family of transcription factors and its clinical relevance in ovarian cancer. Ann N Y Acad Sci. 2006;1091:270-81
11. Baiz D, Dapas B, Farra R, Scaggiante B, Pozzato G, Zanconati F. et al. Bortezomib effect on E2F and cyclin family members in human hepatocellular carcinoma cell lines. World J Gastroenterol. 2014;20:795-803
12. Dyson N. The regulation of E2Fby pRB-family proteins. Genes & development. 1998
13. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220-7
14. Ghazaryan S, Sy C, Hu T, An X, Mohandas N, Fu H. et al. Inactivation of Rb and E2f8 synergizes to trigger stressed DNA replication during erythroid terminal differentiation. Mol Cell Biol. 2014;34:2833-47
15. Moon NS, Dyson N. E2F7 and E2F8 keep the E2F family in balance. Dev Cell. 2008;14:1-3
16. Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H. et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell. 2008;14:62-75
17. Chen H-Z, Tsai S-Y, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nature Reviews Cancer. 2009;9:785-97
18. Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ. E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 2012;26:1533-45
19. Christensen J. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Research. 2005;33:5458-70
20. DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med. 2006;6:739-48
21. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA. et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245-56
22. Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M. et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21:4684-99
23. Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709-16
24. Trimarchi JM LJ. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002. 2002Jan;3(1):11-20
25. Westendorp B, Mokry M, Groot Koerkamp MJ, Holstege FC, Cuppen E, de Bruin A. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 2012;40:3511-23
26. Cohen M, Vecsler M, Liberzon A, Noach M, Zlotorynski E, Tzur A. Unbiased transcriptome signature of in vivo cell proliferation reveals pro- and antiproliferative gene networks. Cell Cycle. 2013;12:2992-3000
27. Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT. et al. E2f8 mediates tumor suppression in postnatal liver development. The Journal of clinical investigation. 2016;126:2955-69
28. Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810-26
29. Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS. et al. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res. 2010;70:782-91
30. Ouseph MM, Li J, Chen HZ, Pecot T, Wenzel P, Thompson JC. et al. Atypical E2F repressors and activators coordinate placental development. Dev Cell. 2012;22:849-62
31. Weijts BG, Bakker WJ, Cornelissen PW, Liang KH, Schaftenaar FH, Westendorp B. et al. E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 2012;31:3871-84
32. Danielian PS, Friesenhahn LB, Faust AM, West JC, Caron AM, Bronson RT. et al. E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene. 2008;27:6561-70
33. Kosugi S OY. E2Ls, E2F-like repressors of Arabidopsis that bind to E2F sites in a monomeric form. J Biol Chem. 2002
34. Vandepoele K, Vlieghe K, Florquin K, Hennig L, Beemster GT, Gruissem W. et al. Genome-wide identification of potential plant E2F target genes. Plant Physiol. 2005;139:316-28
35. Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012;72:571-5
36. Stanelle J, Putzer BM. E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol Med. 2006;12:177-85
37. Stevens C, La Thangue NB. A new role for E2F-1 in checkpoint control. Cell Cycle. 2003;2:435-7
38. Logan N, Graham A, Zhao X, Fisher R, Maiti B, Leone G. et al. E2F-8: an E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene. 2005;24:5000-4
39. Logan N, Delavaine L, Graham A, Reilly C, Wilson J, Brummelkamp TR. et al. E2F-7: a distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene. 2004;23:5138-50
40. Velez-Cruz R, Johnson DG. E2F1 and p53 transcription factors as accessory factors for nucleotide excision repair. Int J Mol Sci. 2012;13:13554-68
41. Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012;72:13-7
42. Udayakumar T, Shareef MM, Diaz DA, Ahmed MM, Pollack A. The E2F1/Rb and p53/MDM2 pathways in DNA repair and apoptosis: understanding the crosstalk to develop novel strategies for prostate cancer radiotherapy. Semin Radiat Oncol. 2010;20:258-66
43. Hu DE, Fan TP. Suppression of VEGF-induced angiogenesis by the protein tyrosine kinase inhibitor, lavendustin A. Br J Pharmacol. 1995;114:262-8
44. Qi QR, Zhao XY, Zuo RJ, Wang TS, Gu XW, Liu JL. et al. Involvement of atypical transcription factor E2F8 in the polyploidization during mouse and human decidualization. Cell Cycle. 2015;14:1842-58
45. Jin Y, Kaluza D, Jakobsson L. VEGF, Notch and TGFbeta/BMPs in regulation of sprouting angiogenesis and vascular patterning. Biochem Soc Trans. 2014;42:1576-83
46. Wheeler T, Elcock CL, Anthony FW. Angiogenesis and the placental environment. Placenta. 1995;16:289-96
47. Eswarappa SM, Fox PL. Antiangiogenic VEGF-Ax: A New Participant in Tumor Angiogenesis. Cancer Res. 2015;75:2765-9
48. Bakker WJ, Weijts BG, Westendorp B, de Bruin A. HIF proteins connect the RB-E2F factors to angiogenesis. Transcription. 2013;4:62-6
49. Fortunato P, Pillozzi S, Tamburini A, Pollazzi L, Franchi A, La Torre A. et al. Irresponsiveness of two retinoblastoma cases to conservative therapy correlates with up- regulation of hERG1 channels and of the VEGF-A pathway. BMC Cancer. 2010;10:504
50. Pan XY, Zhang ZH, Wu LX, Wang ZC. Effect of HIF-1a/VEGF signaling pathway on plasma progesterone and ovarian prostaglandin F2a secretion during luteal development of pseudopregnant rats. Genet Mol Res. 2015;14:8796-809
51. Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell. 2009;17:755-73
52. Weijts BG, van Impel A, Schulte-Merker S, de Bruin A. Atypical E2fs control lymphangiogenesis through transcriptional regulation of Ccbe1 and Flt4. PLoS One. 2013;8:e73693
53. Hogan BM, Herpers R, Witte M, Helotera H, Alitalo K, Duckers HJ. et al. Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development. 2009;136:4001-9
54. Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460-76
55. Bos FL, Caunt M, Peterson-Maduro J, Planas-Paz L, Kowalski J, Karpanen T. et al. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ Res. 2011;109:486-91
56. Pollmann C, Hagerling R, Kiefer F. Visualization of lymphatic vessel development, growth, and function. Adv Anat Embryol Cell Biol. 2014;214:167-86
57. Le Guen L, Karpanen T, Schulte D, Harris NC, Koltowska K, Roukens G. et al. Ccbe1 regulates Vegfc-mediated induction of Vegfr3 signaling during embryonic lymphangiogenesis. Development. 2014;141:1239-49
58. van Impel A, Schulte-Merker S. A fisheye view on lymphangiogenesis. Adv Anat Embryol Cell Biol. 2014;214:153-65
59. Mendola A, Schlogel MJ, Ghalamkarpour A, Irrthum A, Nguyen HL, Fastre E. et al. Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Mol Syndromol. 2013;4:257-66
60. Lee HO, Davidson JM, Duronio RJ. Endoreplication: polyploidy with purpose. Genes Dev. 2009;23:2461-77
61. Gentric G, Desdouets C. Polyploidization in liver tissue. Am J Pathol. 2014;184:322-31
62. Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556-66
63. Paterson AH. Polyploidy, evolutionary opportunity, and crop adaptation. Genetica. 2005;123:191-6
64. Chen HZ, Ouseph MM, Li J, Pecot T, Chokshi V, Kent L. et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol. 2012;14:1192-202
65. Gorla GR, Malhi H, Gupta S. Polyploidy associated with oxidative injury attenuates proliferative potential of cells. J Cell Sci. 2001;114:2943-51
66. Decordier I, Cundari E, Kirsch-Volders M. Survival of aneuploid, micronucleated and/or polyploid cells: crosstalk between ploidy control and apoptosis. Mutat Res. 2008;651:30-9
67. Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19:658-62
68. Hazar-Rethinam M, Endo-Munoz L, Gannon O, Saunders N. The role of the E2F transcription factor family in UV-induced apoptosis. Int J Mol Sci. 2011;12:8947-60
69. Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PC, Cornelissen PW. et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14:1181-91
70. Nemoto K, Ikeda A, Yoshida C, Kimura J, Mori J, Fujiwara H. et al. Characteristics of nobiletin-mediated alteration of gene expression in cultured cell lines. Biochem Biophys Res Commun. 2013;431:530-4
71. Schaal C, Pillai S, Chellappan SP. The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv Cancer Res. 2014;121:147-82
72. Sin-Aye Park JP, Jong Woo Lee, Francesc López-Giráldez, Roy S. Herbst and Ja Seok Koo. E2F8 as a Novel Therapeutic Target for Lung Cancer. JNCI J Natl Cancer Inst. 2015:18 107(9)
73. Ye L, Guo L, He Z, Wang X, Lin C, Zhang X. et al. Upregulation of E2F8 promotes cell proliferation and tumorigenicity in breast cancer by modulating G1/S phase transition. Oncotarget. 2016;7(17):23757-71
74. Lee S, Park YR, Kim SH, Park EJ, Kang MJ, So I. et al. Geraniol suppresses prostate cancer growth through down-regulation of E2F8. Cancer medicine. 2016;5(10):2899-2908
75. Thurlings I, Martinez-Lopez LM, Westendorp B, Zijp M, Kuiper R, Tooten P. et al. Synergistic functions of E2F7 and E2F8 are critical to suppress stress-induced skin cancer. Oncogene. 2016;36(6):829-839
76. Shimada Y, Kuninaga S, Ariyoshi M, Zhang B, Shiina Y, Takahashi Y. et al. E2F8 promotes hepatic steatosis through FABP3 expression in diet-induced obesity in zebrafish. Nutr Metab (Lond). 2015;12:17
77. Suwa A, Yoshino M, Kurama T, Shimokawa T, Aramori I. Glucose regulates RMI1 expression through the E2F pathways in adipose cells. Endocrine. 2011;40:56-61
78. Suwa A, Yoshino M, Yamazaki C, Naitou M, Fujikawa R, Matsumoto S. et al. RMI1 deficiency in mice protects from diet and genetic-induced obesity. FEBS J. 2010;277:677-86
79. Saran U, Humar B, Kolly P, Dufour JF. Hepatocellular Carcinoma and Lifestyles. J Hepatol. 2015
80. Kirstein MM, Vogel A. The pathogenesis of hepatocellular carcinoma. Dig Dis. 2014;32:545-53
81. Hagemann IS, Narzinski KD, Baranski TJ. E2F8 is a nonreceptor activator of heterotrimeric G proteins. J Mol Signal. 2007;2:3
82. Williams JA, Chen X, Sabbatini ME. Small G proteins as key regulators of pancreatic digestive enzyme secretion. Am J Physiol Endocrinol Metab. 2009;296:E405-14
83. Wieland T. Interaction of nucleoside diphosphate kinase B with heterotrimeric G protein betagamma dimers: consequences on G protein activation and stability. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:373-83
Author contact
![]() Corresponding authors: Professors Feiyue Xing and Jing Liu, Department of Immunobiology, Institute of Tissue Transplantation and Immunology, Jinan University, Guangzhou, China. E-mail: tfyxingedu.cn or tjliuedu.cn
Corresponding authors: Professors Feiyue Xing and Jing Liu, Department of Immunobiology, Institute of Tissue Transplantation and Immunology, Jinan University, Guangzhou, China. E-mail: tfyxingedu.cn or tjliuedu.cn