Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(8):1433-1440. doi:10.7150/jca.18435 This issue Cite
Research Paper
Anti-Metastatic Effect of Semi-Purified Nuphar Lutea Leaf Extracts
Janet Ozer1 ![]() , Daniel Fishman2, Brit Eilam1, Avi Golan-Goldhirsh4*, Jacob Gopas1, 3*
, Daniel Fishman2, Brit Eilam1, Avi Golan-Goldhirsh4*, Jacob Gopas1, 3*
1. The Shraga Segal Department of Microbiology, Immunology and Genetics, Faculty of Health Sciences Ben-Gurion University of the Negev, Beer Sheva, Israel;
2. Department of Physiology and Cell Biology, Faculty of Health Sciences Ben-Gurion University of the Negev, Beer Sheva, Israel;
3. Dept. of Oncology, Soroka University Medical Center, Beer Sheva, Israel;
4. The Jacob Blaustein Institutes for Desert Research (BIDR), French Associates Institute for Agriculture and Biotechnology of Drylands, Sede Boqer Campus 84990 Israel.
* AGG and JG contributed equally to this work.
Received 2016-11-21; Accepted 2017-3-21; Published 2017-5-12
Abstract
Nuphar lutea L. SM., leaf and rhizome extracts (NUP), contain nupharidines as active components. Nupharidines belong to the sesquiterpene lactones class of a naturally occurring plant terpenoids. This family of compounds has gained considerable interest for treating infection, inflammation and cancer.
NF-κB is a central, downstream regulator of inflammation, cell proliferation and apoptosis. In our previous work we demonstrated strong inhibition of NF-κB activity and induction of apoptosis by NUP. In addition, NUP exhibited anti-inflammatory properties and partial protection from LPS-induced septic shock by modulating ERK pathway and cytokine secretion in macrophages.
In the present study, we examined the effect of NUP in a B16 melanoma experimental murine lung metastasis model and its ability to affect the ERK and NF-κB pathways in variety of cell lines.
We showed that NUP and cisplatin combined treatment was synergistic and reduced the lung metastatic load. In addition NUP treatment inhibited TNFα-induced IκBα degradation and NF- κB nuclear translocation. We also observed that NUP induced ERK activation. Furthermore, ERK inhibition prevented NF-κB inactivation by NUP.
Overall, our work implies that co-administration of NF-κB inhibitors such as NUP, with standard anti-cancer drugs, may act as “sensitizers” for more effective chemotherapy.
Keywords: Nuphar lutea plant extract, nupharidines, melanoma, ERK, NF-κB.
Introduction
Plants biosynthesize various secondary metabolites such as phenolics, alkaloids and terpenoids, which serve in defense against biotic and abiotic stresses. Sesquiterpen thio-alkaloids constitute a group of secondary metabolites that have not been studied in great detail and bear a great potential for medicinal uses. The nupharidines found in Nuphar lutea belong to this group of compounds [1].
In our previous work we have shown that a partially purified extract from leaves and rhizomes of Nuphar lutea was enriched by the sesquiterpen thio-alkaloids: 6-hydroxythiobinupharidine and 6-hydroxythionuphlutine B [2]. This extract, termed NUP, inhibited the activation of the Nuclear Factor - κB transcription factor (NF-κB).
NUP exhibited anti-inflammatory properties and partial protection from LPS-induced septic shock by modulating cytokine secretion by macrophages [3].
NF-κB is a family of transcription factors (composed of five sub-units, p50, p65, p52, Rel-B and c-Rel, that form dimmers) that is involved in many biological and pathological processes and can both induce and repress gene expression by binding to discrete DNA sequences, known as κB sites [4].
In most unstimulated, non-diseased mammalian cells, NF-κB dimmers are found predominantly in the cytoplasm bound to a member of the IκB (Inhibitor of NF-κB) family of proteins. After stimulation, IκB is subject to phosphorylation and afterward degradation by the proteosome. This leads to translocation of NF-κB to the nucleus where it stimulates the transcription of a wide variety of genes, including cytokines, cell adhesion molecules and acute phase response proteins, which are involved in proliferation and survival as well as the inflammatory response.
There are several pathways, such as Extracellular Signal-Regulated kinase (ERK) pathway, that are known for their ability to modulate proteins upstream of NF-κB [5, 6].
Given the role of NF-κB in cell proliferation, survival and regulation of many genes involved in the promotion of cancer such as metastatic, angiogenic and tumor promoting genes, it is not surprising that constitutive NF-κB signaling has been implicated in many inflammatory processes, oncogenesis and tumor progression [7, 8].
In the present study, we examined the effect of NUP in a B16 melanoma experimental murine lung metastasis model and its ability to affect the ERK and NF-κB pathways in variety of cell lines. We showed that NUP and cisplatin combined treatment was synergistic and reduced the lung metastatic load. In addition, NUP treatment inhibited TNFα-induced IκBα degradation and NF- κB nuclear translocation. We also observed that NUP induced ERK activation. Interestingly, ERK inhibition prevented NF-κB activation by NUP.
Materials and Methods
Preparation of plant extracts for screening assay
Samples (1g) of frozen plant material were ground in a pre-chilled mortar containing liquid nitrogen. Two ml of 50% methanol/ water (v/v) were added, and the slurry was mixed and kept on ice for 15 min. The mixture was then centrifuged at 11,000 rpm for 5 min., at room temperature on a microfuge. The supernatant was stored at -800C for analysis. Since leaves were harvested and extracted at different times, the potency of the different batches varied. The activity of each batch was calibrated by the NF-κB reporter gene assay as described elsewhere [2].
Purification of NUP
Dried Nuphar lutea powder from rhizomes or leaves was methanol 1:10 (w: v) extracted. The supernatant obtained after centrifugation of the extract at 4000 rpm at 40C for 30 min. was rotary evaporated and re- dissolved in chloroform: 1N HCl at ratio of 1:1 (v:v). The mixture was separated in funnel into 2 phases. The lower chloroform phase discarded and the upper aqueous phase was saved and adjusted to pH 9 with 25% NH4OH. The precipitate formed was separated by centrifugation, as before. The precipitate contained the anti NF-κB activity. The precipitate was re-dissolved in methanol and further fractionated on a silica gel column using chloroform: ethyl-acetate: di-ethyl-amine 20:1:1 (v:v:v), as eluant. Column fractions were assayed in the NF-κB luciferase reporter gene assay and the active fraction pooled, re-dissolved in 50% ethanol and tested for activity as before. The identity of the active components was determined by high resolution NMR.
NUP concentrations throughout the experiments were not cytotoxic as established previously [2].
Cell culture
Human alveolar adenocarcinoma A549 cell line and human breast cancer MCF-7 cell line were maintained in DMEM medium supplemented with10% heat-inactivated fetal bovine serum, 1% L- glutamine, and 1% pen-strep (Beit Haemek, Israel). Murine melanoma B16 cell line, L428 human Hodgkin's lymphoma cells (with constitutive NF-κB activity due to mutation in IκBα [9, 10]) and GA cell line (established from a metastatic explant of a melanoma patient [11] were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 1% L- glutamine, and 1% pen-strep (Beit Haemek, Israel).
Antibodies and reagents
Antibodies against ERK, pERK (Tyr 204), IκBα, p65 and p50 were obtained from Santa Cruz Biotechnology; Anti-mouse and anti-rabbit IgG peroxidases were obtained from Jackson ImmunoResearch and the anti-β actin monoclonal antibody from MP Biomedicals.
Human and murine TNFα was purchased from PeproTech, Israel. Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich, Israel. The ERK inhibitor, PD98059 was purchased from Calbiochem-EMD Chemicals, Gibbstown, USA. Cisplatin was purchased from Teva, Israel.
Western blot analysis
Nuclear and cytoplasmatic protein lysates, 6 * 106 cells per sample, were prepared with the NucBuster kit (Novagen). Quantification of the protein was made using the Bradford method (BioRad). Protein extract (30 μg) were separated in 10% SDS-polyacrylamide gel and blotted onto nitrocellulose membranes. The membranes were incubated with several primary antibodies followed by anti-mouse or anti-rabbit peroxide-linked IgG. Protein bands were detected by chemiluminescence with ECL (Amersham).
Cell migration assay
The migration ability of B16 or A549 cells was tested as follows: cells were seeded on 6-well plates (5×106). After the formation of the monolayer (24 h) NUP or vehicle were added and the monolayers were scratched. The cells were photographed after 28 or 48hrs, in order to observe the effect of NUP on cell migration.
Mice
C57BL/6 mice were obtained from Harlan Laboratories Inc. (Israel). All mice were maintained at the Animal Resource Center of the Ben-Gurion University of the Negev and were used at 8 to 12 weeks of age. All animal experiments described in this work were approved by the Ben-Gurion University Committee for the Ethical Care and Use of Animals in Experiments.
Establishment of experimental lung metastasis of murine B16 melanoma cells
C57BL/6 mice were treated with an intravenous injection of B16 melanoma cells (3*105 cells/200 μl PBS). NUP (20mg/kg) and cisplatin (4 mg/kg) in PBS were given by intraperitoneal injection (IP) every other day from day 0 to day 14. Control animals received only vehicle. Lungs were excised on day 18.
Quantification and image analysis
All image analysis was performed by ImageJ - an open source image processing program.
Results
NUP treatment inhibited TNFα-induced IκBα degradation and NF-κB nuclear translocation
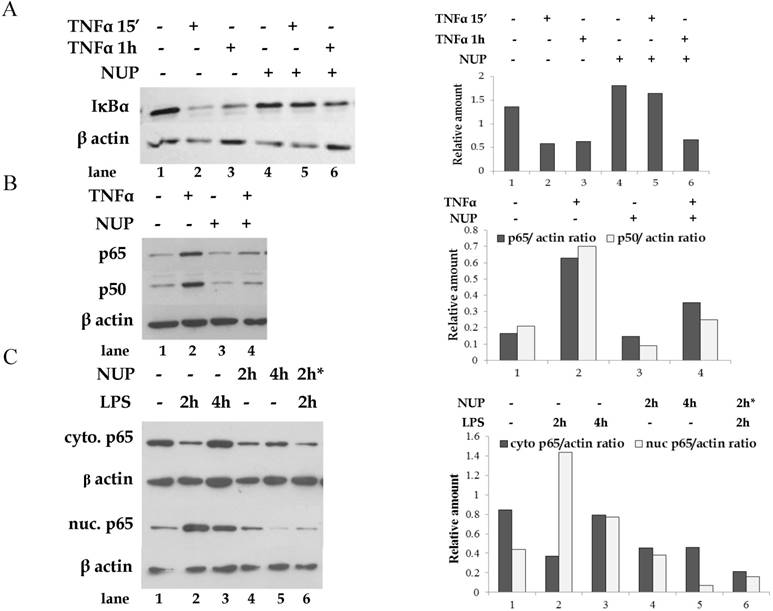
IκBα presence in the cytoplasm and NF- κB presence in the nucleus was examined by western blot analysis. TNFα treatment of A549 cells resulted in rapid IκBα degradation (Fig. 1A) and NF-κB nuclear translocation (as represented by p65 and p50 sub-units) (Fig. 1B). NUP pretreatment prevented IκBα degradation. Incubation with NUP, in the absence of TNFα, had no effect on IκBα levels. Consistently, pretreatment with NUP, prevented NF-κB nuclear translocation. Similarly, NUP inhibited p65 nuclear translocation in LPS activated B16 melanoma cells (Fig 1C).
NUP treatment prevented TNFα-induced IκBα degradation and NF-κB nuclear translocation. (A) A549 cells were treated with TNFα (2.5 ng/ml) for 15 min or 1h with or without NUP pretreatment (12 μg/ml for 2h). Cytoplasmic lysates from treated cells were tested for the presence of IκBα by Western blot. (B) A549 cells were treated with TNFα (2.5 ng/ml) for 15 min. with or without NUP pretreatment (12 μg/ml for 2h). Nuclear lysates from treated cells were tested for the presence of NF- κB sub-units (p65 and p50) by Western blot. Anti-β-actin was used as loading control. (C) B16 cells were treated with LPS (1 µg/ml) for indicated times with or without NUP pretreatment* (6 μg/ml for 2h). Nuclear lysates from treated cells were tested for the presence of p65 NF- κB subunit by Western blot. Anti-β-actin was used as loading control. All experiments were repeated at least three times. A representative blot and its densitometry analysis are shown here, normalized to β-actin.
NUP treatment activates the ERK signaling pathway
The ERK pathway is known for its ability to modulate proteins upstream of NF-κB [5, 6]; therefore we investigated whether the ERK pathway is involved in the inhibition of NF-κB by NUP.
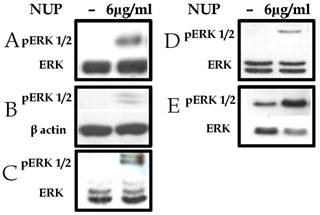
Various cell lines were incubated with NUP, with or without activators of NF-κB. ERK phosphorylation was determined in cell lysates by Western blots. As shown in Figure 2, NUP treatment in all cells tested resulted in ERK phosphorylation (i.e. activation), regardless of whether the activation of NF-κB was inducible or constitutive.
NUP treatment (6 µg/ml for 2h) activated the ERK signaling pathway. (A) L428 Hodgkin's lymphoma cells (with constitutive NF-κB activation) (B) MCF-7 human breast cancer cells (C) A549 human adenocarcinoma cells (D) B16 murine melanoma cells (E) GA human melanoma cells. Cytoplasmic lysates were prepared and pERK was detected. Anti-ERK or anti-β actin was used as loading control. All experiments were repeated at least three times. Representative blots are shown here.
The ERK inhibitor PD98059 prevents NUP dependent down-regulation of NF-κB
PD98059 is a highly selective ERK inhibitor. We used PD98059 to determine whether ERK activation is required for NUP-dependent NF-κB inhibition. As shown in Fig. 3, A549 cells were treated with different combinations of TNFα, NUP or PD98059. Cytoplasmic and nuclear extracts were tested by Western blot. As expected, TNFα treatment alone increased the amount of nuclear p50 (lanes 2-3) and NUP pretreatment prevented this effect (lanes 5-6).
PD98059 ERK inhibitor blocks the effect of NUP on NF-κB translocation. A549 cells were treated with PD98059 (25 μM for 1h) before addition of NUP (6 μg/ml for 2h). Followed by the addition of TNFα (2.5 ng/ml) for the indicated times. The presence of p50 in nuclear extracts and of ERK and pERK in the cells cytoplasmic extracts was detected by Western blot. The experiment was repeated at least three times. A representative blot and its densitometry analysis are shown here.
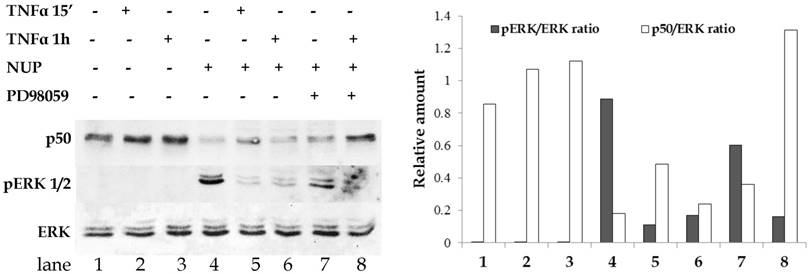
Consistent with previous results, NUP treatment alone resulted in strong ERK activation (lane 4). Incubation with PD98059 prior to NUP and TNFα treatment noticeably reduced NUP-induced ERK phosphorylation. By contrast, ERK inhibition resulted in high amounts of nuclear p50 in spite of NUP pretreatment (lane 8).
NUP inhibits cells migration in vitro
The metastatic potential of tumor cells depends, among other properties, on their migration abilities. Since NUP inhibited the NF-κB pathway, a pathway known to promote the expression of migration proteins in various types of tumors [12-14], we investigate how NUP affects this feature. A549 and B16 cell monolayers were scratched in the presence or absence of NUP. In both cell lines NUP inhibited migration of cells, as compared to control (100% vs. 34% and 86% vs. 45% wound closure, Fig. 4A and 4B respectively).
Inhibition of cell migration and adhesion by NUP. (A) A549 cell monolayers were "scratched" and were grown in the absence (control) or presence of NUP (0.75 μg/ml) for 28 hrs. NUP prevented the cells from closing the wound. (B) B16 melanoma cell monolayers were "scratched" and were grown in the absence (control) or presence of NUP (3.2 μg/ml) for 48 hrs. NUP prevented the cells from closing the wound in a dose dependent manner. All experiments were repeated at least three times. Representative images are shown here.

Treatment of mice with NUP and cisplatin reduced B16 melanoma lung metastasis
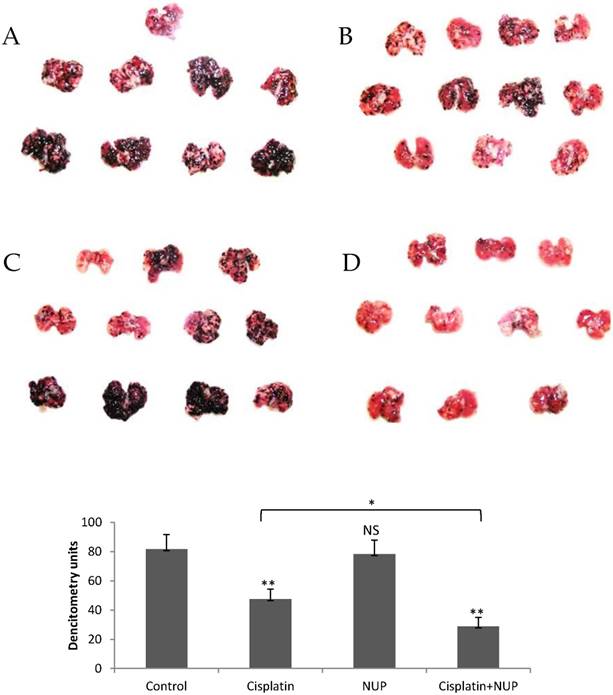
The effect of NUP on experimental lung metastasis of murine B16 melanoma cells, as a single agent or in combination with cisplatin is shown in figure 5. C57BL/6 mice were treated with an intravenous injection of B16 melanoma cells as described in materials and methods. Lungs were excised on day 18 and examined for metastatic black nodules. NUP treatment showed a slight but non-significant nodule reduction. As expected, cisplatin significantly reduced the number of lung metastasis (p<0.01). Treatment with both agents reduced metastasis more efficiently than cisplatin as single agent (p<0.05) as well as when compared to non-treated mice (p< 0.01). These results supported the anticancer and chemotherapy "sensitizer" effect of NUP observed in the in vitro experiments previously published [2].
The anti-metastatic activity of NUP, cisplatin or both in vivo. C57BL/6 mice were given an intravenous injection of B16 melanoma cells (3*105 cells/200 μl PBS). (B) NUP (20 mg/kg), (C) cisplatin (4 mg/kg) or both (D) were given every other day from day 0 to day 14. Control animals received only vehicle (A). Lungs were excised on day 18. NUP in combination with cisplatin treatment reduced metastasis. The average and standard error was determined by densitometry of each group and the significance was determined by t-test (p<0.05 *, p<0.01 **). These results are representative of two similar independent experiments.
Discussion
NF-κB is a central, downstream regulator of inflammation, cell proliferation and apoptosis, not fully tapped for therapeutic purposes. In spite of a vast literature describing NF-κB inhibitors from many natural or synthetic sources [1, 15] the search for effective and specific inhibitors for therapeutic use and minimal side effects is needed and ongoing. Many of the newly reported molecules act as either general inhibitors of NF-κB induction or as inhibitors of specific pathways of induction. Some of these compounds appear to target multiple steps in the NF-κB pathway. Since inhibition of NF-κB activation may occur at various levels in the regulatory cascade, upstream therapeutic targets may be potentially found to have a direct effect on the NF-κB activation pathways. Co-administration of NF-κB inhibitors with standard anti-cancer drugs or radiotherapy, may act as “sensitizers” or as inhibitors of multidrug resistance [16].
An important mechanism of NF-κB regulation is the signal-induced proteolytic degradation of the family of cytoplasmic inhibitors of NF-κB, the IκBs. However, with the discovery of IκB-independent non-canonical or “alternative” pathways of NF-κB activation, the importance of other regulatory mechanisms for the fine-tuning of NF-κB has being recognized [17].
Our results demonstrated that NUP is able to inhibit NF-κB activation both through IκBα dependent (Fig.1 A, B) and independent degradation mechanisms [2].
In addition to the IκBα regulation, NF-κB can be modulated by ERK1/2 through other mechanisms. Xiao et al. demonstrated that LPS stimulation of macrophages led to NF-κB activation. NF-κB activation was indirectly inhibited by activated ERK [18]. In addition, Dhingra and colleagues demonstrated that in rat cardiomyocytes, IL-10 treatment activated ERK 1/2 which in turn blocked NF-κB activation. Addition of PD98059, a selective inhibitor of ERK 1/2, enabled NF-κB activation [5].
Based on our previous results that NUP treatment increased secretion of IL-10, up regulated the phosphorylation of ERK and down regulated the expression of NF-κB subunits p65 and p50 in murine macrophages [3], we investigated whether similar processes also take place in several cancer cell lines.
Our results clearly showed that NUP treatment lead to phosphorylation of ERK in all cell lines tested (Fig. 2).
Furthermore, consistent with Dhingra et al. [5], selective inhibition of ERK 1/2 with PD98059, resulted in TNFα induced NF-κB activation, despite the presence of NUP (Fig.3).
Potent antitumor activity has been reported for compounds belonging to the nupharidine family [19]. Dimeric sesquiterpene thioalkaloids with the 6-hydroxyl group, (6-hydroxythiobinupharidine, 6, 6'-dihydroxythiobinupharidine, and hydroxythionuphlutine B) from the rhizomes of Nuphar pumilum, were shown to inhibit invasion of B16 melanoma cells across collagen-coated filters in vitro. The 6-hydroxythiobinupharidine significantly inhibited lung tumor formation by more than 90%, 10 days after injection of B16 melanoma cells in mice [20].
Since many of the invasion and metastasis genes are dependent on NF-κB activation, the inhibition of the NF-κB pathway can be an important step towards restriction of tumor dissemination [14, 21]. We have shown here that NUP treatment inhibited A549 and B16 cells migration (Fig. 4A, B).
Furthermore, our in vivo experiments demonstrated that mice treated with NUP only slightly reduced the metastatic load while cisplatin was significantly effective as compared to those treated only with vehicle. Most importantly, the lungs of mice treated with a combination of NUP and cisplatin showed a remarkable reduction in the number of metastatic nodules (Fig. 5) as compared to both untreated and cisplatin treated mice. These results support our in vitro findings [2], indicating that NUP indeed serves as a "sensitizer" for conventional chemotherapy.
There are a few possible mechanisms by which NUP may augment the cytotoxicity of cisplatin. Wang et al. [22] demonstrated that ERK activation plays an active role in mediating cisplatin-induced apoptosis of HeLa cells, by inducing PARP cleavage, cytochrome C release and caspase activation. These reports are in line with our previous findings that NUP induced PARP cleavage and caspase-9 activation [2].
It was also reported that inhibition of ERK, enhanced cisplatin-induced NF-κB activation by increasing the level of a nuclear protein phosphatase PP4. Thr 435 phosphorylation of p65 represses the transcriptional potential of p65 [23]. In addition, PP4 have been shown to decrease the threonine phosphorylation status of p65 [24]. Therefore ERK activation may contribute to NF-κB inhibition by maintaining the Thr 435 phosphorylation status of p65.
An alternative explanation can be based on the concept of "NF-κB addiction" in cancer cells, as discussed by Chaturvedi et al. [25]. The addition of NF-κB inhibitors to malignant cells, dependent on NF-κB for survival, led to sensitization of the cells to cytotoxic chemotherapy and apoptosis. Additional research is needed to further define whether NUP can be used effectively in combination with other anti-cancer drugs, to reduce their therapeutic concentration and diminish/prevent side effects.
Taken together our results indicated that NUP's anti-tumor activity is related to its ability to down-regulate the NF-κB pathway, up-regulate the ERK pathway and act as a sensitizer to chemotherapy.
Abbreviations
NUP: Nuphar lutea leaf extract;
TNF-α: Tumor necrosis factor alpha;
MAPK: Mitogen-activated protein kinase;
ERK: Extracellular signal-regulated kinase;
NF-κB: Nuclear factor kappa B.
Acknowledgements
This project was supported by the following grants: The Weinkselbaum Family Medical Research Fund, The Israel Ministry of Health; The Israel Cancer Association through the Miriam and Shlomo Hasid Memorial Fund; The Abraham found through BG Negev and The Richard H. Holzer Foundation.
Author Contributions
Conceived and designed the experiments: JO, AGG, DF, and JG. Performed the experiments: JO, BE, DF.
Analyzed the data: JO, DF, AGG, and JG. Wrote the paper: JO, AGG, JG.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Golan-Goldhirsh A, Gopas J. Plant derived inhibitors of NF-kappa B. Phytochemistry Reviews. 2014;13:107-21
2. Ozer J, Eisner N, Ostrozhenkova E, Bacher A, Eisenreich W, Benharroch D. et al. Nuphar lutea thioalkaloids inhibit the nuclear factor kappaB pathway, potentiate apoptosis and are synergistic with cisplatin and etoposide. Cancer Biology & Therapy. 2009;8:1860-8
3. Ozer J, Levi T, Golan-Goldhirsh A, Gopas J. Anti-inflammatory effect of a Nuphar lutea partially purified leaf extract in murine models of septic shock. J Ethnopharmacol. 2015;161:86-91
4. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes & Development. 2004;18:2195-224
5. Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovascular Research. 2009;82:59-66
6. Lee DF, Hung MC. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clinical Cancer Research. 2008;14:5656-62
7. Amit S, Ben-Neriah Y. NF-kappaB activation in cancer: a challenge for ubiquitination- and proteasome-based therapeutic approach. Semin Cancer Biol. 2003;13:15-28
8. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431-6
9. Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Dohner K. et al. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol. 2003;201:413-20
10. Jungnickel B, Staratschek-Jox A, Brauninger A, Spieker T, Wolf J, Diehl V. et al. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin's lymphoma. J Exp Med. 2000;191:395-402
11. Tanic N, Brkic G, Dimitrijevic B, Dedovic-Tanic N, Gefen N, Benharroch D. et al. Identification of differentially expressed mRNA transcripts in drug-resistant versus parental human melanoma cell lines. Anticancer Res. 2006;26:2137-42
12. Pei X, Li M, Zhan J, Yu Y, Wei X, Guan L. et al. Enhanced IMP3 Expression Activates NF-small ka, CyrillicB Pathway and Promotes Renal Cell Carcinoma Progression. PLoS One. 2015;10:e0124338
13. Wu MH, Lo JF, Kuo CH, Lin JA, Lin YM, Chen LM. et al. Endothelin-1 promotes MMP-13 production and migration in human chondrosarcoma cells through FAK/PI3K/Akt/mTOR pathways. J Cell Physiol. 2012;227:3016-26
14. Tafani M, Pucci B, Russo A, Schito L, Pellegrini L, Perrone GA. et al. Modulators of HIF1alpha and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front Pharmacol. 2013;4:13
15. Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25:6887-99
16. Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297-309
17. Neumann M, Naumann M. Beyond IkappaBs: alternative regulation of NF-kappaB activity. Faseb j. 2007;21:2642-54
18. Xiao YQ, Freire-de-Lima CG, Janssen WJ, Morimoto K, Lyu D, Bratton DL. et al. Oxidants selectively reverse TGF-beta suppression of proinflammatory mediator production. J Immunol. 2006;176:1209-17
19. Modzelewska A, Sur S, Kumar SK, Khan SR. Sesquiterpenes: natural products that decrease cancer growth. Curr Med Chem Anticancer Agents. 2005;5:477-99
20. Matsuda H, Morikawa T, Oda M, Asao Y, Yoshikawa M. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum. Bioorg Med Chem Lett. 2003;13:4445-9
21. Chen CC, Rosenbloom CL, Anderson DC, Manning AM. Selective inhibition of E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression by inhibitors of I kappa B-alpha phosphorylation. J Immunol. 1995;155:3538-45
22. Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435-43
23. O'Shea JM, Perkins ND. Thr435 phosphorylation regulates RelA (p65) NF-kappaB subunit transactivation. Biochem J. 2010;426:345-54
24. Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J Biol Chem. 2004;279:26143-8
25. Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: 'one size does not fit all'. Oncogene. 2011;30:1615-30
Author contact
![]() Corresponding author: Janet Ozer PhD, The Shraga Segal Department of Microbiology, Immunology and Genetics, Faculty of Health Sciences, Ben-Gurion University, Beer Sheva 84105, Israel, Phone: 972-8-6477253; Fax: 972-8-6477626. e- mail: ozerjbgu.ac.il
Corresponding author: Janet Ozer PhD, The Shraga Segal Department of Microbiology, Immunology and Genetics, Faculty of Health Sciences, Ben-Gurion University, Beer Sheva 84105, Israel, Phone: 972-8-6477253; Fax: 972-8-6477626. e- mail: ozerjbgu.ac.il