Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(8):1378-1394. doi:10.7150/jca.17478 This issue Cite
Research Paper
Targeting TPX2 Suppresses the Tumorigenesis of Hepatocellular Carcinoma Cells Resulting in Arrested Mitotic Phase Progression and Increased Genomic Instability
Chao-Wen Hsu1, 5, Yu-Chia Chen2, Hsing-Hao Su3, Guan-Jin Huang4, Chih-Wen Shu6, Tony Tong-Lin Wu2,5 ![]() , Hung-Wei Pan6
, Hung-Wei Pan6 ![]()
1. Division of Colorectal Surgery, Department of Surgery, Kaohsiung Veteran General Hospital, Kaohsiung, Taiwan;
2. Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan;
3. Department of Otorhinolaryngology-Head and Neck Surgery, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan;
4. Department of Pathology, National Chung Kung University Hospital, Tainan, Taiwan;
5. School of Medicine, National Yang-Ming University, Taipei, Taiwan;
6. Department of Medical Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan.
Received 2016-9-6; Accepted 2017-3-13; Published 2017-5-12
Abstract
Hepatocellular carcinoma (HCC) remains one of the most difficult cancers to treat, with chemotherapies being relatively ineffective. Therefore, a better knowledge of molecular hepatocarcinogenesis will provide opportunities for designing targeted therapies. TPX2 (targeting protein for Xklp2) is overexpressed as a consequence of oncogenic alterations and is likely to alter the proper regulation of chromosome segregation in cancer cells. Disrupting the machinery which is responsible for mitosis and chromosome instability in cancer cells can be one of the most successful strategies for cancer therapy. Therefore, we consider the targeting TPX2 could provide novel therapeutic strategies for cancer. In this study, increased TPX2 protein expression was present in 16 (42%) of 38 primary HCCs and was associated with advanced stage, distant metastatic HCCs and poor prognosis. Knockdown of TPX2 inhibited cancer cell growth and downregulation of cyclin A, cyclin E and CDK2 proteins. However, over-expressed EGFP-TPX2 protein enhanced the in vitro tumor spheroid formation and rescued the TPX2 depleted cell growth. Targeting TPX2 caused a rising impaired chromosomal instability resulting in multinuclearity, cell cycle progression arrest, apotosis, senescence and an increased polyploidy in cells. An image-cytometry analysis revealed cell cycle progression arrest after TPX2 inhibition. A correlation was observed between the downregulation of the protein levels of genes related to chromosomal segregation and spindle assembly checkpoint (securin, seprase, Aurora A, Aurora B, Cyclin B1, Cyclin B2, MPS1, BUB1, BUB3, MAD1 and MAD2) and increased cell ploidy, indicating mitotic progression failure and the loss of the balance of genomic instability. In vitro tumor spheroid assay and in vivo xenografts mouse model showed a therapeutic opportunity. Our findings indicate that targeting TPX2 lead to suppress tumorigenicity in liver cancer cells, suggesting that TPX2 is a potential target for anticancer therapy in HCC.
Keywords: TPX2, Hepatocellular carcinoma, cell cycle progression, multinuclearity, polyploidy, anticancer.
Introduction
Hepatocellular carcinoma (HCC) has a very poor long-term prognosis and is the leading cause of death caused by cancer in Taiwan and worldwide [1, 2]. Partial hepatectomy has been one of the most reliable methods of treatment and offers the hope of cure to those with early HCC [3]. Unfortunately, the number of patients who can benefit is limited because of the advanced disease caused by intrahepatic metastasis at the time of diagnosis, and the associated viral hepatitis-induced cirrhosis [4]. Due to late diagnosis and advanced underlying liver cirrhosis, only limited treatment options with marginal clinical benefit are available to most patients. Systemic therapies for patients with HCC using cytologic agents such as tamoxifen, doxorubicin, everolimus and thalidomide have shown marginal success [5]. Therefore, a better knowledge of molecular hepatocarcinogenesis will provide opportunities for the design of targeted therapy.
Cell cycle deregulation is a common event in human cancer and provides several therapeutic strategies to inhibit cancer cell growth. Although most current therapies are designed to arrest cell cycle progression either in G1/S or mitosis, new proposals include targeting the intrinsic chromosomal instability (CIN), an increased rate of gain or loss of chromosomes during cell division that many tumor cells display [6]. CIN leads to genomic instability. Therefore, several factors regulate the assembly and function of the mitotic spindle, such as structural and signaling functions [7-9].
Disrupting the machinery which is responsible for cancer associated genes involved in mitosis and chromosome instability in cancer cells can be one of the most successful strategies for cancer therapy [10]. Hence, gaining a better understanding of mitotic entry, progression and exit is essential, not only for working out the causes of CIN but also for the design of more effective drugs to kill tumor cells [10]. If we can validate these events between the normal and disease states, especially in cancer, we could design specific, complete, and personalized strategies for cancer treatment. For this reason, we suggest that the investigation of chromosome segregation and its consequential events could provide novel therapeutic strategies for cancer.
TPX2 (targeting protein for Xklp2) is a protein required for targeting kinesin-like protein-12 (Xklp2) to the spindle pole in Xenopus egg extracts [11]. TPX2 triggered the nucleation of microtubules that are essential for spindle assembly, in particular, for spindle pole organization in a variety of cell types [12]. Several studies have demonstrated that TPX2 is overexpressed in different types of human cancers, including medullary thyroid cancer [13], esophageal squamous cell carcinoma (ESCC) [14], estrogen receptor-positive metastatic breast cancers [15] and bladder carcinoma [16], and promotes tumor growth and metastasis, including in HCC. The expression level of TPX2 was obviously higher in HCC, and TPX2 overexpression was positively correlated with poor prognosis, metastasis, recurrence and time of recurrence after curative resection [17-19]. TPX2 might be beneficial for T-cell specific HCC immunotherapy [20]. TPX2 knockdown obviously slowed tumor growth in a nude mouse xenograft liver cancer cell model [19]. Otherwise, TPX2 knockdown prominently suppressed HCC cell invasion and migration. These results indicate that TPX2 may serve as a viable cancer target [20] and therapeutic target [19, 21] in HCC patients. However, other new mechanisms of this protein in cancer cell proliferation are now emerging, and further understanding of the functional relevance of TPX2 may facilitate the design of therapeutic approaches in HCC.
Misregulation of these factors leads to abnormal spindle formation or function, and their encoding genes such as TPX2 are increasingly being found to be aberrantly expressed in cancer [22]. This further illustrates the link between mitotic errors, chromosome missegregation, genomic instability and tumorigenesis. However, an outstanding question is how and whether targeting TPX2 leads to genomic instability events, and the answer could provide novel therapeutic strategies for liver cancer. In this study, we present the first evidence and consequent mechanisms that targeting TPX2 arrested G2/M transition causes genomic instability events, such as DNA damage, multinuclearity and ploidy, and suppresses the tumorigenesis of HCC cells resulting in the disruption of the mitotic proteins Cyclin B, CDK1, Securin, Seprase, Aurora A and Aurora B, as well as the deregulation of the S phase proteins Cyclin A, Cyclin E and CDK2.
Materials and Methods
Human tissue samples
RNA samples of paired human hepatocellular carcinoma and adjacent tissue were obtained from the Kaohsiung Veterans General Hospital; these tissues were obtained according to the regulations of the Ethical Committee of the Kaohsiung Veterans General Hospital (IRB protocol: VGHKS13-CT3-009). RNA samples formed the TPX2 mRNA expression level of this study in HCC. The commercial human hepatocellular carcinoma tissue array (Liver: cancer-metastasis-normalIMH-360, © IMGENEX Corporation, USA) formed the TPX2 protein expression in HCC. This tissue array included 38 primary HCCs, 2 cholangiocarcinomas, 8 metastatic hepatocellular carcinomas, 2 metastatic cholangiocarcinomas, and 9 non-neoplastic livers (adjacent to cancer).
Immunohistochemistry and scoring
The HCC tissue array was sent for immunohistochemistry (IHC) staining. Tissue sections stained with an anti-TPX2 antibody (Sigma-Aldrich Corp, St. Louis, MO, USA), followed by HRP-conjugated secondary antibody, to observe the TPX2 protein expression level in both tumor and normal cells by using the mouse/rabbit probe HRP labeling kit (BioTnA, Kaohsiung, Taiwan). All the tumor cells within each microscopic field were counted, and the positive rates of target proteins in cells were then calculated. The immunoreactivity of tumor cells for TPX2 immunostain varied considerably, The proportion of tumor cells positive for TPX2 immunostain varied considerably, ranging from diffuse positive (>50%), heterogeneous, nodular, and trace to negative. In the nontumorous liver, TPX2 protein was detected only in very few isolated liver cells. Hence, scored as 0 (all cells negative; 22 cases), HCC with 1% of tumor cells showing nuclear immunostaining for TPX2 was regarded as positive and scored as 1+ (1-10%; 9 cases), 2+ (10-25%; 4 cases), and 3+ (>50%; 3 cases), respectively. In subsequent data analyses, the expression level of TPX2 scored as 0 was considered to be “negative”, whereas 1+, 2+ and 3+ were categorized as “High”. The difference between the two expression groups of cancer and their prognosis were calculated using statistical SPSS software.
Reverse Transcription-polymerase chain reaction (RT-PCR)
We used RT-PCR assays for semi-quantitative analysis of TPX2 mRNA as described previously [23-26]. mRNA encoding either S26 ribosomal protein (RPS26) was used as an internal control for the initial RNA loading. PCR was performed using a SelectCycler Thermal Cycler SBT9600 (Select BioProducts, Edison, New Jersey, USA). The quantitative of mRNA expression was carried out in an automated DNA thermal cycler, the StepOnePlus Real-Time PCR System (Life Technologies).
Western blotting
Protein samples (60 μg) were separated by SDS-PAGE (10% gels) and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, UK). The membranes were then incubated with primary and secondary antibodies, and immunoreactive signals were detected using an ECL kit (Amersham Pharmacia Biotech, UK).
Small RNA interference (siRNA) and knockdown of gene expression
The TPX2 siRNA oligos pool (1: 5'-GGACGAACCGGUAGUGAU-3'; 2: 5'-GCAUAAGGCAAAUCCAAUA-3'; 3: 5'-GUACCAUUGUUAAGCCUUU-3'; 4: 5'-GAAAUUCUACCCUCUAAGA-3') and the si-TPX2 3'-UTR siRNA oligos (5'-GGACCGTCTTGCTTTGTCA-3') were synthesized by Sigma-Aldrich Company (St. Louis, MO, USA). All transient transfections of the TPX2 siRNA oligos pool at a final concentration of 20 nM were accomplished with LipofectAMINE RNAiMAX (Invitrogen, Carlsbad, CA, USA) by following the manufacturer's protocols.
Cell proliferation, colony formation, anchorage-independent (spheroid) growth and in vitro invasion
The initiated cell density for TPX2 siRNA transfection was 1.5×105 cell per 2-mL suspension. For cell proliferation analysis, 1000 living cells were plated on the 96-well plates after transfection with the 20 nM siRNA oligos pool. The luminescence units indicating cell growth were determined at 0, 1, 2, and 3 days using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI, USA). For colony formation assays, 2500 cells were seeded in six-well plates and incubated for 2 weeks. The colonies were then fixed with 2% formaldehyde and stained with 0.5% crystal violet. Photographs were taken, and the number of colonies in each well was counted. For spheroid assays, 1000 living cells were seeding in an Ultra Low Attachment 96-well Microplate (Corning Incorporated, NY, USA), and cell spheroids were visualized under a microscopic low-power field. For the in vitro invasion assay, we used Corning Transwell chambers (Corning Incorporated, NY, USA) and Growth Factor Reduced Matrigel Matrix (BD Biosciences, MA, USA). Briefly, Matrigel (20 μL, 2 μg/μL in serum free medium) was added to the upper side of a transwell chamber and allowed to polymerize for 30 min at 37 °C. Cells (2 x 104) in 100 μL of serum free medium were added to the upper chamber, and 500 μL of growth medium with 10% FBS was added to the lower chamber. After 24 h of incubation, the noninvading cells on the upper side of the chamber membranes were removed. The invading cells migrating to the opposite of the chamber membranes were stained with 0.5% crystal violet in methanol and counted at a low-power field (X10 magnifications, 12 fields were counted and averaged). The experiments and readings were repeated and analyzed using the two-sided Student's t test.
Primary tumour cell and hepatocyte culture
The generation of single-cell suspensions was thorough dissociator from HCC specimens. Briefly, the tissue was washed and minced with fine scissors into fragments of 1x1x1 mm³ and apply to gentleMACS™ Dissociator (Miltenyi Biotec). Trypan blue staining confirmed more than 80% viability after the procedure. The single-cell suspensions were addressed to followed experiments. For tumor cell line establish, the single-cell suspensions were cultured in DMEM/F12 (1:1) medium, supplemented with FCS, glutamine, antibiotics and non-essential amino acids (all from Sigma Aldrich, St Louis, MO, USA), 15 ng/ml basic firbroblast growth factor (bFGF), 20 ng/ml epidermal growth factor (EGF), 2mM/l L-glutamine, 4 U/l insuline growth factor (IGF) and B 27 supplement (1:50) (Sigma Aldrich). Cells were cultured in a humidified amosphere at 37 ˚C and 7% CO2.
Expression vector and stable transfection
RT-PCR amplified full-length TPX2 cDNA was subcloned into expression vectors pEGFP-C1 (Clontech, CA). HCC cell line (SkHep-1) was grown in Dulbecco's modified Eagle's medium (DMEM). We used lipofectamine 3000 reagent (Invitrogen, CA) for transfection. The EGFP-TPX2 stable expression cells were selected by medium with G418 (800 μg/mL) more than 2 weeks. The EGFP-H2B (human histone H2B protein) as the control vector.
In vivo anti-cancer assay
Male Nude mice (BALB/c Nude; 7 weeks old) were purchased from the BioLASCO Taiwan Co., Ltd. Cells were suspended in matrigel and injected s.c. (5×106 cells, total volume 0.2 ml) into the right flank. Animals were observed daily for 2 to 3 weeks. Tumors were allowed to develop more than 0.5 x 0.5 mm, the tumor-bearing nude mice were randomly divided into control and treatment groups. For mimicking the in vivo therapeutic effect, the synthesized siRNA pool molecules (15 μg per injection) were directly delivered into the tumor via in vivo jetPEI reagent (Polyplus-transfection, New York, NY, USA) every 7 days following the manufacturer's protocols. Tumor volume was determined every 7 days using the formula V = 1/2 (A × B2), where A and B represent the largest and smallest dimensions of the tumor, respectively. All animal work was performed under protocols approved by the Institutional Animal Care and Use Committee of Kaohsiung Veterans General Hospital.
Cell cycle analysis (Image-flow cytometry assay)
The cells were harvested and washed with PBS and then fixed in 0.5% formaldehyde for 1 h at 0 °C. After a second wash with PBS, the cells were re-suspended in 50% ethanol in PBS for 1 h at -20 °C. Cells were washed at least once with cold PBS, and the cells were then re-suspended in 300 to 500 μl of PI/Triton X-100 staining solution (to 10 ml of 0.1 % (v/v) Triton X-100 (Sigma-Aldrich Company, St. Louis, MO, USA) in PBS and 0.40 ml of 500 mg/ml DAPI (Sigma-Aldrich Company, St. Louis, MO, USA). Data were acquired for analysis using the NucleoCounter NC-3000 machine (Chemometec, Denmark) and Nucleoview NC-3000 Software for automated Image-flow cytometry analysis. The cell cycle distribution was determined using Nucleoview NC-3000 Software.
Immunofluorescence staining
Cells cultured on coverslips were fixed in 4% paraformaldehyde in 1X PBS for 10 min at room temperature. After washing in PBS, the cells were incubated with 1X PBST (0.2% Triton X-100 in 1X PBS) for 20 min and then incubated in 5% FBS blocking solution for 30 min. The cells were incubated with α-tubulin and TPX2 antibodies at 4 °C overnight. After washing with 1X PBST three times for 5 min each, the coverslips were incubated with secondary antibody (Alexa Fluor-488 or 568 -conjugated IgG; (Invitrogen, Carlsbad, CA, USA) and DAPI (2.5 μg/ml) for 60 min and rewashed in 1X PBST three times for 5 min. The cells were mounted in 90% glycerol in Tris buffer (pH 10.0) and observed with a fluorescence microscope.
Comet assays
Neutral comet assays were performed using the CometAssay kit (Trevigen, Gaithersburg, MD, USA) and the recommended protocol. Images were acquired with an Olympus IX81F-3 fluorescence microscope mounted with an Olympus DP70 digital camera. The comet tail lengths were measurement using ImageJ software.
Annexin V detection
For Image-flow cytometry assay, Annexin V-FITC/propidium iodide (PI) staining assay (Strong Biotech Corporation, Taipei, Taiwan) was employed to detect cells in early apoptotic and late apoptotic stages. Cells were transfected siRNA oligos for 72 hours and were harvested after incubation and washed in cold PBS. Cells were resuspended in 100 μl of Annexin V-FITC/propidium iodide (PI) staining solution and incubate for 10-15 min at 15 to 25°C. After incubation for 15min, the cells were collected and analyzed in a NucleoCounter NC-3000 machine (Chemometec, Denmark) and Nucleoview NC-3000 Software for automated Image-flow cytometry analysis. A total of 20,000 cells were analyzed per sample. Data were later analyzed using the Nucleoview NC-3000 Software.
Senescence associated β-galactosidase assay
Senescence associated β-galactosidase assay (SA-β-gal), a widely used marker for identifying senescent cells, was done as described manufacturer's protocols (Senescence Cells Histochemical Staining Kit, SIGMA). Briefly, cells were fixed in 4% paraformaldehyde and stained with β-gal staining solution containing X-Gal (1 mg/ml), citric acid/sodium phosphate, pH 6.0 (40 mM), potassium ferrocyanide (5 mM), potassium ferricyanide (5 mM), NaCl (150 mM) and MgCl2 (2 mM). The plates ere incubated in a dry incubator and monitored every 2 h for development of blue color. The reaction was then stopped by addition of 50% glycerol solution and then photographed under light microscope. 12 photos (10X magnification) of cells from different fields were counted for β-gal positivity for quantifying the number of senescent cells at each treatment.
Statistical analysis
For all bar plots, two-sided T-tests were used to determine the significance of the data with a normal distribution and equal variances. The boxplot was plotted using SigmaPlot.v10, the bottom and top of the box correspond to the 25th and 75th percentiles, respectively, and whiskers represent data points. The gray line extending through the boxplot indicates the mean value, and the black line contained within the box plot represents the median value. The prognosis was analyzed using the Mann-Whitney rank sum test, which was calculated using statistical SPSS software. Groups with P-values less than an alpha of 0.05 were considered significantly different.
Results
TPX2 is highly expressed in G2/M phase, human hepatocellular carcinoma tissues and cancer cell lines
We obtained 9 pairs of hepatocellular carcinoma (T) and non-tumorous liver (N) tissue samples from patients who underwent surgical resection. Using RT-PCR measurements, we found that TPX2 mRNA was expressed at high levels in the tumor portion. In contrast, the TPX2 mRNA level was very low or undetectable in the normal liver portion (Figure 1A). Using RT-PCR at the linear range, we observed that TPX2 mRNA was expressed dynamically in several human cancer cell lines, including liver, colon, breast and prostate cancer types (Figure 1B). The TPX2 protein was also expressed dynamically in liver cancer cell lines and primary cultured HCCs, but not detected in primary hepatocytes (Figure 1C). To elucidate the biological role of TPX2, we analyzed the TPX2 gene mRNA expression during cell cycle progression using synchronized HeLa cells. TPX2 expression was high in the G2/M phase, but decreased dramatically upon the entry into G1 phase, increased upon entry into S phase and again peaked at the next G2/M phase (Figure 1D).
The expression profile of TPX2 in HCCs, human cancer cell lines and cell cycle progression. Using an RT-PCR assay in the linear range for gene expression detection, (A) TPX2 mRNA was expressed at a high level in hepatocellular carcinoma (T), but expression was very low or undetectable in non-tumorous liver (N) tissue samples. (B), TPX2 mRNA expression was detected dynamically in several human cancer cell lines, including liver, colon, breast and prostate cancer types. (C) The TPX2 protein expression in liver cancer cell lines, primary cultured HCCs and hepatocytes were detected by western blot. (D) HeLa cells were arrested at the mitotic phase by nocodazole and then released for the time period indicated. The phase of cell fractions during cell cycle progression was monitored by flow cytometry analysis and protein expression evolution of Cyclin B1 was indicated G2/M phase progression. TPX2 mRNA levels were measured by an RT-PCR assay. For all RT-PCR assays, TPX2 was at 32 cycles; RPS26, s26 ribosomal protein mRNA, was amplified at 22 cycles as an internal control.
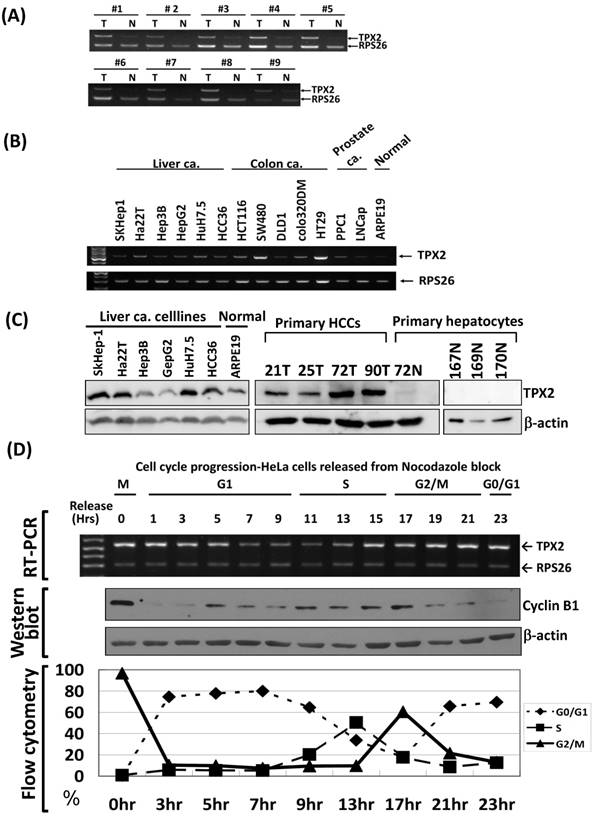
The pathological features of TPX2 protein in liver cancer tissue
To understand the correlation between TPX2 expression and clinical characteristics of liver cancer, we performed IHC staining for the TPX2 protein using a commercial HCC tissue array section for the evolution of TPX2 expression in liver cancer. There were differential expression proportions in the individual core of the tissue array section (Figure 2A). In 42% (16 of 38) of the cases, the positive staining (score +1 to +3) of TPX2 in the tumor cells resulted in an immuno-positive signal in the nucleus. Nevertheless, no expression was observed in the adjacent nontumorous liver tissues (0% (0 of 9) of cases were positive for TPX2) or in either of the 2 cholangiocarcinomas. However, TPX2 was expressed in all distant metastasis liver cancers (Figure 2B). Our data also showed that TPX2 expression correlated with a high stage (stages II and III~IV; p=0.047, 0.024, respectively) (Figure 2C) and with poor prognosis (Figure 2D). In the cases of distant metastasis liver cancer, there were 2 cholangiocarcinomas that metastasized to soft tissue and lymph node, and 8 HCCs that metastasized to the adrenal gland, lumbar vertebra, soft tissue, lymph node, lung and colon (Figure 3). These data indicate a pathogenesis role for elevated expression of TPX2 in HCC.
TPX2 protein expression in HCC. (A) Using the commercial HCC tissue array section, the TPX2 expression level in HCC was determined by immunohistochemistry. The proportion score was categorized into 3 grades (score +1 to +3) based on the TPX2 protein level as described in the materials and methods section. (B) The proportions of TPX2 protein expression in HCC, distance metastasis liver cancers, cholangiocarcinoma and adjacent nontumorous liver tissues. The positive staining (score +1 to +3) of TPX2 in the tumor cells resulted in an immuno-positive signal in the nucleus. (C) The clinicopathological correlation of TPX2 expression in 38 patients with resected primary hepatocellular carcinoma. (D) The cumulative curves for 38 patients with resected primary hepatocellular carcinoma in relation to TPX2 expression.
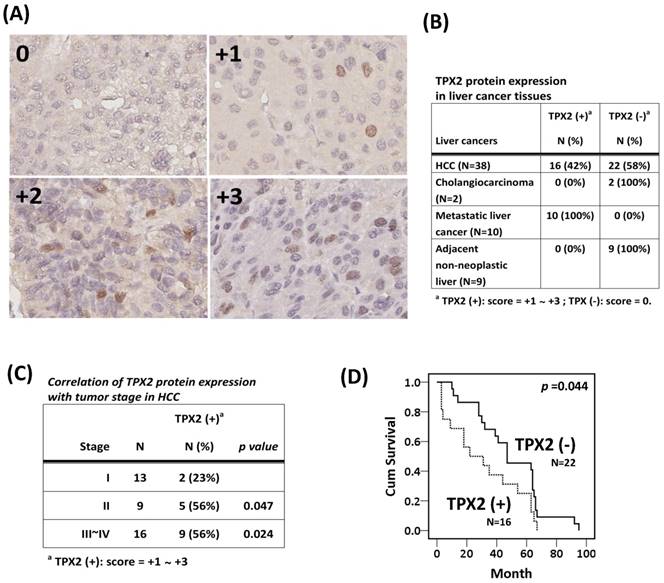
Elevated expression of TPX2 in cases with distance metastasis liver cancer cases. TPX2 protein detected in HCCs metastasized to the adrenal gland, lung and colon. TPX2 protein was present in 2 distant metastatic cholangiocarcinomas and HCCs cases (table).
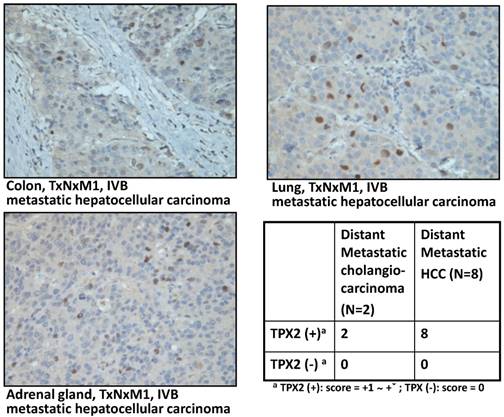
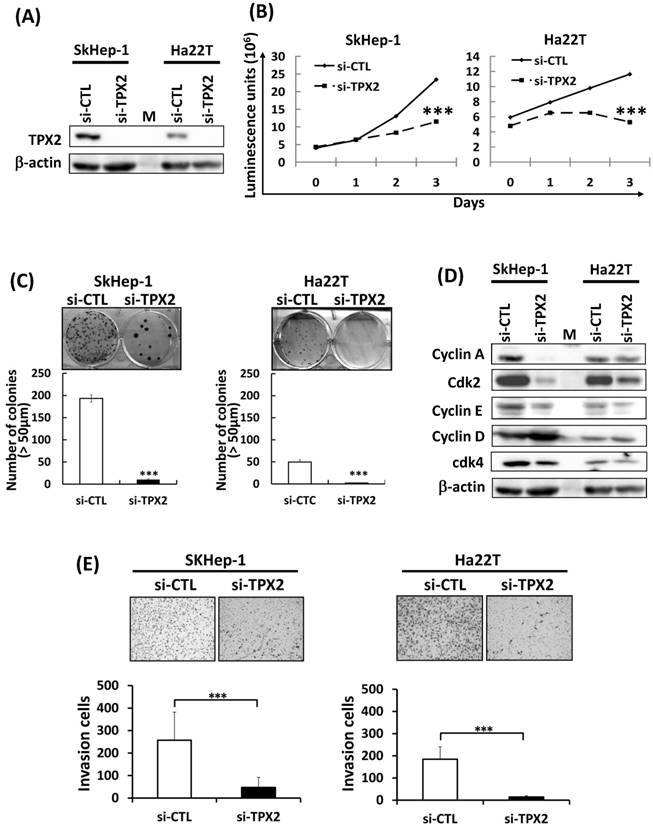
Silencing TPX2 inhibits liver cancer cell growth, in vitro invasion and reduced G1 to S phase related proteins
To investigate how TPX2 is involved in liver cancer cells, we synthesized a 4 commercial siRNA oligos for TPX2, and to evaluate TPX2 functions in HCC cells. To evaluate the efficiency for these 4 individual siRNAs (#1, #2, #3 and #4), we transfected the individual siRNA into the Skhep-1 and Ha22T cells. In addition to si-TPX2 #4 siRNA treatment, the others (#1, #2, #3) showed an efficiently in TPX2 protein depletion and cell growth inhibition (Figure 4A, 4C). The cells were represented large size and multinucleate, and the morphology was similarly in si-TPX2 siRNA #1, #2 and #3 treatment (Figure 4B, 4D). These results indicated the targeting effects were correlated with knockdown efficiency. Moreover, the si-TPX2 pool siRNA (combined 4 individual siRNAs) had concordance efficiency with those siRNAs (#1~#3). In order to minimize off-target effects of siRNA transfection, the si-TPX2 pool siRNA was used for the following analysis. Using the cell proliferation analysis (Figure 5B) and clonogenic formation assay (Figure 5C), we found that knockdown of TPX2 protein expression (Figure 5A) potently inhibited anchorage-dependent cell growth in Ha22T and SkHep-1 liver cancer cells. Because of the observed effect on cell growth inhibition, we evaluated the expression of Cyclin A, Cyclin E and CDK2, which are important and tightly initiate DNA replication in S phase, and Cyclin D1 and CDK4, which serve as sensors for cell cycle transition from the G1 to the S phase. We found that the protein expression of Cyclin A, Cyclin E, CDK2, and CDK4 was reduced by TPX2-silencing, but not the Cyclin D (Figure 5D). Since TPX2 expression was tightly correlated to metastasized liver cancers, we then performed a transwell assay to study the potential involvement of TPX2 in HCC cell motility. We found that TPX2 depletion dramatically compromised the invasion in both SkHep-1 and Ha22T cells (Figure 5E).
The efficiency of individual siRNA. (A, C) Scrambled siRNA (20 nM, si-Ctrl), four individual siRNA oligos against TPX2 (20 nM, #1 ~ #4) or siRNA oligos pool against TPX2 (20 nM, si-TPX2) were transfected into human liver cancer Skhep-1 and Ha22T cells for 72 h. The cells were harvested and lysed for immunoblotting to determine the protein levels of TPX2. β-actin was used as a loading control (upper panels). Cell viability was measured with the CellTiter-Glo® Luminescent Cell Viability Assay and the luminescence units indicating cell growth were measured and plotted as the growth plot (lower panels). (B, D) The cell morphology of individual siRNA transfected Skhep-1 and Ha22T cells. Representative data and quantitative results were shown. All representative graphs from two independent experiments. Values represent means ± SDs (Student's t-test, n.s.: non-significant, *P < 0.05, **P < 0.01, ***P < 0.001,).
Silencing TPX2 inhibits liver cancer cell growth and in vitro invasion and reduced the expression of G1 to S phase related proteins in two human liver cancer cell lines. (A) Scrambled siRNA (20 nM, si-Ctrl) or siRNA oligos pool against TPX2 (20 nM, si-TPX2) was transfected into human liver cancer Skhep-1 and Ha22T cells for 72 h. The cells were harvested and lysed for immunoblotting to determine the protein levels of TPX2. β-actin was used as a loading control. (B) Cell viability was measured with the CellTiter-Glo® Luminescent Cell Viability Assay and the luminescence units indicating cell growth are measured and plotted as the growth curve. (C) Two human liver cancer cells were transfected with scrambled and TPX2 siRNA for the clonogenic assay. Representative data and quantitative results are shown. (D) Scrambled siRNA (20 nM, si-Ctrl) or siRNA oligos pool against TPX2 (20 nM, si-TPX2) was transfected into human liver cancer Skhep-1 and Ha22T cells for 72 h, and the cells were harvested and lysed for immunoblotting to determine the protein levels of Cyclin A, Cyclin E, CDK2, Cyclin D1 and CDK4. β-actin was used as a loading control. (E) Two human liver cancer cell lines were transfected with scrambled and TPX2 siRNA for the in vitro invasion assay. Representative data and quantitative results were shown. All representative graphs from three independent experiments. Values represent means ± SDs (Student's t-test, *P < 0.05, **P < 0.01, ***P < 0.001,).
TPX2 promoted tumorigenesis and liver cancer cell growth
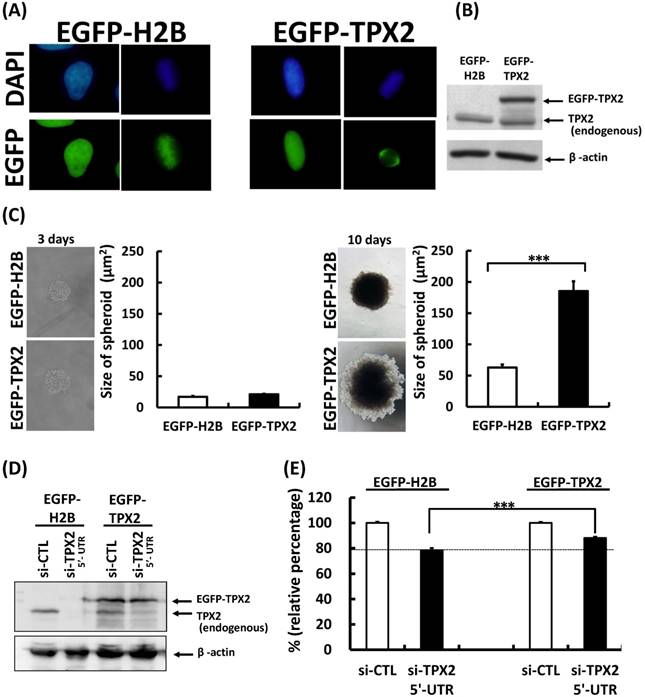
To further evaluate the biological function of TPX2 protein, we stable expressed EGFP-TPX2 protein in Skhep-1 cells. The EGFP-TPX2 represented nucleus localization in interphase cells and spindle pole localization in mitotic cell. The EGFP-H2B control protein showed the chromatin associated phenotype (Figure 6A). The EGFP-TPX2 protein level was measured by western blot (Figure 6B). Ectopic expression of TPX2 increase tumor spheroid (Figure 6C; 10 days) and diminished cell growth inhibition caused by silencing endogenous TPX2 using the si-TPX2 3'-UTR siRNA in Skhep-1 cells (Figure 6D, E). These results indicated TPX2 contribute to the tumorigenesis in liver cancer cells.
TPX2 promoted tumorigenesis and liver cancer cell growth. (A) SkHep-1 cells were stable transfected with the vectors expressing EGFP-H2B or EGFP-TPX2. The EGFP-fusion proteins were observed by fluorescence microscopy. (B) The protein level was detected by immunoblotting. (C) Skhep-1 cells with stable expressed EGFP-fusion protein were seeded on an ultra-low attachment 96-well microplate for spheroid formation. The cells were fixed to observe spheroid formation (left panels) at 3 days and 10 days, and the spheroid sizes were measured with micrometer ruler under a microscopic low-power field. The quantitative results are shown in the right panels. (D) Scrambled siRNA (20 nM, si-Ctrl) or siRNA 3'-UTR oligos against TPX2 (20 nM, si-TPX2 3'-UTR) was transfected into EGFP-fusion protein stable expressed Skhep-1 cells for 72 h. The cells were harvested and lysed for immunoblotting to determine the protein levels of TPX2. β-actin was used as a loading control. (E) Cell viability was measured with the CellTiter-Glo® Luminescent Cell Viability Assay and the luminescence units indicating cell growth are measured and plotted as the bar plot. Representative data and quantitative results were shown. All representative graphs from two independent experiments. Values represent means ± SDs (Student's t-test, *P < 0.05, **P < 0.01, ***P < 0.001,).
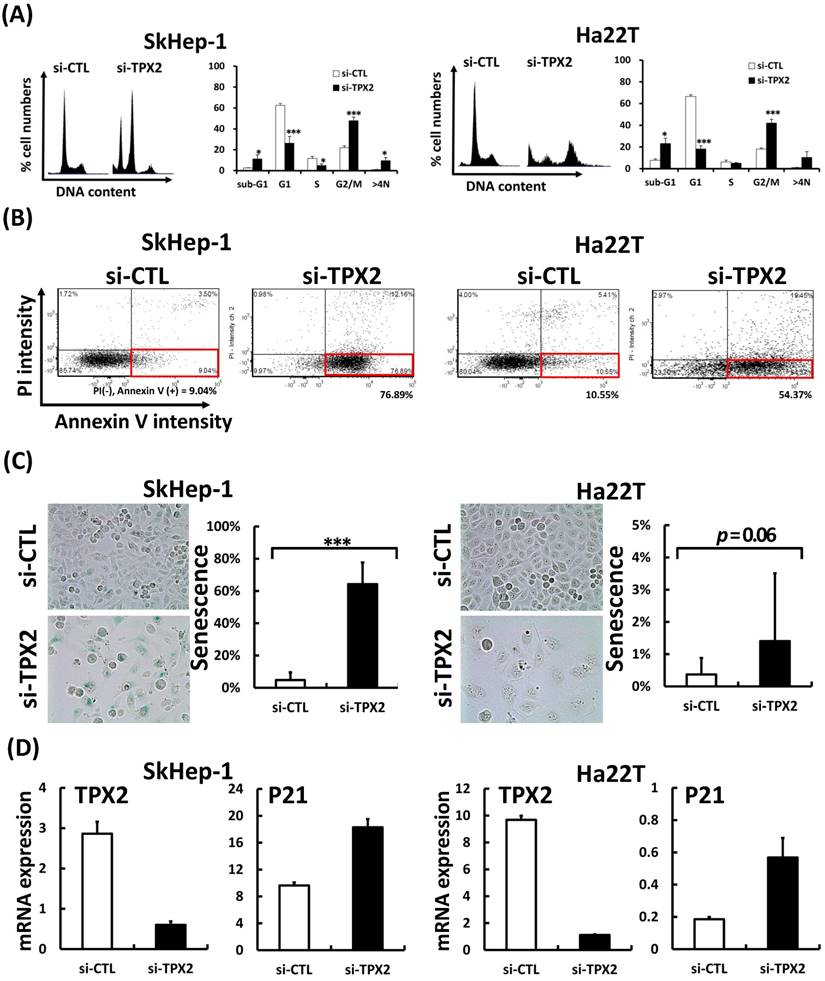
Depletion of TPX2 increased the proportion of polyploid cells, apotosis, senescence and arrested cell cycle progression
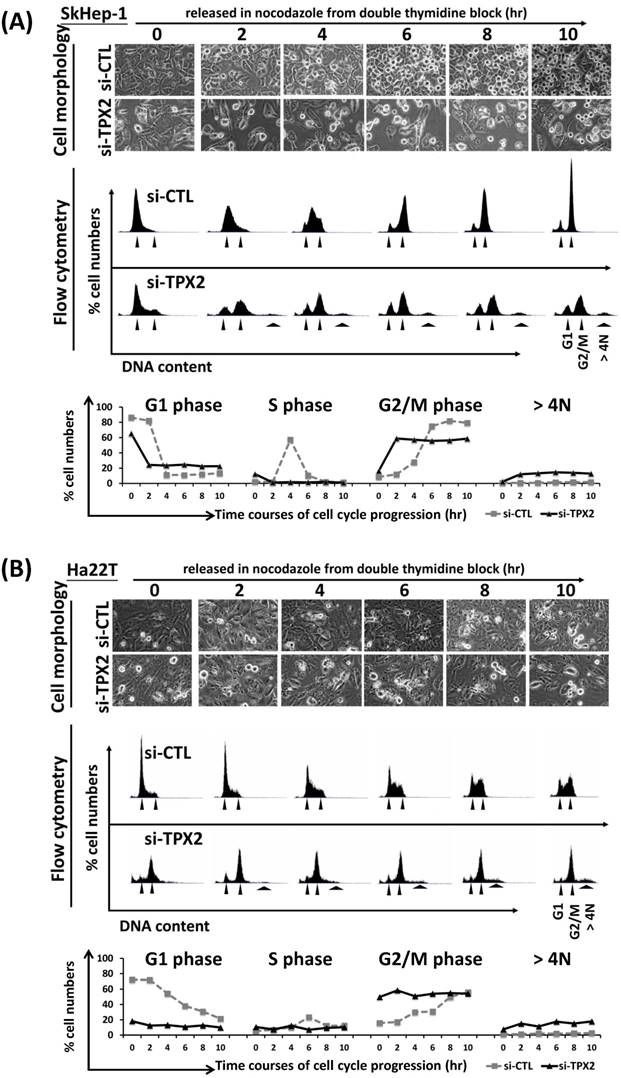
Since TPX2 expression was regulated by cell cycle progression, we further examined whether TPX2 is involved in the control of the cell cycle and contributes to the tumor growth of liver cancer cells. The cell cycle profiles of TPX2 knockdown cells were measured using an image-flow cytometry assay. Depletion of TPX2 increased the G2/M phase and significantly increased the sub-G1 and G2/M phase proportions in Ha22T and SkHep-1 cancer cells. In contrast, TPX2 depletion decreased the G1 phase in both cell lines. Practically, TPX2 depletion raised the polyploidy in SkHep-1 and Ha22T cells (Figure 7A). By Annexin V staining assay, targeting TPX2 increased 8-folds in SkHep-1 and 5-folds in Ha22T with annexin V positive cells (Figure 7B). Cells that undergo senescence due to ectopic overexpression of p21 or p16INK4a experience growth arrest. TPX2 depletion presented an increased senescence cell population and increased p21 mRNA expression (cell cycle arrest indicator) in both HCC cell lines (Figure 7C, D). These results showed a possibility of G2 to M phase progression error. We next analyzed the dynamics of cell cycle progression by TPX2 depletion. SkHep-1 and Ha22T cells were cell cycle synchronized by a double thymidine block and released into nocodazole medium, led cells from G1/S boundary phase progressed to G2/M phase. Through the time courses of cell cycle progression, we found that the TPX2-depleted cells could not be sybchrinized in G1/S boundary phase, and presented a cell cycle progression arrest event, particularly in 4N DNA (as G2/M fraction) proportion arrest. At the same time, TPX2 depletion also increased the polyploidy (>4N DNA) proportion in both cell lines. These results indicated that there was G2/M transition and mitotic error in the TPX2-depleted cells (Figure 8).
Depletion of TPX2 increased the proportion of polyploid cells, apotosis, senescence and arrested cell cycle progression. (A) Depletion of TPX2 increases the proportion of sub-G1, G2/M phase and polyploidy cells; in contrast, the G1 phase in human liver cancer cells is decreased. Scrambled siRNA (20 nM, si-Ctrl) or siRNA oligos pool against TPX2 (20 nM, si-TPX2) was transfected into human liver cancer Skhep-1 and Ha22T cells for 72 h; then, the cells were harvested. The knockdown cells were fixed and stained with DAPI to examine proportions of the cell cycle using an image-flow cytometry assay as described in the materials and methods section. The data were analyzed and quantitated with Nucleoview NC-3000 Software. (B) Using the annexin V expression analysis for apotosis cell detection. The siRNA transfected HCC cells were stained with annexin V and PI without fixed. The proportions of the cell cycle using an image-flow cytometry assay. The lower right quadrant (red box) were indicated the annexin V positive (early apotosis) cells. The representative and quantitative frequency data were shown in below. (C) Senescence associated β-galactosidase assay (SA-β-gal) used for identifying senescent cells, the representative and quantitative frequency data were shown in right panel. (D) The mRNA expression levels of TPX2 and P21 were measured by real-time RT-PCR in two siRNA transfected HCC cells. The results are expressed as the mean ± SD from 2 individual experiments (Student's t-test, *P < 0.05, **P < 0.01, ***P < 0.001).
The G2/M progression arrest by TPX2 depletion. SkHep-1 (A) and Ha22T (B) cells were cell cycle synchronized by a double thymidine block and released into nocodazole medium for the indicated times. The status of mitotic round-up cells was observed at the indicated time courses (upper panel). The cell cycle proportions were measured by image-flow cytometry and DNA contents are indicated by the arrowhead (G1, G2/M and > 4N) (middle panel). Quantitated values of cell cycle phase for each time point in the diagram of curves (bottom panel).
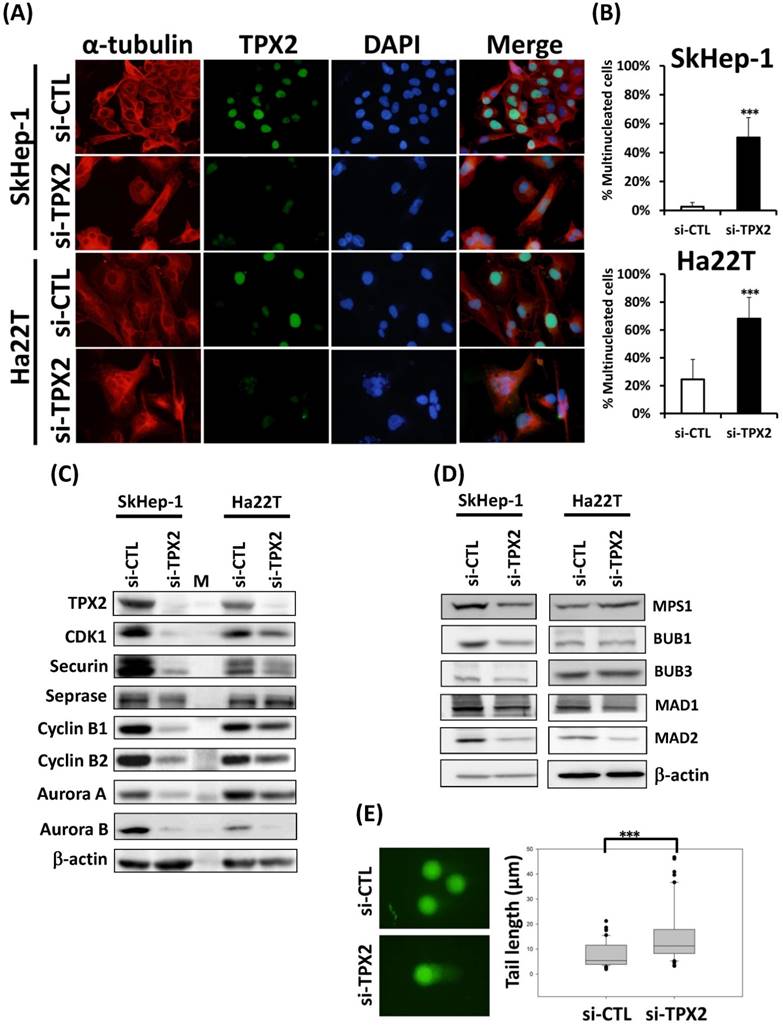
TPX2 depletion increases genomic instability resulting in multinucleation and DNA damage in liver cancer cells
Since TPX2 silencing caused the G2/M transition and mitotic error and increased the DNA content, this finding indicated that the cell nuclei were varied. Morphological examination of the cell nucleus revealed that transfection with the TPX2 siRNA oligos led to multinucleated cells (Figure 9A), an almost 16-fold (4% vs 21%) increase in SkHep-1 cells and a 2.7-fold (25% vs 68%) increase in Ha22T cells compared with cells transfected with the control RNAi oligos (Figure 9B). To define the role of TPX2 depletion in genomic instability, we assessed the DNA damage in SkHep-1 cells transfected with siRNAs targeting TPX2. DNA damage was measured by comet assay (a single cell gel electrophoresis assay). Undamaged DNA remained in the nucleoid and was observed as the comet head, whereas damaged DNA migrated through the agarose gel and formed a comet tail. We measured the tail length and found a significant increase in the mean tail length in TPX2-depleted cells (Figure 9E), indicating that TPX2 inhibition resulted in increased DNA damage. TPX2 depletion resulted in increased polyploidy, multinucleation and DNA damaged cells, indicating mitotic progression errors in the TPX2-depleted cells. Therefore, we followed up on certain chromosome segregation related genes, which also contribute to chromosomal instability (CIN), and we found the CDK1, securin, separase, cyclin B1, B2, Aurora A and Aurora B proteins were downregulated in TPX2-targeted cells (Figure 9C). In order to understand the effect of TPX2 in causing mitotic errors and increasing polyploidy cells, we then check the markers of spindle assembly checkpoint (SAC) in TPX2 depleted cells. The MPS1, BUB1, BUB3, MAD1 and MAD2 were downregulated in TPX2-targeted SkHep-1 cells, but only MAD1 and MAD2 were downregulated in Ha22T cells (Figure 9D).
TPX2 depletion increases genomic instability resulting in multinucleation and DNA damage in liver cancer cells. (A) TPX2 and α-tubulin immunofluorescence in TPX2-depleted SkHep-1 and Ha22T cells. Representative images are shown. TPX2 expression was decreased in si-TPX2 treated cells. (B) Histograms of multinucleation in both liver cancer cell lines. Morphological examination of the cell nucleus (DAPI-stain) revealed that transfection with the TPX2 RNAi oligo led to multinucleated cells. (C) Western blot analysis to determine the protein levels of CDK1, securin, separase, cyclin B1, B2, Aurora A and Aurora B proteins in TPX2-targeted cells. β-actin was used as a loading control. (D) Western blot analysis to determine the protein levels of MPS1, BUB1, BUB3, MAD1 and MAD2 proteins in TPX2-targeted cells. β-actin was used as a loading control. (E) Neutral comet assay of SkHep-1 cell 3 days after transfection with siRNAs. Representative nuclei are shown. Dot plots of Tail moments are shown (right panel) (si-CTL, n=100; si-TPX2, n=100).
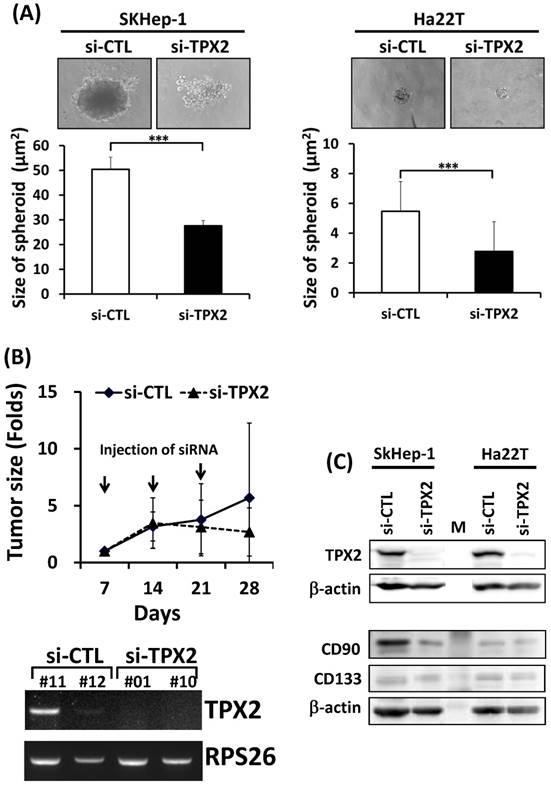
The therapeutic potential of TPX2 targeting in HCC
To investigate the role of TPX2 in tumorigenesis, we performed a spheroid formation assay and found that TPX2 inhibition reduced the spheroid size in both liver cancer cell lines (Figure 10A). Moreover, we performed an in vivo experiment to evaluate the feasibility of TPX2 as a therapeutic target. siRNA against TPX2 mixed with in vivo jetPEI reagent was injected into xenografted tumors. Treatment with TPX2 siRNA injection, given once a week for 3 weeks, resulted in tumor growth suppression. The measurement of TPX2 expression was used the resection tumors from mouse xenograft model at the endpoint date, the expression levels of TPX2 were validated by RT-PCR. The expression of TPX2 mRNA were detected in two si-CTL siRNA injected tumors, but not detected in si-TPX2 siRNA injected tumors (Figure 10B). Moreover, the suppression of tumorigenesis by TPX2 depletion resulted in decreased CD90 protein but not CD133 in both SkHep-1 and Ha22T cell lines (Figure 10C).
The therapeutic potential of TPX2 targeting in HCC. (A) Scrambled siRNA (20 nM, si-Ctrl) or siRNA oligos pool against TPX2 (20 nM, si-TPX2) was transfected into human liver cancer Skhep-1 and Ha22T cells and seeded on an ultra-low attachment 96-well microplate for spheroid formation. The cells were fixed to observe spheroid formation (upper panels), and the spheroid sizes were measured with micrometer ruler under a microscopic low-power field. The quantitative results are shown in the lower panels. (B) Chronological changes in the tumor volume after two injections of the indicated siRNA. The volume of tumors was determined every 7 days as described in Materials and Methods. At the day 28, the mRNA measurements of TPX2 in resection tumors from mouse xenograft model were validated by RT-PCR (lower panel). (C) Western blot analysis to determine the protein levels of TPX2, CD90 and CD133 proteins in TPX2-targeted cells. β-actin was used as a loading control.
Discussion
Cell cycle deregulation is a common feature of human cancer. Tumor cells accumulate mutations that result in unscheduled proliferation, genomic instability and chromosomal instability [27]. Chromosome instability plays a dual role in cancer; low chromosome instability results in a slight growth advantage in tumor promotion and transformation, whereas high chromosome instability leads to cell death and acts as a tumor-suppressor mechanism [28, 29]. Therefore, inducing massive aneuploidy at mitotic division has been proposed as a therapeutic strategy to selectively eliminate highly proliferating tumor cells [30-32]. The present study describes TPX2 depletion that led to G2/M phase stagnation and mitotic error, increased the polyploidy of cells and the genomic instability, resulting in multinucleation and DNA damage in liver cancer cells. We suggest a potential therapeutic benefit of TPX2 targeting in HCC.
Sister chromatid cohesion ensures accurate chromosome segregation and pro- motes faithful DNA repair by homologous recombination. Thus, cohesin is essential for genome stability [33]. At the onset of mitosis, most cohesin is released from chromatin to allow proper sister chromatid resolution and efficient segregation during anaphase. This process is known as the prophase dissociation pathway and requires the action of three protein kinases, cyclin-dependent kinase 1 (CDK1), aurora kinase B (AURKB) and polo-like kinase 1 (PLK1) [33]. Aurora-A also appears to be involved in microtubule formation and/or stabilization at the spindle pole during chromosomal segregation [34]. Aurora-A associates with the centrosome and spindle microtubules during mitosis and plays a critical role in various mitotic events, including the establishment of the mitotic spindle, the duplication of centrosomes, centrosomal separation and maturation, chromosomal alignment, spindle assembly checkpoint, and cytokinesis [34].
The anaphase program is induced by the activity of the anaphase-promoting complex or cyclosome (APC/C). Upon the release from spindle assembly checkpoint (SAC) arrest, the APC/C begins to promote proteolysis of securin and cyclin B1. Degradation of cyclin B1 causes a decline in cdk1 activity, and that of securin induces activation of separase, thereby promoting dissociation of sister chromatid cohesion and poleward movement of sisters [35]. During the prometaphase, unattached kinetochores catalyse the formation of the mitotic checkpoint complex (MCC) composed of MPS1, Bub1, Bub3, Mad2 and Cdc20, leading to inhibition of the APC/C. Once all the chromosomes are aligned with their kinetochores attached to the spindle, generation of the MCC ceases, allowing Cdc20 to activate the APC/C, leading to the ubiquitylation and degradation of securin and cyclin B1 than cleaves the cohesin ring structure; this opens the ring, allowing sister chromatids to separate [36]. Molecular and structural studies indicate that Aurora-A is activated fully and localized properly during mitosis through interaction with the spindle regulator TPX2 [37], which protects Aurora-A from degradation during interphase and mitosis in human cells. Specifically, Aurora-A levels are reduced in G2 and prometaphase cells in which TPX2 expression is silenced, and this reduction in Aurora-A levels requires proteasome activity and the Cdh1 protein, which activates the APC/C ubiquitin ligase [38]. Aurora-A interacts with cyclin B1, which is also essential for mitosis, and Aurora-A overexpression reduces the interaction of cyclin B1 with APC subunits and protects cyclin B1 against degradation; conversely, Aurora-A knockdown through RNAi enhances cyclin B1 degradation [39]. Moreover, in the investigation of Giubettini et al., TPX2 plays a major role in regulating the activity and localization of Aurora-A in mitotic cells. The cyclin B1 expression had no difference in control and TPX2 depletion U2OS cells [38]. This finding was very different than our results, however, in TPX2 depleted U2OS cells, the Aurora A protein was not complete downregulation, similar to our TPX2 depleted Ha22T cells. This suggested that remain sufficient Aurora A should be protecting the cyclin B1 against degradation. Our study shows that the downregulation of TPX2 increased polyploidy, multinucleation and DNA damage in cells, indicating mitotic progression errors in TPX2-depleted liver cancer cells that resulted in reduced MPS1, BUB1, BUB3, MAD1, MAD2, CDK1, securin, separase, cyclin B1, B2, Aurora A and Aurora B protein expression in TPX2-targeted cells. These results indicated that depletion of this protein expression disrupted the machinery involved in the proper orientation of sister kinetochores and the separation of sister chromatids during mitosis and led to a multinucleated nucleus and G2/M transition delay in TPX2-depleted liver cancer cells.
Cyclin E, a G1 cyclin essential for G1-S phase transition, is known to have a profound effect on tumorigenesis and is a therapeutic target [40, 41]. Cyclin E/CDK2 is responsible for initiating the assembly of the pre-replication complex. As the cell passes from G1 into S phase, cyclin A associates with CDK2, replacing cyclin E. This sequential event is important and tightly initiated in DNA replication in S phase [40]. Our results showed that TPX2 depletion led to reduced liver cancer cell growth and inhibition of tumorigenesis. The disruption of S phase cyclin proteins, cyclin A, E and CDK2 protein, indicated that the DNA synthesis process was blocked, and cell proliferation was reduced. Moreover, the expression levels of Cyclin D were not suppressed, instead even increase in TPX2 silencing cells. It suggested the cell growth inhibition not only caused by apotosis, M phase arrest but also G1/S phase transition error.
The therapeutic relevance of targeting TPX2 has already been shown by using TPX2 siRNA. Suppressing TPX2 gene expression in esophageal cancer EC9706 cells affected esophageal cancer cell growth and invasion ability [42]. TPX2 siRNA transfection significantly reduced tumor growth of cervical cancer [43] and pancreatic cancer [44] xenografts in nude mice. Our results also show that TPX2 depletion by TPX2 siRNA transfection reduced in vitro HCC tumor spheroid formation, and TPX2 siRNA injection inhibited in vivo HCC xenograft tumorigenesis. HCC remains one of the most difficult cancers to treat, with chemotherapies being relatively ineffective. These results demonstrated that TPX2 is important in the regulation of tumor growth and could thus be a potential therapeutic target as a novel treatment strategy for hepatocellular carcinoma.
In conclusion, targeting CIN or aneuploidy for cancer therapy is being considered, although further molecular and in vivo research are required to establish clear pathways and properly validate targets. Our findings indicated that TPX2 depletion results in DNA damage, mitotic progression failure and multinucleation, thereby arresting the G2/M transition in cancer cells, which suggests TPX2 may be a potential new target for the treatment of liver cancer.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST103-2320-B-075B-001-MY3 to Hung-Wei Pan), Kaohsiung Veterans General Hospital Research Program, Taiwan (VGHKS103-G01-4, VGHKS104-G01-1 to Hung-Wei Pan, VGHKS103-040 to Chao-Wen Hsu).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen CH, Chen YY, Chen GH, Yang SS, Tang HS, Lin HH. et al. Hepatitis B virus transmission and hepatocarcinogenesis: a 9 year retrospective cohort of 13676 relatives with hepatocellular carcinoma. Journal of hepatology. 2004;40:653-9
2. Chen PJ, Chen DS, Lai MY, Chang MH, Huang GT, Yang PM. et al. Clonal origin of recurrent hepatocellular carcinomas. Gastroenterology. 1989;96:527-9
3. Wang CH, Wey KC, Mo LR, Chang KK, Lin RC, Kuo JJ. Current trends and recent advances in diagnosis, therapy, and prevention of hepatocellular carcinoma. Asian Pacific journal of cancer prevention: APJCP. 2015;16:3595-604
4. Tung-Ping Poon R, Fan ST, Wong J. Risk factors, prevention, and management of postoperative recurrence after resection of hepatocellular carcinoma. Annals of surgery. 2000;232:10-24
5. Waghray A, Murali AR, Menon KN. Hepatocellular carcinoma: From diagnosis to treatment. World journal of hepatology. 2015;7:1020-9
6. Perez de Castro I, Malumbres M. Mitotic Stress and Chromosomal Instability in Cancer: The Case for TPX2. Genes Cancer. 2012;3:721-30
7. Gadde S, Heald R. Mechanisms and molecules of the mitotic spindle. Curr Biol. 2004;14:R797-805
8. Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. Int Rev Cytol. 2008;265:111-58
9. Manning AL, Compton DA. Structural and regulatory roles of nonmotor spindle proteins. Curr Opin Cell Biol. 2008;20:101-6
10. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nature reviews Cancer. 2005;5:773-85
11. Wittmann T, Boleti H, Antony C, Karsenti E, Vernos I. Localization of the kinesin-like protein Xklp2 to spindle poles requires a leucine zipper, a microtubule-associated protein, and dynein. J Cell Biol. 1998;143:673-85
12. Brunet S, Dumont J, Lee KW, Kinoshita K, Hikal P, Gruss OJ. et al. Meiotic regulation of TPX2 protein levels governs cell cycle progression in mouse oocytes. PLoS One. 2008;3:e3338
13. Yang X, Liu G, Xiao H, Yu F, Xiang X, Lu Y. et al. TPX2 Overexpression in Medullary Thyroid Carcinoma Mediates TT Cell Proliferation. Pathol Oncol Res. 2014
14. Hsu PK, Chen HY, Yeh YC, Yen CC, Wu YC, Hsu CP. et al. TPX2 expression is associated with cell proliferation and patient outcome in esophageal squamous cell carcinoma. J Gastroenterol. 2014;49:1231-40
15. Geiger TR, Ha NH, Faraji F, Michael HT, Rodriguez L, Walker RC. et al. Functional analysis of prognostic gene expression network genes in metastatic breast cancer models. PLoS One. 2014;9:e111813
16. Yan L, Li S, Xu C, Zhao X, Hao B, Li H. et al. Target protein for Xklp2 (TPX2), a microtubule-related protein, contributes to malignant phenotype in bladder carcinoma. Tumour Biol. 2013;34:4089-100
17. Liang B, Jia C, Huang Y, He H, Li J, Liao H. et al. TPX2 Level Correlates with Hepatocellular Carcinoma Cell Proliferation, Apoptosis, and EMT. Digestive diseases and sciences. 2015;60:2360-72
18. Liu Q, Yang P, Tu K, Zhang H, Zheng X, Yao Y. et al. TPX2 knockdown suppressed hepatocellular carcinoma cell invasion via inactivating AKT signaling and inhibiting MMP2 and MMP9 expression. Chinese journal of cancer research = Chung-kuo yen cheng yen chiu. 2014;26:410-7
19. Huang Y, Guo W, Kan H. TPX2 is a prognostic marker and contributes to growth and metastasis of human hepatocellular carcinoma. International journal of molecular sciences. 2014;15:18148-61
20. Aref AM, Hoa NT, Ge L, Agrawal A, Dacosta-Iyer M, Lambrecht N. et al. HCA519/TPX2: a potential T-cell tumor-associated antigen for human hepatocellular carcinoma. OncoTargets and therapy. 2014;7:1061-70
21. Satow R, Shitashige M, Kanai Y, Takeshita F, Ojima H, Jigami T. et al. Combined functional genome survey of therapeutic targets for hepatocellular carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:2518-28
22. Perez de Castro I, de Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899-912
23. Pan HW, Ou YH, Peng SY, Liu SH, Lai PL, Lee PH. et al. Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer. 2003;98:119-27
24. Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell cycle. 2006;5:2676-87
25. Lee YC, Pan HW, Peng SY, Lai PL, Kuo WS, Ou YH. et al. Overexpression of tumour-associated trypsin inhibitor (TATI) enhances tumour growth and is associated with portal vein invasion, early recurrence and a stage-independent prognostic factor of hepatocellular carcinoma. European journal of cancer. 2007;43:736-44
26. Lin SY, Pan HW, Liu SH, Jeng YM, Hu FC, Peng SY. et al. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4814-20
27. Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell death and differentiation. 2012;19:369-77
28. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO reports. 2012;13:528-38
29. Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO reports. 2012;13:501-14
30. Janssen A, Medema RH. Genetic instability: tipping the balance. Oncogene. 2013;32:4459-70
31. Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19108-13
32. Duijf PH, Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32:4727-36
33. Losada A. Cohesin in cancer: chromosome segregation and beyond. Nature reviews Cancer. 2014;14:389-93
34. Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70:661-87
35. Shindo N, Kumada K, Hirota T. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Developmental cell. 2012;23:112-23
36. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Current biology: CB. 2012;22:R966-80
37. Asteriti IA, Rensen WM, Lindon C, Lavia P, Guarguaglini G. The Aurora-A/TPX2 complex: a novel oncogenic holoenzyme? Biochim Biophys Acta. 2010;1806:230-9
38. Giubettini M, Asteriti IA, Scrofani J, De Luca M, Lindon C, Lavia P. et al. Control of Aurora-A stability through interaction with TPX2. Journal of cell science. 2011;124:113-22
39. Qin L, Tong T, Song Y, Xue L, Fan F, Zhan Q. Aurora-A interacts with Cyclin B1 and enhances its stability. Cancer Lett. 2009;275:77-85
40. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature reviews Cancer. 2009;9:153-66
41. Butt AJ, Caldon CE, McNeil CM, Swarbrick A, Musgrove EA, Sutherland RL. Cell cycle machinery: links with genesis and treatment of breast cancer. Advances in experimental medicine and biology. 2008;630:189-205
42. Liu HC, Zhang GH, Liu YH, Wang P, Ma JF, Su LS. et al. TPX2 siRNA regulates growth and invasion of esophageal cancer cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2014
43. Jiang P, Shen K, Wang X, Song H, Yue Y, Liu T. TPX2 regulates tumor growth in human cervical carcinoma cells. Mol Med Rep. 2014;9:2347-51
44. Miwa T, Kokuryo T, Yokoyama Y, Yamaguchi J, Nagino M. Therapeutic potential of targeting protein for Xklp2 silencing for pancreatic cancer. Cancer medicine. 2015;4:1091-100
Author contact
![]() Corresponding authors: Hung-Wei Pan, Department of Medical Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan. Tel: +886 7 342 2121 ext. 1501; Fax: +886 7 346 8056; E-Mail: hwpangov.tw; d89444001com Tony Tong-Lin Wu, Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan. E-Mail: tonywugov.tw
Corresponding authors: Hung-Wei Pan, Department of Medical Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan. Tel: +886 7 342 2121 ext. 1501; Fax: +886 7 346 8056; E-Mail: hwpangov.tw; d89444001com Tony Tong-Lin Wu, Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan. E-Mail: tonywugov.tw