Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(9):1598-1608. doi:10.7150/jca.18744 This issue Cite
Research Paper
GSK-3β suppresses HCC cell dissociation in vitro by upregulating epithelial junction proteins and inhibiting Wnt/β-catenin signaling pathway
Jing-Hua Zhang1, Li-Yan Jiao2*, Tie-Jun Li3, 4, York Yuanyuan Zhu3, 4, Jian-Wei Zhou5, Jian Tian3, 5, 6 ![]()
1. Department of Surgery, Tangshan People's Hospital/Tangshan Cancer Hospital, Tangshan 063001, China;
2. School of Medicine, Nantong University, Nantong 226001, China;
3. Small RNA Technology and Application Institute, Nantong University, Nantong 226016, China;
4. Biomics Biotechnologies Co., Ltd., Nantong 226016, China;
5. Department of Oncology, Henan People's Hospital, Zhengzhou University, Zhengzhou, Henan 45003, China;
6. Cancer Institute, Tangshan People's Hospital/Tangshan Cancer Hospital, Tangshan 063001, China.
* Contributing to this manuscript equally.
Received 2016-12-13; Accepted 2017-3-31; Published 2017-6-3
Abstract
Glycogen synthase kinase-3β (GSK-3β) is required in the expression of epithelial junction proteins. It was found downregulated in hepatocellular carcinoma (HCC) tissues. The purpose of this study was to investigate the role of GSK-3β in modulating the metastatic behaviors of human HCC cell lines in vitro. In this study, the expression level of GSK-3β was measured in 4 human HCC cell lines, and the small interfering RNA (siRNA) vectors against or plasmids encoding GSK-3β were used to evaluate the responses of target cells to the knockdown or overexpression of this kinase, respectively. Our results showed that GSK-3β expression was significantly lower in human HCC cell lines with high metastatic potential than that in HCC cell lines without metastatic characteristics or in a normal human liver cell line. The knockdown of GSK-3β by siRNA led to a decreased expression of the epithelial junction molecules (ZO-1, E-cadherin) and an increase in the expression of a mesenchymal cell marker (α-SMA) and a gene transcription factor (β-catenin), resulting in enhanced tumor cell dissemination. In contrast, gain-of-function studies revealed that ectopic expression of GSK-3β reduced invasive and migratory abilities of HCC cells accompanied by decreased HCC cell proliferation and induced apoptosis. More importantly, downregulation of GSK-3β led to an increase in the expression and accumulation of β-catenin in the nuclei, promoting gene transcription. In conclusion, GSK-3β might play a vital role in suppressing HCC dissociation by preventing the disassembly of cancer cell epithelial junctional complex via the GSK-3β/β-catenin pathway.
Keywords: Glycogen synthase kinase-3β, hepatocellular carcinoma, metastatic behaviors, GSK-3β/β-catenin signaling pathway, epithelial junctional complex.
Introduction
Hepatocellular carcinoma (HCC) accounts for 70-85% of primary liver cancers and ranks as the third most lethal cancer worldwide [1, 2]. Even though liver resection and transplant may prolong survival, the long-term post-treatment prognosis remains poor due to the highly metastatic potentials of HCC cells [3].
The cancer metastasis is a complex process that requires multiple steps. The first and foremost step is the disassociation of cancer cells from a primary tumor mass. Disrupting epithelial cancer cell junctions is thought to be a main factor leading to cancer cell shedding. Zonula occludens-1 (ZO-1, also known as tight junction protein-1) and E-cadherin are the two main proteins in epithelial junctions. Their expressions have been shown to be significantly reduced in many types of cancers.
Glycogen synthase kinase-3β (GSK-3β), a member of the GSK-3 family with highly conserved serine/threonine kinases, was initially identified in the regulation of glycogen synthesis. Severson et al.'s study showed that downregulation, inactivation or inhibition of GSK-3 resulted in the decrease in the expression of Occluding, Claudin-1 and E-cadherin at both mRNA and protein levels in human colon epithelial cells SKCO-15 and canine renal epithelial cells MDCKO [4], suggesting that GSK-3β family may play a role in maintaining the epithelial architecture by stabilizing cell membrane proteins. In addition, GSK-3β can also inhibit several factors, including a mesenchymal marker--α-smooth muscle actin (SMA), and negatively regulate β-catenin expressions [5]. Mishra et al. showed that inhibition of GSK-3β caused the acquisition of mesenchymal phenotype [6], which facilitates cell disassociation. A recent study performed on 80 HCC patients' surgical specimens showed that significantly lower expression of GSK-3β protein was found in HCC tissues compared to normal liver and peri-cancerous tissues [7].
β-catenin is a dual function protein; it regulates both the coordinated cell-cell adhesion as well as gene transcription. β-catenin has two sub-cellular locations, either bound to E-cadherin on the cell membrane in adherens junctions in intercalated disc structures or free floating in cytoplasm. It regulates gene expression via the Wnt/β-catenin signaling pathway when it enters the nucleus, which induces transition from an epithelial phenotype to a mesenchymal one. GSK-3β protein inhibits Wnt/β-catenin pathway in two ways: one is by anchoring β-catenin to E-cadherin on the cell membrane; the other is by phosphorylating β-catenin, targeting it for ubiquitination and proteasomal degradation [8], consequently, gene transcription cannot be initiated [9, 10]. Therefore, we hypothesized that GSK-3β inhibits HCC disassociation by maintaining epithelial junction proteins and preventing cell phenotype transition by blocking Wnt/β-catenin signaling pathway.
In this study, GSK-3β was significantly downregulated in human HCC cell lines with high metastatic potential, indicating that this kinase may act as a suppressor gene for HCC metastasis. Moreover, we found that ectopic expression of GSK-3β by plasmid DNA could inhibit migration of HCC cells, promote degradation of β-catenin, and enhance the expression of junction proteins. Therefore, the re-expression of GSK-3β in malignant cells could block Wnt/β-catenin signaling pathway and regain epithelial junction proteins, in turn preventing cell disassociation.
Materials and Methods
Cell culture and transfection
Human HCC cell lines SMCC-7721, Huh7 and a normal human liver cell line LO2 were obtained from Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (China); human HCC cell lines MHCC-97H with high metastatic potential and MHCC-97L without metastatic characteristic were obtained from Liver Cancer Institute, Fudan University (Shanghai, China). MHCC-97H and MHCC-97L cell strains were cloned from the same parent cell line MHCC-97 with high metastatic potential [11], therefore they have similar biological and genetic background. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, USA) in a humidified atmosphere containing 5% CO2 at 37 °C.
Cells were transiently transfected with the expression plasmids encoding GSK-3β gene (pGSK-3β, GeneCopoeia, China). The same empty plasmids were used as a control (vector). In order to select a siRNA that is most prominently to silence GSK-3β, four siRNAs with different sequences against GSK-3β were compared for their capability to knockdown GSK-3β gene in HCC cells. Lipofectamine® 2000 (Thermo Fisher Scientific, USA) was used to transfect the above siRNAs into HCC cells separately according to the manufacturer's recommendations. A siRNA (NC_siR) that did not contain any sequences against GSK-3β was used as a negative control. siRNAs were synthesized and provided by Biomics Biotechnologies Co., Ltd (Biomics Biotech, China). The sequences for four GSK-3β siRNAs were listed in Table 1.
The sequences of four siRNAs targeting GSK-3β
| siRNAs | Sequence (5'-3') | |
|---|---|---|
| GSK-3β_siR1 | Sense | CCACAAGAAGUCAGCUAUAdTdT |
| Antisense | UAUAGCUGACUUCUUGUGGdTdT | |
| GSK-3β_siR2 | Sense | GGAUCAGUUGGUAGAAAUAdTdT |
| Antisense | UAUUUCUACCAACUGAUCCdTdT | |
| GSK-3β_siR3 | Sense | CUCAAGAACUGUCAAGUAAdTdT |
| Antisense | UUACUUGACAGUUCUUGAGdTdT | |
| GSK-3β_siR4 | Sense | GGAUCCUGAUACUGCUGUAdTdT |
| Antisense | UACAGCAGUAUCAGGAUCCdTdT | |
Real-time quantitative PCR (RT-qPCR)
RT-qPCR was performed to determine relative mRNA levels. Total RNA of four HCC cell lines and a normal liver cell strain was isolated using a RISOTM (Biomics Biotech, China) according to the manufacturer's recommendation. RT-qPCR was performed in the StepOnePlusTM Real-Time PCR System (ABI, USA). One μg of total RNA was used as a template for cDNA synthesis at 42 °C for 30 min. Subsequently, PCR amplifications were carried out at 95 °C for 15 sec for denaturation, followed by annealing at 60 °C for 30 sec, and extension at 72 °C for 30 sec for 45 cycles using EzQuickTM SYBR Green OneStep qPCR Kit (Biomics Biotech, China). The primers used for RT-qPCR as shown in Table 2. The mRNA levels were analyzed by 2-ΔΔCt method [12], normalized by internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
The sequences of RT-qPCR primers
| Gene | Sequences (5'-3') |
|---|---|
| GSK-3β | F: ACTCTTCAACTTCACCACTCAAG |
| R: TCCACGGTCTCC AGTATTAGC | |
| E-cadherin | F: GCTGGACCGAGAGAGTTTCC |
| R: CGACGTTAGCCTCGTTCTCA | |
| ZO-1 | F: GGAAGAGGATGAAGATGAAGATG |
| R: AGGTGGAAGGATGCTGTTG | |
| α-SMA | F: GAGTTACGAGTTGCCTGATG |
| R: GGTCCTTCCTGATGTCAATATC | |
| β-catenin | F: ACAGCTCCTCTGACAGAGTT |
| R: GAGAGCTGGTCAGCTCAACT | |
| GAPDH | F: GAAGGTGAAGGTCGGAGTC |
| R: GAAGATGGTGATGGGATTTC |
Western blot
Cell protein lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China) containing proteases and phosphatase inhibitors on ice. The supernatants of the lysates were obtained by centrifugation. Equal amounts of proteins (about 25 μg) from each sample were loaded on 5%, 8% or 12% sodium dodecyl sulfate (SDS) polyacrylamide gels to separate proteins, and then the separated proteins on the gels were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA). Nonspecific protein antigens on the membranes were blocked using 5% skim milk, followed by incubating them with primary antibodies at 4 ºC overnight. The primary antibodies were against GSK-3β (1:10000 dilution, Abcam, USA), E-cadherin (1:2000 dilution, Abcam, USA), ZO-1 (1:500 dilution, Thermo Fisher Scientific, USA), β-catenin (1:5000 dilution, Abcam, USA), α-SMA (1:10000 dilution, Abcam, USA), or β-actin (1:5000 dilution, Abcam, USA). Subsequently, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:2000 dilution, Boster, China) at room temperature for 2 h, then washed in tris-buffered saline-Tween (TBST). All the target proteins were detected by enhanced chemiluminescence (ECL) system (Beyotime, China). For quantification, the optical density of individual bands was analyzed by ImageJ software (NIH, USA) and the values were normalized to β-actin.
Cell migration assay
The migratory ability was used to determine the dissociation and therefore metastatic potential of cancer cells. In this study, the effect of GKS-3β on cell migration was assessed by the time taken to refill the gaps of scratches by cancer cells. Briefly, SMCC-7721 cells were grown on 24-well culture plates allowed to reach appropriate confluence, and then the cells were treated with GKS-3β siRNAs or plasmids before the scratches on cell monolayer were made using 1 mL pipette tips. Time-lapse images were observed under a microscope for at least 24 h. Cell migratory ability (as measured by scratch recovery) was calculated using Tt-T0. Here Tt was the distance at 24 h or 48 h, and T0 was the distance at 0 h after scratch. Data were calculated from three independent experiments, each was performed in triplicate.
Transwell invasion assay
Invasion assay was performed to assess the effect of GKS-3β on cell invasion in 24-well BioCoat Matrigel Invasion Chambers (BD, USA) according to the manufacturer's instructions. Briefly, after 48 h transfection, SMCC-7721 cells (1×105) with different treatments were seeded into 8 μm pore inserts, respectively, and cultured for 24 h. Then, the cells invading and penetrating the membranes were fixed in 4% paraformaldehyde for 20 min, and matrigel (BD, USA) was removed from the filter with a cotton swab. The cells invading and adhering to the under-side of the filter were stained with crystal violet (Sigma, USA) and cell numbers was counted under a microscope (5 fields per chamber). Data were calculated from three independent experiments, each was performed in triplicate.
Cell proliferation assay
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric assay was applied to assess the effect of GSK-3β on the cell proliferation. Briefly, the cells treated with different transfection vectors were seeded in 96-well plates at 4×104 cells/well containing 200 μL of medium and cultured up to 72 h. At last, 20 μL of MTT solution (5 mg/mL) was added into each well, and then incubated at 37 °C. After 4 h, the MTT solution was replaced with 200 μL of dimethyl sulfoxide for 10 min to terminate MTT action. The optical density values were measured using a microplate reader (Bio-Rad, USA) at 490 nm. Data were calculated from three independent experiments, each was performed in triplicate.
Cell apoptosis assay
Annexin V-FITC/PI double staining and flow cytometry (FCM) analysis was performed to evaluate the ability of GSK-3β to induce cell apoptosis. Briefly, SMCC-7721 cells were seeded in 6-well plates and treated as described above. After 48 h, cells were collected and re-suspended in 195 μL binding buffer (Beyotime, China). Then, cells were double stained with FITC-conjugated Annexin V and PI (Beyotime, China) at room temperature in the dark for 15 min; samples were immediately analyzed by flow cytometry (BD, USA).
Then, Hoechst staining was used to further confirm cell apoptosis. SMCC-7721 cells (1×105) were seeded on collagen-coated glass coverslips overnight, followed by transfection with the expression plasmids pGSK-3β for 48 h, and then cells were fixed with 4% paraformaldehyde for 15 min. Cell nuclei was stained with Hoechst33258 (Thermo Fisher Scientific, USA). The fluorescence staining was visualized under a fluorescence microscope (5 fields per coverslip).
Immunocytochemistry
Cells on glass coverslips were treated as indicated above for 48 h, and then fixed in pre-cold acetone for 1 min followed by washing three times with Phosphate buffered saline (PBS). Before performing immunostaining, cells were permeabilized in 0.1% Triton X-100 solution, and blocked by blocking buffer (50 mM glycine, 2% bovine serum albumin (BSA), 0.01% Na-azide in PBS) for 1 h. Subsequently, the cells were incubated with a rabbit monoclonal antibody against β-catenin (1:250, Abcam, USA) at 4 °C overnight, the membranes were washed in PBS and incubated with HRP-conjugated goat anti-rabbit IgG (1:50 dilution, Boster, China) for 30 min at 37 °C. After washed by PBS, 3,3-diaminobenzidin (DAB) was used for colouration of immunostaining. The nuclei were counterstained with Hematoxylin Staining Solution (Beyotime, China). Images were taken under a microscope at 400× magnification.
Immunofluorescence staining
Cells were cultured on collagen-coated glass coverslips overnight. After 48 h siRNA transfection, cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100 for 15 min, then incubated with blocking buffer (50 mM glycine, 2% BSA, 0.01% Na-azide in PBS) for 1 h and probed with β-catenin antibody (1:250, Abcam, USA) for 2 h. After being washed with PBS, cells were incubated with FITC-conjugated goat anti-rabbit β-catenin antibody (diluted 1:60, Boster, China). The fluorescence staining was visualized under a fluorescence microscope (5 fields per slip). Hoechst33258 (Thermo Fisher Scientific, USA) was used to counter-stain cell nuclei.
Statistical analysis
All experiments were performed independently in triplicates and data were presented as mean ± standard deviation (SD). All statistical analyses were performed with SPSS19.0 software. Differences in the results between two groups were evaluated using either two-tailed Student's t test or one-way ANOVA followed by post hoc Dunnett's test. The differences with P <0.05 were considered statistically significant.
Results
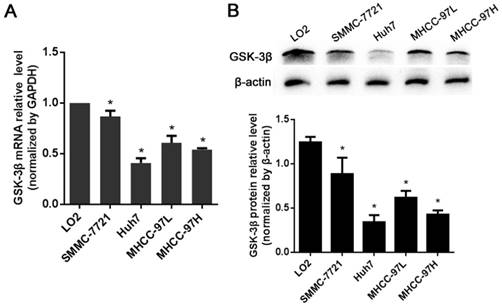
The expression levels of GSK-3β in HCC cell lines
In this study, GSK-3β mRNA levels were detected in all four human HCC cell lines (SMCC-7721, Huh7, MHCC-97H and MHCC-97L) and in a normal human liver cell line LO2. However, GSK-3β mRNA levels in 4 HCC cell lines were shown to be lower than that in a normal liver cell line LO2 based on RT-qPCR levels (P<0.05, Fig. 1A). The differences in GSK-3β expression were also confirmed at the protein level by Western blot analysis (P<0.05, Fig. 1B). Among the subgroups of HCC cell lines, levels of GSK-3β was significantly reduced in Huh7 and MHCC-97H, which are the two HCC cell lines with metastatic potential, compared with that in SMCC-7721 and MHCC-97L cells (P<0.05).
The expression of GSK-3β in HCC cell lines. (A) The mRNA levels of GSK-3β in HCC cell lines (SMMC-7721, Huh7, MHCC-97L and MHCC-97H) and a normal human liver cell LO2 detected by RT-qPCR. (B) The protein levels of GSK-3β in HCC cell lines (SMMC-7721, Huh7, MHCC-97L and MHCC-97H) and a normal human liver cell LO2 detected by Western blot. *P<0.05 versus LO2.
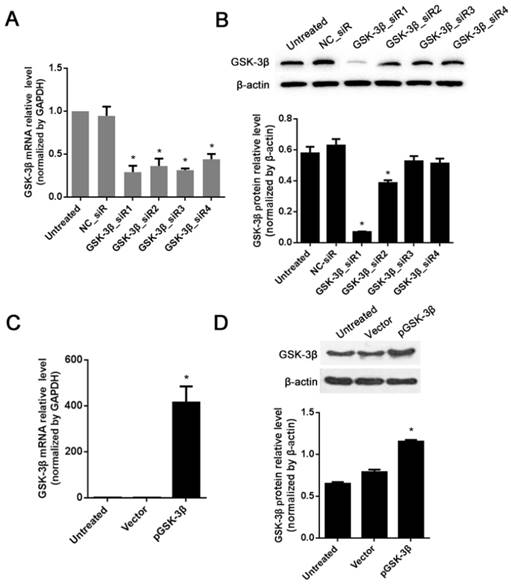
GSK-3β levels in treated HCC cells with siRNAs and pGSK-3β plasmids
To further explore the impact of GSK-3β gene deletion or ectopic expression on the phenotypes of HCC cells, four siRNAs against GSK-3β were transfected into HCC cells to knockdown of GSK-3β and pGSK-3β plasmids were transfected to overexpress of GSK-3β. In this study, human HCC cell line SMCC-7721 was selected for gene transfection due to their relatively higher level of endogenous GSK-3β and higher transfection efficiency. Four pre-designed siRNAs with different nucleotide sequences targeting GSK-3β (GSK-3β_siR1~4) were transfected individually into SMCC-7721 cells to suppress GSK-3β expression. Compared with NC_siR (a negative control) treated and untreated HCC cells, all siRNAs exhibited strong inhibition effects on the expression of GSK-3β at both mRNA and protein levels, but the effect of GSK-3β_siR1 was the most prominent (P<0.05, Fig. 2A and B). Therefore, GSK-3β_siR1 was used for gene knockdown experiments. In addition, in order to evaluate the effect of ectopic expression of GSK-3β on the inhibition of HCC cell metastatic behaviors, namely invasion, migration and dissociation, the plasmids encoding GSK-3β gene (pGSK-3β) were transfected into SMCC-7721 cells allowed to overexpress of GSK-3β. As expected, as compared with empty vector control and untreated cells, the cells treated with pGSK-3β plasmids revealed significantly high mRNA and protein levels of GSK-3β (P<0.05, Fig. 2C and 2D).
The expression of GSK-3β in SMCC-7721 HCC cells treated with siRNA or plasmids encoding GSK-3β gene. (A) The mRNA levels of GSK-3β in SMCC-7721 cells silenced by 4 different siRNAs were analyzed by RT-qPCR. (B) The protein levels of GSK-3β knocked down by 4 different siRNAs were measured in SMCC-7721 cells by Western blot. (C) The mRNA levels of GSK-3β were detected by RT-qPCR in SMCC-7721 cells treated with transfected with the plasmids encoding or non-encoding GSK-3β gene. (D) The protein levels of GSK-3β were analyzed by Western blot in SMCC-7721 cells transfected with the plasmids encoding or non-encoding GSK-3β gene. *P<0.05 versus NC_siR or an empty vector control.
Effects of GSK-3β on HCC cell migration
A wound-healing assay was used to investigate the effects of GSK-3β overexpression or deletion on HCC cell motility and migration. SMCC-7721 cells were seeded in cell culture dishes, then transfected with pGSK-3β or GSK-3β_siR1, and allowed to grow to reach confluence prior to a scratch made on the cell monolayer. The results showed that the cells treated with pGSK-3β required a longer time to re-fill the gap compared to the cells untreated or treated with empty vectors. By contrast, the time required for the cells to re-fill the gap was significantly less after knockdown of GSK-3β compared with NC_siR treated and untreated cells (P<0.05, Fig. 3A).
Effects of GSK-3β on migration and invasion of SMCC-7721 HCC cells. (A) Images of the cell scratch test showing the migration ability of HCC cells with different treatments. (B) Transwell invasion assay showing the invasion ability of HCC cells treated with siRNA or pGSK-3β plasmids. (C) MTT assay showing the viability of HCC cells expressing different levels of GSK-3β. *P<0.05 versus NC_siR or vector control. Each was performed in triplicate.
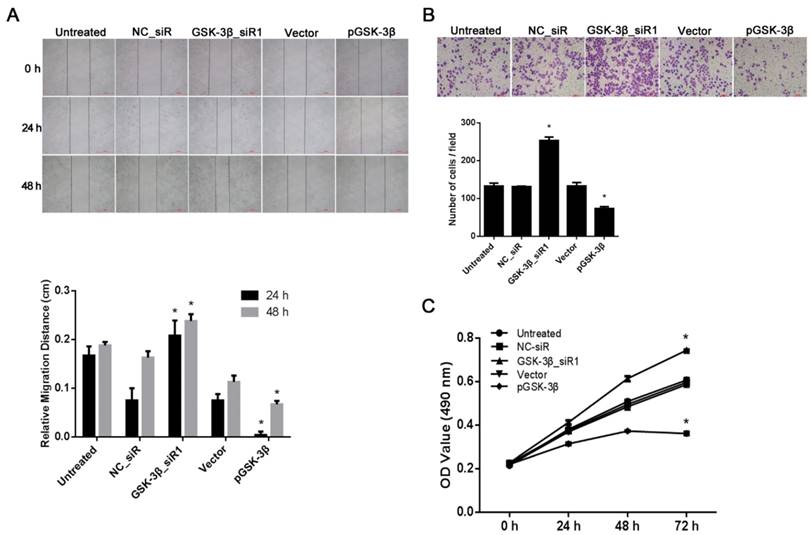
Effects of GSK-3β on HCC cell invasion
Generally, tumor cells that have high aggressive phenotype possess and high invasive capability, which facilitates dissociation from primary tumor to seed distant organs. The invasion ability of SMCC-7721 cells transfected with pGSK-3β or GSK-3β_siR1 was evaluated by a transwell invasion assay. The number of the cells invading and penetrating insert filters covered with matrigel was significantly decreased in SMMC-7721 cells with pGSK-3β treatment compared with the empty vector control. However, when GSK-3β was knocked down by GSK-3β_siR1, SMMC-7721 cells showed an extraordinary invasive potential (P<0.05, Fig. 3B).
Effects of GSK-3β on HCC cell proliferation
To determine whether overexpression or knockdown of GSK-3β affects SMCC-7721 cell proliferation, MTT assay was used to reveal cell viability. Cell proliferation was greatly increased after GSK-3β gene knockdown by siRNA, and significantly decreased after GSK-3β upregulation by pGSK-3β as compared with NC_siR and empty vector controls (P<0.05, Fig. 3C).
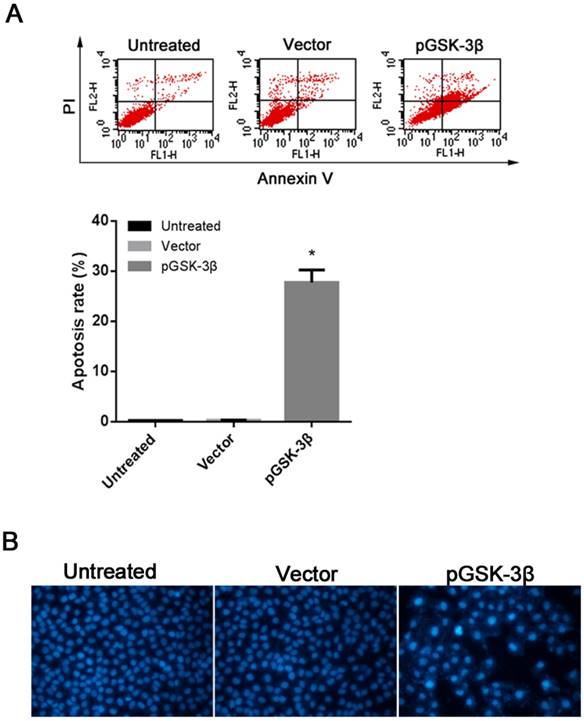
Effects of GSK-3β on HCC cell apoptosis
GSK-3β induced apoptosis of HCC cells was further determined by Annexin V-FITC/PI double staining and FCM analysis. Indeed, pGSK-3β induced greater apoptosis of SMCC-7721 cells up to 27-fold compared with the empty vector control (P<0.05, Fig. 4A). Hoechst staining assay further confirmed the above finding in SMCC-7721 cells treated by pGSK-3β (Fig. 4B).
Effects of GSK-3β on viability of SMCC-7721 cells. (A) Annexin V-FITC/PI double staining and FCM analysis revealing apoptosis rates of HCC cells. (B) Images of HCC cell nuclei with Hochest staining. *P<0.05 versus NC_siR or vector control. Each was performed in triplicate.
The expression of epithelial junction molecules and a mesenchymal marker in HCC cells
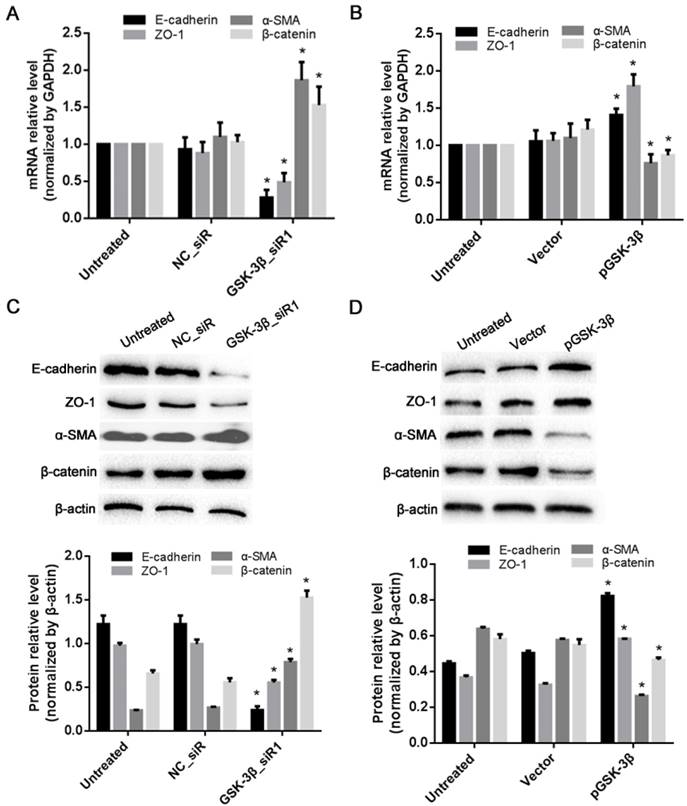
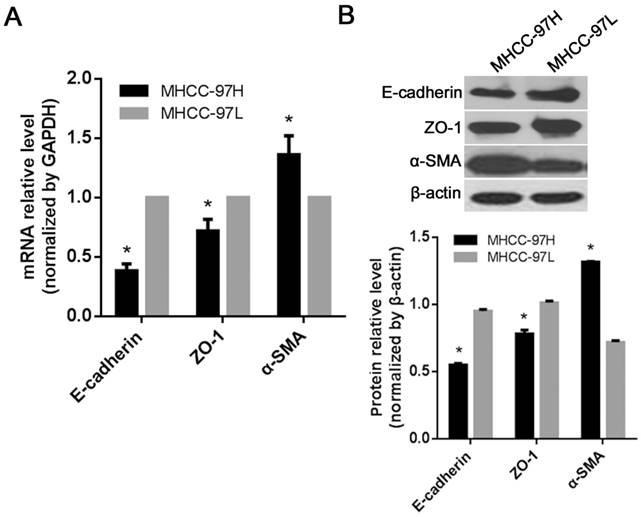
To verify the effects of GSK-3β on the expression of HCC epithelial junction molecules, the expression levels of ZO-1 (epithelial tight junction molecule) and E-cadherin (epithelial adheren cadherin) and α-SMA (mesenchymal cell marker) were evaluated in SMCC-7721 cells treated with the GSK-3β_siR1. Both RT-qPCR and Western blot assays revealed that the expressions of E-cadherin and ZO-1 were significantly downregulated (P<0.05). In contrast, the level of α-SMA was significantly increased (P<0.05, Fig. 5A and C). Ectopic expression of GSK-3β in SMCC-7721 cells also resulted in decreased expression of a mesenchymal marker and increased expression of epithelial junction molecules (P<0.05, Fig. 5B and D). Moreover, MHCC-97H (a high metastatic cell line) exhibited lower levels of GSK-3β while MHCC-97L (without metastatic potential) expressed higher GSK-3β. In addition, E-cadherin and ZO-1 expression were higher and α-SMA was lower in MHCC-97L than those in MHCC-97H (P<0.05, Fig. 6).
Effects of GSK-3β on the expression of EMT biomarkers in SMCC-7721 HCC cells. (A) The mRNA levels of EMT biomarkers in HCC cells when knockdown of GSK-3β analyzed by RT-qPCR. (B) The mRNA levels of EMT biomarkers in HCC cells when overexpression of GSK-3β analyzed by RT-qPCR. (C) The protein levels of EMT biomarkers in HCC cells when knockdown of GSK-3β analyzed by Western blot. (D) The protein level of EMT biomarkers in HCC cells when overexpression of GSK-3β analyzed by Western blot. *P<0.05 versus NC_siR or empty vector control. Each factor was analyzed three times.
The expression of EMT biomarkers in HCC cells with high or low metastatic potential. (A) The mRNA levels of E-cadherin, ZO-1 and α-SMA in HCC cell line with high (MHCC-97H) or low (MHCC-97L) metastatic potential detected by RT-qPCR. (B) The protein levels of E-cadherin, ZO-1 and α-SMA in MHCC-97H and MHCC-97L analyzed by Western blot. *P<0.05 versus MHCC-97L. Each factor was analyzed three times.
Expression and distribution of β-catenin in HCC cells
β-catenin is a subunit of the cadherin protein complex and acts as an intracellular signal transducer in the Wnt signaling pathway; its rapid turnover depends on the GSK-3β phosphorylation. To investigate the effect of GSK-3β on β-catenin in HCC cells, RT-qPCR and Western blot were used to observe the mRNA and proteins levels of β-catenin in SMCC-7721 cells treated with siRNA against or plasmids encoding GSK-3β. Results showed increased expression of β-catenin in SMCC-7721 cells at mRNA and protein levels after knockdown of GSK-3β by GSK-3β_siR1. Conversely, β-catenin level was decreased with overexpression of GSK-3β induced by pGSK-3β (P<0.05, Fig. 4). Immunocytochemistry also validated these results. Immunocytochemistry showed that strongly positive reaction for β-catenin was seen in SMCC-7721 HCC cells treated with siRNA; in contrast, staining for β-catenin was markedly decreased after treatment with GSK-3β plasmids (Fig. 7A).
Images of the expression of β-catenin in HCC cells. (A) The expression of β-catenin in SMCC-7721 cells detected by immunocytochemical staining. (B) The expression of β-catenin in SMCC-7721 cells detected by immunofluorescence staining, FITC-conjugated antibody (green) against β-catenin, and Hoechst staining (blue) for nuclei labeling. The intensity of β-catenin immunefluorescence was stronger in GSK-3β_siR1 cells than that of untreated or NC_siR group.
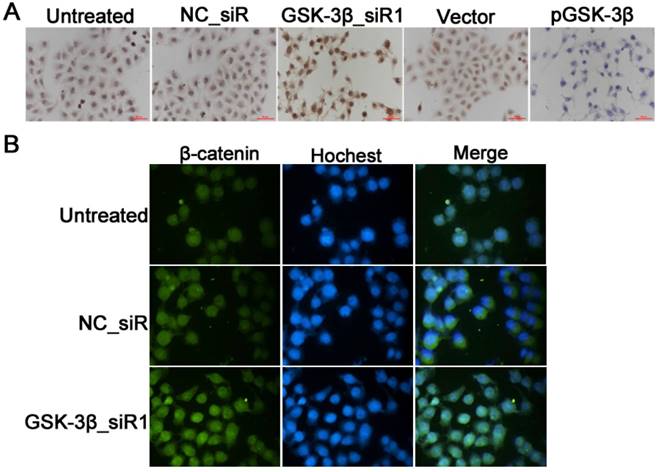
Furthermore, immunofluorescence assay revealed that treatment of SMCC-7721 cells with GSK-3β_siR1 led to accumulation of β-catenin in the nuclei (Fig. 7B). The elevated levels of intranuclear β-catenin may activate Wnt/β-catenin signaling pathway and promoted gene transcription.
Discussion
There is no doubt that HCC progression is a multistep process involving both the activation of oncogenes and the silencing of tumor suppressor genes [13-15]. Several molecular mechanisms have been shown to directly link GSK-3β to cancer cell metastasis. Cell disassociation from the primary tumor mass is a pre-requisite for cancer metastasis. When epithelial junctions on cell membranes are disrupted, cell disassociation occurs. During disassociation, epithelial phenotype transits into a mesenchymal one; as a result, cell motility and migration are enhanced. This process is called epithelial-mesenchymal transition (EMT). In prostate cancer, EMT-positive cancer cells express enhanced levels of phosphorylated GSK-3β (its inactive state) and β-catenin compared with EMT-negative cancer cells [16]. Several studies on HCC have also demonstrated that EMT is a key process driving the metastatic cascade [17]. In this study, we established direct mechanisms of GSK-3β in inhibition of cancer migration in vitro, which is its ability to maintain cancer cell-cell junction complex integrity and decrease the expression of a mesenchymal marker.
The genetic and/or epigenetic aberration of GSK-3β have been found in many types of tumor, such as breast cancer, renal cancer, prostate cancer, gastric cancer and leukemia [18-21]. However, the expression and molecular function of GSK-3β in HCC has not been explored in great detail. In this study, a lower level of GSK-3β in 4 different human HCC cell lines compared to that in a normal human liver cell strain LO2 indicated that this kinase is downregulated in HCC. The higher apoptosis rate and lower cell viability of SMCC-7721 cells transfected with plasmids encoding GSK-3β further suggested that GSK-3β serves as a tumor suppressor gene. In the present study, the elevated expression of ZO-1 and E-cadherin in pGSK-3β SMCC-7721 cells and longer time for such treated cells to refill the gap implies that cancer cell dissociation could be prevented by GSK-3β overexpression. In contrast, the downregulation of GSK-3β by siRNA caused an increase in α-SMA and a decrease in epithelial junction proteins, indicating an epithelial-mesenchymal transition. This is especially true in highly mestastic MHCC-97H cells. Conversely, a loss of GSK-3β expression rendered SMCC-7721 cell to gain higher migratory and invasive capabilities. Therefore, GSK-3β may play a pivotal role in maintaining HCC cell phenotypes through elevating expression of epithelial junction proteins. GSK-3β's metastatic suppression is also evidenced in low-levels GSK-3β in MHCC-97H cells with high metastatic potential and high levels of GSK-3β in MHCC-97L cells without metastatic phenotype.
Amongst the various biomarkers of EMT, β-catenin has been shown to be especially prominent. It is either bound to epithelial junction protein E-cadherin located on cell membrane or involved in Wnt/β-catenin signaling [22]. When β-catenin enters the nucleus, Wnt/β-catenin signaling is activated. GSK-3β has been recognized as a primary kinase involved in both the sub-cellular distribution and the expression of the β-catenin protein [23, 24]. It is therefore plausible to hypothesize that the loss of GSK-3β could lead to upregulation of β-catenin expression and/or a change of β-catenin in sub-cellular location. In this study, both immunocytochemtry and immunefluorescent staining showed that ectopic expression of GSK-3β by pGSK-3β led to a decreased expression of β-catenin in SMCC-7721 cells. By promoting degradation of β-catenin, GSK-3β effectively prevents intranuclear accumulation of β-catenin and subsequent gene transcription [8]. This is confirmed in our study with enhanced immunereactivity for β-catenin in the nuclei after treatment with GSK-3β_siR1.
In summary, our study demonstrated for the first time that GSK-3β acts as a potential tumor suppressor gene in HCC. GSK-3β is capable of preventing cancer cell dissociation likely by maintaining epithelial junction complex and inhibiting Wnt/β-catenin signaling pathway. Further studies on more HCC cell lines that are treated with siRNA against or plasmids encoding GSK-3β will be conducted to demonstrate the mechanisms of GSK-3β in prevention of cell disassociation in vitro. Since cell migration and invasion in vitro may not necessarily be the same as it is in vivo, HCC animal models will be used to confirm the role of GSK-3β in inhibiting HCC metastasis in future in vivo study.
Acknowledgements
This work was supported by Nantong University. We would like to greatly appreciate Mr. Ying Meng, Tangshan Jianhua Industrial Group, Tangshan, China, and Mr. Yu-Yin Li, Guangzhou Senchang Industrial Co., Ltd. for both generous financial supports.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sun J, Lu H, Wang X, Jin H. MicroRNAs in hepatocellular carcinoma: regulation, function, and clinical implications. Scientific World J. 2013;2013:924206
2. M.C. Kew. Epidemiology of chronic hepatitis B virus infection, hepatocellular carcinoma, and hepatitis B virus-induced hepatocellular carcinoma. Pathol Biol (Paris). 2010;58:273-7
3. Samuel D, Colombo M, El-Serag H, Sobesky R, Heaton N. Toward optimizing the indications for orthotopic liver transplantation in hepatocellular carcinoma. Liver Transpl. 2011;17:S6-S13
4. Severson EA, Kwon M, Hilgarth RS, Parkos CA, Nusrat A. Glycogen synthase kinase 3 (GSK-3) influences epithelial barrier function by regulating occluding, claudin-1 and E-cadherin expression. Biochem Biophys Res Commum. 2010;397:592-7
5. Popova AP, Bentley JK, Anyanwu AC, Richardson MN, Linn MJ, Lei J, Wong EJ, Godlsmith AM, Pryhuber GS, Hershenson MB. Glycogen synthase kinase-3β/ β-catenin signaling regulates neonatal lung mesenchymal stromal cell myofibroblastic differentiation. Am J Physiol Lung Cell Mol Physiol. 2012;303(5):L439-448
6. Mishra P, Senthivinayagam S, Rana A, Rana B. Glycogen synthase kinase-3beta regulates Snail and beta-catenin during gastrin-induced migration of gastric cancer cells. J Mol Signal. 2010;5:9
7. Huang KT, Huang YH, Li P, He B, Chen ZK, Yu X. et al. The correlation between TSC2 and GSK3β levels, and outcomes of patients with hepatocellular carcinoma treated by hepatectomy. Hepatol Res. 2013;44:1142-50
8. van Noort M, Meeldijk J, van der zee R, Destree O, Vlevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901-5
9. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9-26
10. Lu FI, Sun YH, Wei CY, Thisse C, Thisse B. Tissue-specific derepression of TCF/LEF controls the activity of the Wnt/β-catenin pathway. Nat Commun. 2014;5:5368
11. Tian J, Tang ZY, Ye SL, Liu YK, Lin ZY, Chen J, Xue Q. New human hepatocellular carcinoma (HCC) cell line with highly metastatic potential (MHCC97) and its expressions of the factors associated with metastasis. Br J Cancer. 1999;81(5):814-21
12. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
13. Kanda M, Sugimoto H, Kodera Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J Gastroenterol. 2015;21:10584-97
14. Lachenmayer A, Alsinet C, Chang CY, Llovet JM. Molecular approaches to treatment of hepatocellular carcinoma. Dig Liver Dis. 2010;3:S264-72
15. Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795-806
16. Jiang YG1, Luo Y, He DL, Li X, Zhang LL, Peng T, Li MC, Lin YH. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancerinduced by hypoxia-inducible factor-1alpha. Int J Urol. 2007;14(11):1034-9
17. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117-34
18. Gavilán E, Sánchez-Aguayo I, Daza P, Ruano D. GSK-3β signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis. 2013;4:e572
19. Pal K, Cao Y, Gaisina IN, Bhattacharya S, Dutta SK, Wang E. et al. Inhibition of GSK-3 induces differentiation and impaired glucose metabolism in renal cancer. Mol Cancer Ther. 2014;13:285-96
20. Tang X, Zheng D, Hu P, Zeng Z, Li M, Tucker L. et al. Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the b-Catenin/TCF/ LEF-1 pathway in gastric cancer cells. Nucleic Acids Res. 2014;42:2988-98
21. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G. et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 2014;28:15-33
22. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192-205
23. Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I. et al. Wnt signaling requires msequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136-48
24. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL. et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881-911
Author contact
![]() Corresponding author: Jian Tian, Ph.D., Department of Oncology, Henan People's Hospital, Zhengzhou University, Zhengzhou, Henan 45003, China +086 0371 65580014, Fax: +086 0371 65964376, Email: 1398710910com
Corresponding author: Jian Tian, Ph.D., Department of Oncology, Henan People's Hospital, Zhengzhou University, Zhengzhou, Henan 45003, China +086 0371 65580014, Fax: +086 0371 65964376, Email: 1398710910com