Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(8):1425-1432. doi:10.7150/jca.18371 This issue Cite
Research Paper
Upregulation of Coxsackie Adenovirus Receptor Sensitizes Cisplatin-Resistant Lung Cancer Cells to CRAd-Induced Inhibition
Ali Sakhawat1*, Yanan Liu2*, Ling Ma1, Tahir Muhammad1, Shensen Wang1, Lina Zhang1, Xianling Cong3, Yinghui Huang1 ![]()
1. College of Life Sciences and Bio-Engineering, Beijing University of Technology, China;
2. Basic Medical College, Jilin University, China;
3. China-Japan Union Hospital, Jilin University, China.
* The first two authors contributed equally to the paper.
Received 2016-11-16; Accepted 2017-3-7; Published 2017-5-12
Abstract
Objective. Conditionally replicating adenoviruses (CRAds) have been proven potent oncolytic viruses in previous studies. They selectively replicate in the tumor cells because of incorporated survivin promoter and ultimately lead to their killing with minimal side effects on normal tissue. Chemotherapy with cisplatin is commonly employed for treating tumors, but its cytotoxic effects and development of resistance remained major concerns to be dealt with. The aim of this study was to explore the anticancer potential of survivin regulated CRAd alone or in combination with cisplatin in the A549 lung cancer cell line and cisplatin-resistant lung cancer cell line, A549-DDPR.
Methods. CRAd was genetically engineered in our laboratory by removing its E1B region and adding survivin promoter to control its replication. A549, H292, and H661 lung cancer cell lines were procured from the CAS-China. The anti-tumor effectiveness of combined treatment (cisplatin plus CRAd) was evaluated in vitro through MTS assays and in vivo through mouse model experimentation. RT- PCR was used to assess MDR gene and mRNA expression of coxsackie adenoviral receptor (CAR).
Results. Results of in vitro studies established that A549 lung cancer cells were highly sensitive to cisplatin showing dose-dependent inhibition. The resistant cells of A549-DDPR exhibited very less sensitivity to cisplatin but were infected with CRAd more efficiently as compared to A549. A549-DDPR cells exhibited higher expression of MDR gene and CAR in the RT-PCR analysis. The nearly similar rise in the CAR expression was seen when lung cancer cell lines received cisplatin in combined treatment (cisplatin plus CRAd). Combined anti-cancer therapy (cisplatin plus oncolytic virus) proved more efficient than monotherapy in the killing of cancer cells. Results of in vivo experiments recapitulated nearly similar tumor inhibition activities.
Conclusion. This study highlighted the significant role of survivin in gene therapy as it has the potential to render CRAd more tumor specific. It also establishes that higher CAR expression plays a vital role in the success of adenovirus-based therapies. Furthermore, a careful combination of chemotherapy drugs and oncolytic viruses can culminate in significant therapeutic achievements against cancer.
Keywords: Lung cancer, Resistance, Conditional replication Adenovirus, Chemotherapy, Cisplatin, Survivin, CAR.
Introduction
Administration of chemotherapeutic agents coupled with exposure to radiation and surgical removal of tumor mass are treatment strategies widely used to combat cancer. A chemotherapeutic agent cisplatin or DDP is administered either alone or in combination with other chemo drugs to treat cancer, but its clinical applications are affected by severe chemotoxic side effects and increasing drug resistance issues [1, 2]. Lung cancer is ranked second amongst all malignant tumors with the highest cancer-related mortality rate [3]. Small cell lung carcinoma is less common as compared to non-small cell lung carcinoma but more severe [4]. Beside achievements have been made in the treatment of lung cancer, recurrence has remained the issue nearly in all patients after initial treatment. This issue is may be due to drug resistance and metastasis of cancer [5, 6]. Research had been conducted to devise a combined therapeutic strategy by using cisplatin and adenoviruses together to obtain better results and to overcome the drawbacks of standard treatments [7, 8].
Oncolytic adenoviruses have been proven clinically as potent anticancer agents owing important advantages like broad cell targets, fewer side effects [9], inordinate gene carrying ability, and relatively lower capacity to alter host genes [10]. ONYX-015 was the first reported adenoviral vector that showed cancer-specific activity [11]. Although the monotherapy of adenovirus (Adv) was discouraged [12], it has been extensively engineered for combined therapies against cancer. Recent studies explained that CAR has a significant role in the entry of adenoviral vector into tumor cells. Investigations have established that the success of an Adv based gene therapy is dependent on higher expression of coxsackievirus/adenovirus receptor (CAR) on cancer cell surface because it is necessary for Adv internalization [13]. On the cell surface, following recognition, Adv is attached to CAR receptor. Interaction of viral pentone base motif with integrins, αvβ3, and αvβ5 facilitates its internalization into the cell [14]. Then through nuclear pore complex, the viral genome is translocated to nucleoplasm where its replication takes place [15].
Ads are genetically engineered to generate CRAds, which specifically replicate in and lyse the tumor cells [16]CRAds are generated either through gene deletion, as in Onyx-015 [17] or induction of promoter sequences like survivin, hTERT, and prostate specific antigen [18]. CRAds internalization into the cells depends on CAR expression which is usually quite low on tumor cell surfaces [19, 20]. Alteration in Adv tropism [21] and introduction of polylysine and heparin sulfate sequences in fiber knot domain has increased the internalization of CRAds [17]
p53, a tumor suppressor gene, is found to be altered in many types of human cancer [22]. In normal cells, its expression is low due to the absence of oncogenic activation [23]. World's first commercially available gene therapy product of China in 2003, gendicine, also employed a wt p53 gene and delivered it via Adv 5 to treat head and neck squamous cell carcinoma, HNSCC [24]. The working principle of such gene-based therapies has been criticized by some studies which showed that the functional p53 genes might be present in some tumors [25] and Adv replication may be independent of p53 status [26].
Survivin, which is a protein of inhibitor of apoptosis (IAP) family, exhibited many advantages like high expression specifically in tumor cells, broad spectrum anti-tumor effects, and very little expression in normal cells. Owing to these qualities, the use of survivin in gene therapy is encouraged to acquire high tumor specificity [27, 28]. Many studies have reported that survivin expression is regulated at the transcriptional level [29, 30]. It has placed survivin among top five transcriptomes which are highly expressed in cancerous cells with minimal expression in normal cells of same tissue [31].
Currently, one of the major issues faced by the clinical oncologists which impede effective cancer treatment is rapidly developing resistance in tumor cells against chemotherapy agents. The synergistic inhibitory action of combination therapies, involving an oncolytic virus and a chemotherapy agent, has already been established by many investigations [32, 33]. We developed an oncolytic Adv, genetically engineered with a promoter (Sur-P) of IAP member, survivin. In this study, we also developed a cisplatin-resistant lung cancer cell line, A549-DDPR, to examine the impact of our treatment on the cancer cells resistant to chemotherapy. We performed in vitro and in vivo experiments to investigate the therapeutic effect and mechanism of Sur-P controlled CRAd alone or in combination with cisplatin in lung cancer cell lines, A549 and A549-DDPR.
Materials and Methods
Generation of cisplatin resistant cell line (A549-DDPR) and adenovirus
Cisplatin resistant lung cancer cell line, A549-DDPR, was developed from lung cancer cells of A549. Clinically relevant doses of cisplatin were given to A549 cells in a serum free medium for 60 minutes, and the dose was raised stepwise (0-76 µM), afterward, cells were cloned. A549-DDPR cell clone was selected, further treated with cisplatin (76 µM) and propagated for 40 passages with same cisplatin sensitivity. The ability of CRAds to replicate specifically in cancer cells has proven their use as effective anticancer agents. In this study, survivin responsive CRAds were prepared in which the survivin promoter controls the regulation of adenoviral E1A region while the adenoviral E1B region was deleted. This virus showed efficient cancer selective phenotypes absent reduced anticancer activity. Firefly luciferase expressing Ad-Luc virus was used as a control.
Cell culture
Lung cancer cell lines A549, H661, and H292 used in this study were obtained from the Cell Collection Center, Shanghai (CAS-China). CRAd was multiplied by using a cell line which contains E1A region (HEK-293). RPMI-1640 medium (Gibco-BRL, HyClone) and DMEM containing 10 % FBS were used for culturing and propagation of lung cancer cell lines and HEK-293 cell line.
In vitro analysis of tumor inhibition
In 24 wells plate, cells were placed at a concentration of 1×105cells in each well. After 24 hours, different concentrations of CRAds viruses and DDP were given to cells in each well. By the addition of both cisplatin and CRAds serially, two treated cell groups were prepared. Serially treated cells were shifted to a 96 well plate with a concentration of 5 × 103 cells in each well. Then MTS/ PMS reagents were added in the treated cells and cells were incubated at standard conditions (37C°, 5 % CO2 for 180 minutes). Cells were monitored for five days then spectrophotometrically the absorbance was taken at 490 nm.
Semiquantitative reverse transcription-PCR
Total RNA was extracted by using TRIzol reagent (Ambion, life technologies) RNA samples were subjected to RT-PCR to obtain cDNA by using an RT-reagent kit (TakaRa). The primers employed in this study to amplify CAR (PCR product size 218bp), MDR (417bp) and GADPH control (371bp) are given below:
GAPDH: S: 5'GATTGTTGCCATCAACGACC3' AS: 5'GTGCAGGATGCATTGCTGAC3'
CAR: S: 5'CCACCTCCAAAGAGCCGTAC3' AS: 5'ATCACAGGAATCGCACCC3'
MDR: S: 5'TCGTAGGAGTGTCCGTGGAT3' AS: 5'CATTGGCGAGCCTGGTAG3'
Amplified PCR products were confirmed by performing 1% agarose gel electrophoresis. After electrophoresis, the gel was stained with ethidium bromide and visualized under UV light.
Tumor model
Six to eight-week-old female BALB/C nude mice were procured from the Chinese Academy of Sciences. Experimentations on animals were conducted by following the ethical guidelines from the NIH. A549 and A549-DDPR cells (4×106) were inoculated into the right flanks of the mice subcutaneously (SC). Tumors were visible on the 15th day after inoculation in both A549 and A549-DDPR cells.
In vivo analysis of tumor inhibition
Nude Mice with tumors were divided into four groups a, b, c, d; each group consisted of six mice. Each group was treated differently. Group 'a' was treated with only DDP, group 'b' was treated with only CRAds, and 'c' group was treated with both DDP and CRAds (DDP followed by CRAds order), and group 'd' received PBS. Ad-Luc control virus was added into each treatment group. After the one-month volume of tumor in each treatment group was measured to evaluate the effects of treatment.
Statistical analysis
Obtained data was subjected to Student's t-test to find out the statistical significance, and data values are presented as with standard deviation. Data value at p < 0.05 was considered statistically significant.
Results
Sensitivity of A549 and A549-DDPR to cisplatin
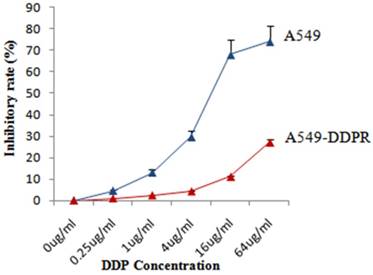
Both cell lines, A549 and A549-DDPR, were treated with different concentrations of cisplatin and for cytotoxicity assessment, MTS/PMS assay was performed after 72 hr of the treatment. Dose-dependent inhibition of cancer cells was observed in both cell lines (Fig. 1) with maximum inhibition in A549 cells (75 %) at 64 ug/ml of cisplatin dose. Figure 1 shows that inhibitory rate in A549-DDPR cells is very low even at higher cisplatin doses owing to their reduced sensitivity and high resistance to cisplatin. Cisplatin resistant cell line (A549-DDPR) remained stable throughout the period of observation. Severe cytotoxicity towards normal cells at higher chemo doses and increasing incidence of chemoresistance are the major concerns of oncologists worldwide [34, 35]. This study of combined treatment approach was aimed to get higher tumor inhibition rates with minimum damage to healthy cells.
Sensitivity of A549 and A549-DDPR to cisplatin. Both cell lines, A549 and A549-DDPR, showed a large difference in response to variable concentrations of cisplatin when MTS/PMS assay was performed.A549-DDPR exhibit very less sensitivity to cisplatin due to resistance. The data in the line graph and error bars represent the mean of triplicate experiments and SD values respectively.
Enhanced sensitivity of A549-DDPR cells to adenoviral infection
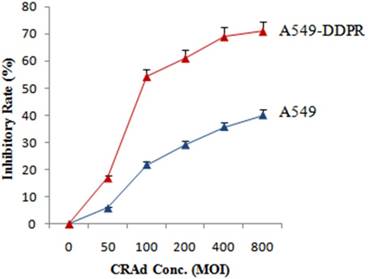
Likewise, to assess susceptibility to CRAds infection and to find optimum MOI, both cancer cell lines (A549 and A549-DDPR) were injected with different MOIs of adenovirus. The results of MTS/PMS assay showed that cisplatin resistantA549-DDPR cells exhibited much higher sensitivity to adenoviral infection as compared to A549 cells. Figure 2 indicates that at 100 MOI nearly 22 % tumor inhibition is achieved in A549 cell line while in cisplatin resistant cell line A549-DDPR, the inhibition rate was almost 2.5 folds higher (55 %). A similar trend was observed at higher doses of CRAd. These observations established that A549-DDPR cells were infected with CRAd more efficiently as compared to non-resistant cells of A549. This significant difference in CRAd transducing ability among two lung cancer cell lines is may be the result of variable CAR expression on their surfaces [2, 36, 37]
In vitro study to assess the sensitivity of A549-DDPR cells to CRAd. A549 and A549-DDPRcells were given different CRAd concentrations. A549-DDPRcells show comparatively very high inhibition rate which is most probably due to higher CAR expression. The data in the line graph and error bars represent the mean of triplicate experiments and SD respectively.
Cisplatin increases the cancer cell-killing potential of CRAd in combined treatment
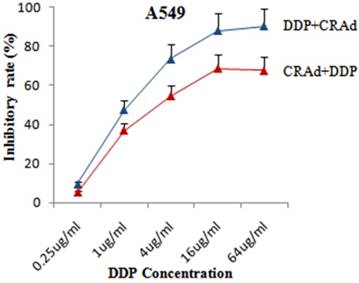
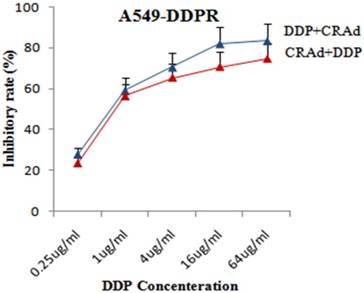
To evaluate that whether cisplatin could augment the tumor suppression capacity of Sur-p regulated CRAd in vitro, both lung cancer cell lines, A549 and A549-DDPR were infected with CRAd at various concentrations of cisplatin (0.25 ug - 64 ug/ml). All treatment groups in in vitro studies were infected with CRAd at 100 MOI. In combined treatment strategy following two sequence approaches were adopted: (a) infecting for 4 hours with CRAd at 100 MOI followed by cisplatin injections for 3 hours; (b) injecting for 3 hours various concentrations of cisplatin followed by infecting for4 hours with CRAd at 100 MOI. MTS/PMS assay revealed that tumor suppressing effect of the combination treatment was more powerful as compared to monotherapy of DDP and CRAd. Figure 3 indicated that the inhibitory effect on A549 cell proliferation obtained by infecting with 100 MOI of CRAd plus cisplatin (1 ug/ml) was higher than that achieved with CRAd (800 MOI) alone. Synergistic therapeutic activity in cancer cells suppression was observed in combined treatment (Fig. 3, 4). Figures 3 and 4 showed that sequence-based approach b (DDP+CRAd) was more effective in tumor suppression as compared to approach a (CRAd+DDP). The difference was more prominent in non-resistant lung cancer cells of A549. The observations of in vitro studies established that cisplatin augmented the tumor inhibitory effect of CRAd-mediated biotherapy.
Enhanced tumor retarding efficacy of combined treatment in A549 cells. Sequence approach b (DDP+CRAd) appeared more successful as compared to sequence approach a. The data in the line graph and error bars represent the mean of triplicate experiments and SD respectively. p < 0.05 value was set as statistically significant for data.
Enhanced tumor retarding efficacy of combined treatment in A549-DDPR cells. High initial inhibition rate was achieved even at a lower dose of DDP and a continuous increase in inhibition was seen with increasing cisplatin doses. The data in the line graph and error bars represent the mean of triplicate experiments and SD respectively. p < 0.05 value was set as statistically significant for data.
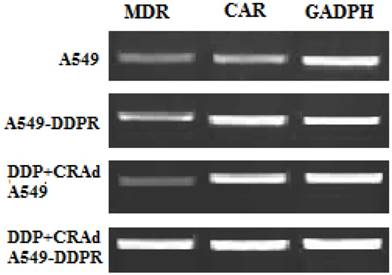
Raised expression of CAR and MDR
We performed RT-PCR analysis to evaluate mRNA levels of MDR and CAR. The aim of RT-PCR experiments was to confirm the cisplatin resistance in A549-DDPR cells and furthermore to investigate the molecular mechanism which raised the anti-cancer activity of combined treatment (DDP plus CRAd). Specific primers for MDR, CAR, and GADPH (internal control) were employed for PCR amplifications. This experiment pointed out that CAR expression was much higher in cisplatin-resistant A549-DDPR cells as compared to non-resistant lung cancer cells of A549. A similar trend was observed in lung cancer cell lines which received cisplatin treatment in combined therapy (DDP+CRAd) (Fig. 5). It can be hypothesized from these outcomes that lung cancer cell lines may be sensitized to adenoviral transduction by cisplatin. These results which linked enhanced transduction efficacy of adv with raised CAR expression were also previously confirmed by some other studies [36, 37]. Lung cancer cell line, A549-DDPR, showed higher expression of MDR gene which confirmed that it had developed resistance to cisplatin (Fig. 5).
RT-PCR analysis to examine the expression of MDR and CAR. Higher expression of both MDR and CAR was found in A549-DDPR cells. Also, cells which received combined treatment (DDP+CRAd), showed higher CAR expressions. GADPH was used as the internal control.
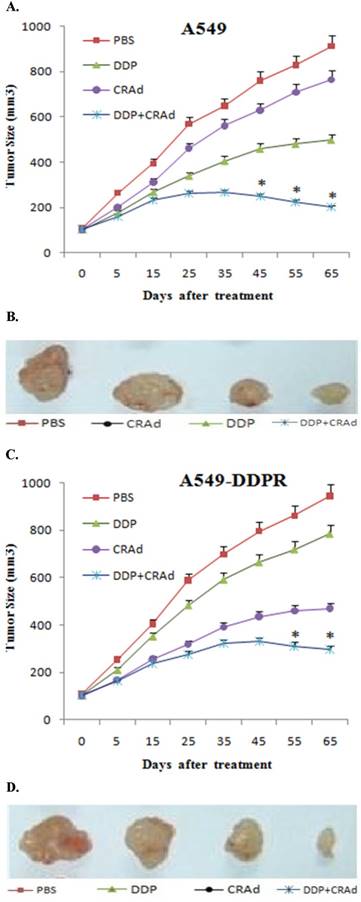
Synergistic therapeutic activity of combined therapy
Our in vitro experiments showed that combined treatment has synergistic inhibitory action on cancer cell proliferation, and the significance of sequence in which combined treatment is given was also established. To discover whether the similar trends can be recapitulated in vivo, we employed 6-8 week old mice, and 8×106 A549 and A549-DDPR cells were injected in their right flank subcutaneously. When tumor volume reached 100-150 mm3, mice were divided into four groups (n=6).We used 4 mg/kg of DDP intratumorally and 100 MOI of CRAd intraperitoneally in our in vivo studies. Three groups were treated with monotherapies of PBS, DDP, CRAd and one group received combined therapy of DDP plus CRAd. A line graph obtained from the observations of in vivo experiments evidenced an antitumor efficacy nearly similar to that was shown by in vitro studies. Figures 6A-D indicated noticeable differences in cancer size reduction efficacy among four treatment groups. The combined treatment group (DDP+CRAd) remained most powerful tumor inhibitor exhibiting synergistic therapeutic activity.
(A, C) In vivo study in female nude mice to evaluate antitumor efficacy of combined treatment. 8×106 cells of A549 and A549-DDPR were injected into nude mice. Upon development of appreciably sized tumors, four mice groups were treated with PBS, CRAd, DDP, and DDP+CRAd respectively. Combined treatment strategy appeared the most powerful in tumor suppression in both cell lines. p < 0.05 value was set as statistically significant for data. 6 (B, D) In vivo images of solid tumors comparing the anti-tumor impact of different treatments. Combined treatment group exhibited the highest antitumor potential.
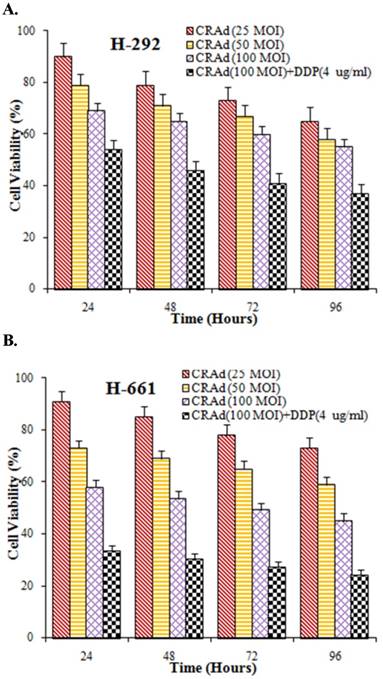
Lung cancer is a group of highly heterogenetic diseases. To further strengthen the results of our study we verified the tumor-inhibiting efficacy of CRAd and cisplatin in vitro in other lung cancer lines also. Two lung cancer cell lines, H292 and H661, were infected with CRAd at different MOIs, alone or with cisplatin. The inhibitory pattern observed in these cell lines was nearly similar to that was seen in A549 cells. The inhibitory effect of combined treatment (CRAd plus cisplatin) was found more potent than the effect of CRAd or cisplatin alone (Fig. 7A, B). Moreover, the decrease in the number of viable cells in the lung cancer cell lines obviously was time and dose-dependent.
(A, B) In vitro study to assess the inhibitory efficacy of CRAd alone or in combination with cisplatin. Cell viability in both lung cancer cell lines H292 and H661 was decreased. Maximum tumor inhibition was observed when CRAd was injected in conjunction with DDP.
Discussion
Gene therapy, manipulating adenoviral vectors to deliver therapeutic gene, has attracted many researchers and oncologists. Genetically engineered adenoviral vector was also employed to transport tumor suppressor gene p53 in world's first commercially available gene therapy product, gendicine. Cancer therapies involving p53 gene has been criticized for being less potent and tumor protective [25, 38-40]. Chemotherapy, despite its severe damage to normal cells and resistance issues, has been the most efficient warhead against cancer so far. Cisplatin is a potent antineoplastic drug. It is well known for the formation of intrastrand crosslink adducts to damage DNA and the activation of apoptosis which culminate in cancer cell killing [2]. Now, the trend has shifted towards combined treatment strategies involving gene therapy, virotherapy, and chemotherapy.
In the present study, instead of the p53 gene, we used survivin promoter (Sur-P) to modify adenoviral vector for specifically targeting cancer cells. Replication of ONYX-015 may not depend on the status of p53 [41], but its anti-cancer activity is beyond doubt. Our Sur-P incorporated adenoviral vector, CRAd, was nearly similar to ONYX-015 but was more tumor cell specific because Sur-P is overexpressed in almost all cancers with limited expression in normal cells [42, 43]. Results of this study showed that CRAd was successful in achieving high cancer cell specificity with very little toxicity towards normal cells. It is in agreement with many recent studies which supported Sur-P role in cancer gene therapy [29, 44, 45].
The results of our experiments established that a combined anti-cancer therapy (cisplatin plus oncolytic virus) was more efficient in the killing of cancer cells than the monotherapies of respective treatments. Many previous experiments have published the similar results [46, 47]. One of the major obstacles in the use of adenoviruses in gene therapy is their interaction and dependency on a transmembrane protein CAR for cell internalization. Many studies have pointed out that CAR has a very low expression in cancer cells [37,48, 49]. In our study, RT-PCR analysis confirmed that cisplatin has the potential to enhance CAR expression on cancer cells. Cisplatin resistant lung cancer cell line, A549-DDPR, also exhibited high expression of CAR as well as MDR genes. Many late studies showed similar results [36, 50].
The objective of our study was to enhance tumor-specific killing with minimum toxicity towards normal cells and moreover to unveil the molecular mechanisms involved. Our strategy of combined treatment (DDP+CRAd) to combat lung cancer appeared synergistic in action to suppress tumor growth; this finding is in agreement with many recent studies [46, 47]. The viable number of mice was raised, and high rate of tumor inhibition was attained. It was because of the highly tumor-targeted oncolytic virus, CRAd, and reduced cisplatin dose.
Against lung cancer, it was the first study of this kind which used Sur-P regulated oncolytic virus in combination with cisplatin. A cisplatin-resistant lung cancer cell line, A549-DDPR, was also produced and employed in this study to examine the impact of treatment on resistant lung cancer cell line. Approximately 2.5 folds higher tumor inhibition was achieved in cisplatin resistant cell line as compared to non-resistant cell line, A549. These results suggested that adenoviral transgenic expression was probably enhanced in A549-DDPR because of drug resistance. We also witnessed in RT-PCR data that levels of CAR messenger RNA were comparatively higher in cisplatin-resistant lung cancer cell line. Recent studies reported similar results for cisplatin-resistant cells of other organs [36, 51]. This study has highlighted the significant role of the survivin promoter in gene therapy which is not restricted to lung cancer only. It further showed that the dose adjustment is very critical in combined therapy as the cells which are pre-exposed to cisplatin before adenoviral-mediated gene therapy and cisplatin-resistant cells showed enhanced adenoviral transduction.
It is concluded from this study that an anti-cancer therapy can be designed by carefully combining cisplatin and genetically engineered oncolytic virus to make significant therapeutic achievements in the war against cancer. Furthermore, adenovirus transduction efficiency should be carefully evaluated before performing gene therapy to adjust doses of a chemotherapy agent and adenovirus, because additive or synergistic action of combined therapy may culminate in the incidence of cytotoxicity or immune response activation.
Drug resistance has emerged as an unavoidable issue with chemotherapy. This study not only indicated the anticancer potential of combined treatment but also suggested a novel treatment approach of using CRAd as monotherapy in chemotherapy-resistant cancer patients. This research offered a highly tumor targeting oncolytic virus, CRAd, for further investigation as lethal warhead against cancer.
Acknowledgements
This research was supported by Natural Science Foundation of China 81472209; Beijing Science Foundation K2015311201501 (China); Jilin Science Foundation 20140414061GH(China).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arany I, Safirstein RL. Cisplatin nephrotoxicity. Seminars in nephrology. Elsevier. 2003:460-4
2. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature Reviews Cancer. 2007;7:573-84
3. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62:10-29
4. Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu Rev Pathol Mech Dis. 2006;1:331-48
5. Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A. et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. Journal of clinical oncology. 2006;24:4539-44
6. Jackman DM, Johnson BE. Small-cell lung cancer. The Lancet. 2005;366:1385-96
7. Egami T, Ohuchida K, Miyoshi K, Mizumoto K, Onimaru M, Toma H. et al. Chemotherapeutic agents potentiate adenoviral gene therapy for pancreatic cancer. Cancer science. 2009;100:722-9
8. Yasui T, Ohuchida K, Zhao M, Cui L, Onimaru M, Egami T. et al. Adenoviral therapy is more effective in gemcitabine-resistant pancreatic cancer than in gemcitabine-sensitive cells. Anticancer research. 2011;31:1279-87
9. Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proceedings of the National Academy of Sciences. 1994;91:8802-6
10. Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. Journal of virology. 1998;72:926-33
11. Gregory RJ, Armentano D, Couture LA, Smith AE. Adenovirus vector for gene therapy. Google Patents. 1997
12. Kirn D, Hermiston T, McCormick F. ONYX-015: clinical data are encouraging. Nature medicine. 1998;4:1341-2
13. Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L. et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nature medicine. 2000;6:879-85
14. Varga MJ, Weibull C, Everitt E. Infectious entry pathway of adenovirus type 2. Journal of virology. 1991;65:6061-70
15. Whittaker GR, Helenius A. Nuclear import and export of viruses and virus genomes. Virology. 1998;246:1-23
16. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ. et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99-102
17. Greenblatt M, Bennett WP, Hollstein M, Harris C. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer research. 1994;54:4855-78
18. Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Research. 1997;57:2559-63
19. Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G. et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. Journal of virology. 1998;72:9706-13
20. Jounaidi Y, Waxman DJ. Use of replication-conditional adenovirus as a helper system to enhance delivery of P450 prodrug-activation genes for cancer therapy. Cancer research. 2004;64:292-303
21. Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA. et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. Journal of the National Cancer Institute. 2003;95:652-60
22. Liu Y, Kulesz-Martin M. P53 regulation and function in normal cells and tumors. MEDICINA-BUENOS AIRES-. 2000;60:9-11
23. Peng Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Human gene therapy. 2005;16:1016-27
24. Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer research. 1993;53:2736-9
25. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. Journal of virology. 1998;72:9470-8
26. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer-a mechanism for early oncogenic pathway addiction? Nature Reviews Cancer. 2006;6:107-16
27. Asanuma K, Kobayashi D, Furuya D, Tsuji N, Yagihashi A, Watanabe N. A role for survivin in radioresistance of pancreatic cancer cells. Japanese Journal of Cancer Research. 2002;93:1057-62
28. Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271-8
29. Bao R, Connolly DC, Murphy M, Green J, Weinstein JK, Pisarcik DA. et al. Activation of cancer-specific gene expression by the survivin promoter. Journal of the National Cancer Institute. 2002;94:522-8
30. Fengzhi L, ALTIERI DC. Transcriptional analysis of human survivin gene expression. Biochemical Journal. 1999;344:305-11
31. Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C. et al. Analysis of human transcriptomes. Nature genetics. 1999;23:387-8
32. Chu RL, Post DE, Khuri FR, Van Meir EG. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clinical Cancer Research. 2004;10:5299-312
33. Heise C, Sampson-Johannes A, Williams A, Mccormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nature medicine. 1997;3:639-45
34. Fillastre J, Raguenez-Viotte G. Cisplatin nephrotoxicity. Toxicology letters. 1989;46:163-75
35. You Z, Fischer DC, Tong X, Hasenburg A, Aguilar-Cordova E, Kieback DG. Coxsackievirusąadenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression1. Cancer gene therapy. 2001;8:168-75
36. Ambriović-Ristov A, Gabrilovac J, Čimbora-Zovko T, Osmak M. Increased adenoviral transduction efficacy in human laryngeal carcinoma cells resistant to cisplatin is associated with increased expression of integrin αvβ3 and coxsackie adenovirus receptor. International journal of cancer. 2004;110:660-7
37. Qin M, Chen S, Yu T, Escuadro B, Sharma S, Batra RK. Coxsackievirus adenovirus receptor expression predicts the efficiency of adenoviral gene transfer into non-small cell lung cancer xenografts. Clinical cancer research. 2003;9:4992-9
38. Bertheau P, Espié M, Turpin E, Lehmann J, Plassa L-F, Varna M. et al. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75:132-9
39. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413-31
40. Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? The lancet oncology. 2003;4:415-22
41. Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nature medicine. 1997;3:917-21
42. Chen J-S, Liu J-C, Shen L, Rau K-M, Kuo H-P, Li YM. et al. Cancer-specific activation of the survivin promoter and its potential use in gene therapy. Cancer gene therapy. 2004;11:740-7
43. Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T. et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer research. 1998;58:5315-20
44. Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S. et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613-22
45. Zhu ZB, Makhija SK, Lu B, Wang M, Kaliberova L, Liu B. et al. Transcriptional targeting of tumors with a novel tumor-specific survivin promoter. Cancer gene therapy. 2004;11:256-62
46. Li D, Zhang Y, Xie Y, Xiang J, Zhu Y, Yang J. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer gene therapy. 2013;20:251-9
47. Zhong S, Yu D, Wang Y, Qiu S, Wu S, Liu XY. An armed oncolytic adenovirus ZD55-IL-24 combined with ADM or DDP demonstrated enhanced antitumor effect in lung cancer. Acta Oncologica. 2010;49:91-9
48. Kim M, Zinn K, Barnett B, Sumerel L, Krasnykh V, Curiel D. et al. The therapeutic efficacy of adenoviral vectors for cancer gene therapy is limited by a low level of primary adenovirus receptors on tumour cells. European Journal of Cancer. 2002;38:1917-26
49. Ma B, Wang Y, Zhou X, Huang P, Zhang R, Liu T. et al. Synergistic suppression effect on tumor growth of hepatocellular carcinoma by combining oncolytic adenovirus carrying XAF1 with cisplatin. Journal of cancer research and clinical oncology. 2015;141:419-29
50. Hemminki A, Kanerva A, Liu B, Wang M, Alvarez RD, Siegal GP. et al. Modulation of coxsackie-adenovirus receptor expression for increased adenoviral transgene expression. Cancer research. 2003;63:847-53
51. Shirakawa T, Sasaki R, Gardner TA, Kao C, Zhang ZJ, Sugimura K. et al. Drug-resistant human bladder-cancer cells are more sensitive to adenovirus-mediated wild-type p53 gene therapy compared to drug-sensitive cells. International journal of cancer. 2001;94:282-9
Author contact
![]() Corresponding author: Yinghui Huang, College of Life Sciences and Bio-Engineering, Beijing University of Technology (BJUT), 100124, P.R. China Tel: (0086)-10-67396342 E-mail: yhuangedu.cn
Corresponding author: Yinghui Huang, College of Life Sciences and Bio-Engineering, Beijing University of Technology (BJUT), 100124, P.R. China Tel: (0086)-10-67396342 E-mail: yhuangedu.cn