Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(10):1892-1899. doi:10.7150/jca.17638 This issue Cite
Research Paper
TBK1 Promote Bladder Cancer Cell Proliferation and Migration via Akt Signaling
Wei Chen*1, Kewang Luo*1, Zhiyi ke1, Bin Kuai2, Shiyang He, Wei Jiang1, Weiren Huang ![]() 1, Zhiming Cai
1, Zhiming Cai ![]() 1
1
1. Key Laboratory of Medical Reprogramming Technology, Shenzhen Second People's Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen 518039, China.
2. HYK High-throughput Biotechnology Institute, Shenzhen 518060, China
* These authors contributed equally to this work.
Received 2016-9-19; Accepted 2017-5-2; Published 2017-7-3
Abstract
Bladder cancer is a challenging and fatal malignancy and the improvement in prognosis is limited over years. Deep understanding the mechanism of bladder cancer tumorigenesis and progression will help to discover novel and effective treatment strategies. In this study, we identify non-canonical IkB kinase TBK1 is up-regulated in bladder cancer tissue and cell lines. Knockdown of TBK1 markedly inhibits cell proliferation and migration. Inhibition of TBK1 kinase activity by BX795 significantly attenuates bladder cancer cell proliferation and migration. Mechanistic study shows that overexpression of TBK1 promoted the phosphorylation of Akt, whereas knockdown of TBK1 reverses this action. Taken together, our data suggest that TBK1 modulates the malignant behaviors of bladder cancer cell via Akt signaling, revealing new insights in discovering new therapy target for bladder cancer.
Keywords: bladder cancer, TBK1, Akt signaling
Introduction
Bladder cancer (BC) is one of the most common malignancies worldwide and the second leading cause of cancer-related death among genitourinary tumors [1]. Risk factors for BC development include smoking, male sex and inflammation [2]. Despite recent advances in conventional and targeted therapy strategy, the prognosis of bladder cancer is extremely poor with a low 5-year survival rate [3], and the mechanism underlying the tumorigensis and progression of bladder cancer is not clear [4]. Therefore, it is highly desirable to illuminate the molecular mechanisms of bladder cancer as well as discovery new strategies for the diagnosis and therapy of bladder cancer.
TBK1, a non-canonical member of the IKK kinase family, plays a critical role in innate immunity and inflammation [5]. TBK1 could activate the transcription factor IRF3, leading to the production of type I IFNs and other cytokines of the immediate early host defense response [6]. Mounting evidence implicates TBK1 play a role in oncogenic signaling and tumorigenesis [7]. The expression of TBK1 is increased in breast, lung and colon cancers [8, 9]. Silencing of TBK1 was able to induce the apoptosis of Ras-transformed cells. Activation of TBK1 by RalB/Sec5 effector complex is required to maintain cancer cell survival and support oncogenic Ras-induced transformation [10]. Systematic RNA interference screening reveals that TBK1 is crucial for survival of KRAS mutant primary lung cancers [11]. In addition, TBK1 could inhibit mTOR function and accentuate drug resistance in prostate cancer [12]. However, the exactly role of TBK1 in human bladder cancer has not been defined, and the potential therapeutic application of TBK1 remains unknown.
Here, we investigate the anti-tumor activity and the molecular mechanisms of TBK1 in bladder cancer cells in vitro. We find that TBK1 is consistently up-regulated in both bladder cancer tissues and cell lines. Our data further prove that TBK1 functions as an oncogene of bladder cancer progression via Akt signaling. Our finding will help to identify a new diagnostic marker and therapeutic target for bladder cancer.
Material and methods
Cell cultures
Normal bladder epithelial cell SV-HUC-1, bladder cancer cell lines, including T24, SW780, 5637 and UM-UC-3 were obtained from ATCC (American Type Culture Collection). T24, SW780 and UM-UC-3 were cultured in DMEM medium (Invitrogen), 5637 was cultured in 1640 medium (Invitrogen), SV-HUC-1 was cultured in F12K medium (Invitrogen), plus with 10% FBS and 1% penicillin/streptomycin (Invitrogen) at 37°C with 5% CO2. Transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
RNA extraction and real-time PCR
Total RNA was isolated from indicated cells by using TRIzol reagent (Invitrogen) and RNA was reverse transcribed by using RT-PCR Quick Master Mix (Toyobo). The quantification of indicated gene transcripts was performed with ABI Vii7 Sequence Detection System by using ReverTra Ace Qpcr RT Master Mix (Toyobo), and GAPDH served as an internal control. PCR primers of indicated target genes are shown as below:
TBK1: sense (5'-ACG CAT GGG CAC ATC AAG AA-3'),
antisense (5'-GTG CGT CAT AGC TTT TGT GG-3');
GAPDH: sense (5'-GAA GGG CTC ATG ACC ACA GT-3'),
antisense (5'-GGA TGC AGG GAT GAT GTT CT-3').
siRNA
The siRNAs duplexes were synthesized from GenePharma. The sequences of siRNAs are shown as follows:
TBK1 siRNA 425, 5'-CCAUGUGGGAGUUUAUACATT-3';
TBK1 siRNA 663, 5'-GCAGUUUGUUUCUCUGUAUTT-3';
The nonspecific siRNA (NC), 5'-UUC UCC GAA CGU GUC ACG UTT-3'.
Western blotting analysis
Total protein was extracted from bladder cancer cells and 40 μg of isolated protein was subjected to SDS-PAGE. The separated proteins were then electrically transferred to a PVDF membrane and probed with the following primary antibodies: TBK1 (abcam), actin (Cell Signaling Technology, Inc.) and indicated second antibody. The protein bands were visualized by using a SuperSignal West Pico chemiluminescence ECL kit (Pierce).
CCK8 assay
After transfection of siRNA for 24 hours, 5x103 cells/well were seeded into 96-well plates. After incubation of cells for 24, 48, 72 hours, the cell proliferation was assessed by CCK8 assay (TransGen Biotech) according to manufacturer's instructions. The amount of viable cells was assessed by measurement of OD450 values, and the experiments were performed in triplicate and repeated three times.
Edu incorporation assay
Briefly, cells were seeded in 12-well plates and transfected indicated siRNA for 48 hours. Then the cells were incubated with Edu for 2 hours and stained with anti-Edu antibody according to manufacturer's instruction. Images were acquired under a fluorescent microscope at 488nm excitation.
Wound healing assay
Cells were seeded in 6-well plates and transfected indicated siRNA for 48 hours. Then the cells were scratched with a 300 μl pipette tip to create a wound and washed twice with PBS to remove floating cells. Migration of the cells into the wound was examined with a fluorescence microscope at 0 and 16 hours.
Transwell assay
2 × 105 cells added with 0.5% FCS were seeded in each Transwell top chamber with 8.0-μm pores (BD Bioscience, USA) and 600μl with 10% FCS medium in the bottom 24-well plate. 24 hours later, the medium in the transwell chamber was removed, and the cells were fixed in 4% paraformaldehyde and stained with crystalline violet solution. Three randomly selected fields were captured and the cell number were counted.
Immunohistochemistry
In brief, sections were incubated with anti-TBK1 antibody (Abcam) at 4°C overnight. After washing with PBS, sections were incubated with enzyme labelled goat anti-rabbit IgG (MXB) for 1 h at room temperature. After PBS wash, colors were developed by DAB kit (MXB) for 5 min and stopped by PBS washing. Hematoxylin was used for counter stain.
Results
TBK1 is highly expressed in bladder cancer tissue and bladder cancer cell line
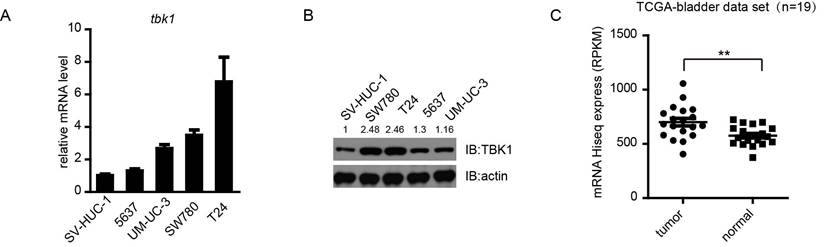
To explore the role of TBK1 in bladder cancer tumorigenesis, we first examined the expression of TBK1 in some bladder cancer cell lines (T24, SW780, 5637 and UM-UC-3) and a normal bladder epithelial cell SV-HUC-1, using quantitative real-time PCR and Western blotting analyses. Compared to human normal bladder epithelial cell SV-HUC-1, the expression of TBK1 was increased in all four bladder cancer cell lines, especially in T24 and SW780, therefore T24 and SW780 should be used for further study (Fig. 1A and 1B). To make it more physiologically relevant, we analyzed data from a cohort of bladder cancer patients (19 paired) provided by The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov/). The TBK1 level was up-regulated in bladder cancer tissues compared with adjacent normal tissues (Fig. 1C). Consistently, the higher expression level of TBK1 in tumor specimens was also confirmed by Immunohistochemistry (IHC) (Fig. S1). These data indicated that TBK1 is markedly up-regulated in bladder cancer.
TBK1 is overexpressed in bladder cancer cell lines and tissues. (A and B). Expression of TBK1 in normal bladder epithelial cells SV-HUC-1 and a panel of four human bladder cancer cell lines was measured by RT-qPCR(A) or Western blot (B) analysis. (C). Expression of TBK1 in human bladder cancer clinical specimens of human bladder cancer using the TCGA mRNA HiSeq expression array data.
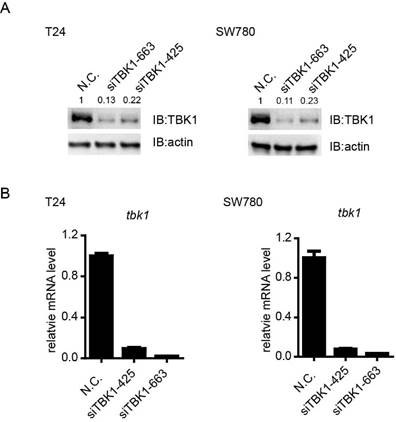
Knockdown efficiency of TBK1 in bladder tumor cell line
To investigate the potential biological function of TBK1 in bladder tumorigenesis, we screened out the specific and effective siRNAs against TBK1, the TBK1 siRNA siTBK-425 and siTBK-663 could obviously attenuate the expression of TBK1 at mRNA and protein level (Fig. 2A and 2B).
Knockdown efficiency of TBK1 in T24 and SW780 cells. (A). T24 (left) or SW780 (right) cells was transfected with the negative control (NC) or TBK1 siRNAs. Cell lysates were immunoblotted with the TBK1 or β-actin antibody. (B). The nonspecific control (NC) or TBK1 siRNA were transfected into T24 (left) or SW780 (right) cells. Expression of TBK1 mRNA was measured by quantitative PCR.
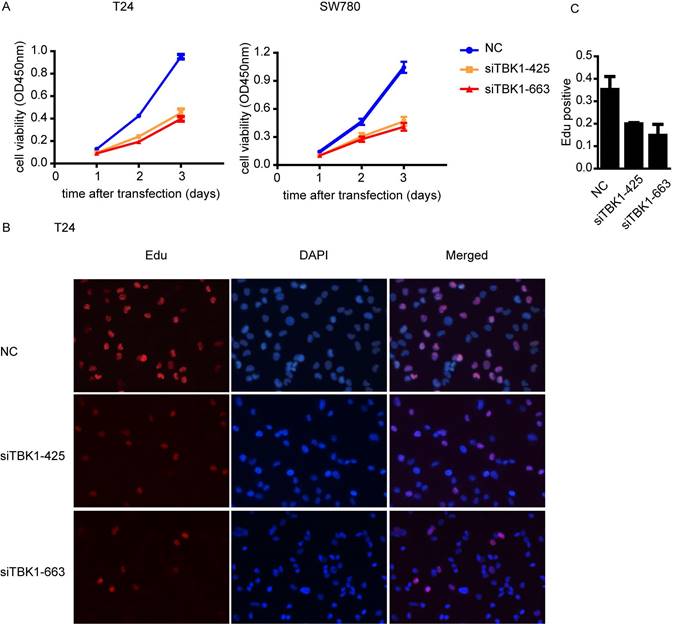
Knockdown of TBK1 inhibit the proliferation of the bladder cancer cell
To investigate whether TBK1 is involved in the progression of the malignant behavior of bladder cancer cells, we performed the CCK-8 cell proliferation assay and found that the proliferation of bladder cancer cell T24 and SW780 was drastically decreased after TBK1 knockdown (Fig. 3A). Furthermore, EdU incorporation assays also showed that the number of EdU-positive cell was lower in TBK1 knockdown cells relative to control cells (Fig. 3B and 3C). Taken together, these data above suggested that silencing of TBK1 significantly suppresses the proliferative ability of bladder cancer cells.
Knockdown of TBK1 inhibited bladder cancer cell proliferation. (A). T24 (left) or SW780 (right) was transfected with the NC or TBK1 siRNA and the cell proliferation was determined by CCK-8 assay. (B). The nonspecific control (NC) or TBK1 siRNA were transfected into T24 cells and the cell proliferation was determined by Edu assay. (C). Quantitative analysis of the Edu positive cells in (B).
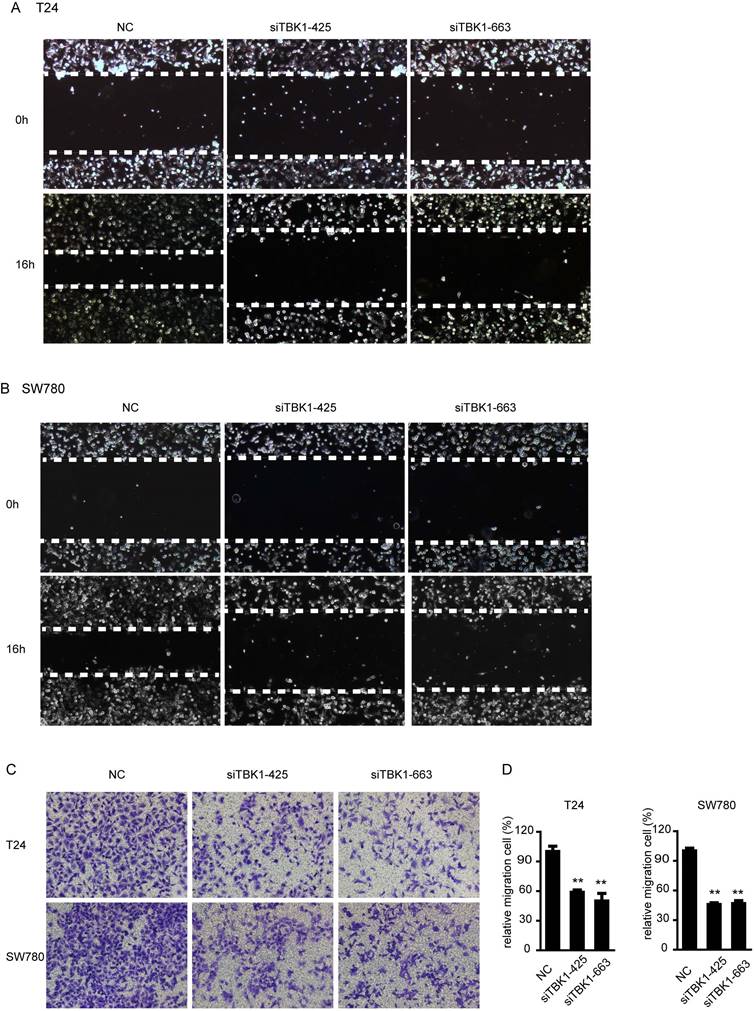
Knockdown of TBK1 inhibit the migration of the bladder cancer cell
We next examined the contribution of TBK1 to migration of bladder cancer cells. In wound healing assays, we found that TBK1 knockdown cells migrated more slowly from the wound edge compared with control cells in T24 and SW780 cells (Fig. 4A and 4B). Similar results was obtained from transwell migration assays, both T24 and SW780 cell lines showed that silencing of TBK1 attenuated cell migration (Fig. 4C and 4D). These data above indicated an enhancing role of TBK1 in regulating migration of bladder cancer cells.
Silencing of TBK1 inhibit the migration and invasion of bladder cancer cells. (A to C). T24 or SW780 was transfected with the NC or TBK1 siRNA. Wound healing assay (A and B) and transwell assay (C) were performed to evaluate the migration capacity of TBK1. (D) The relative cell number of migration (C) in each well was statistically analyzed.
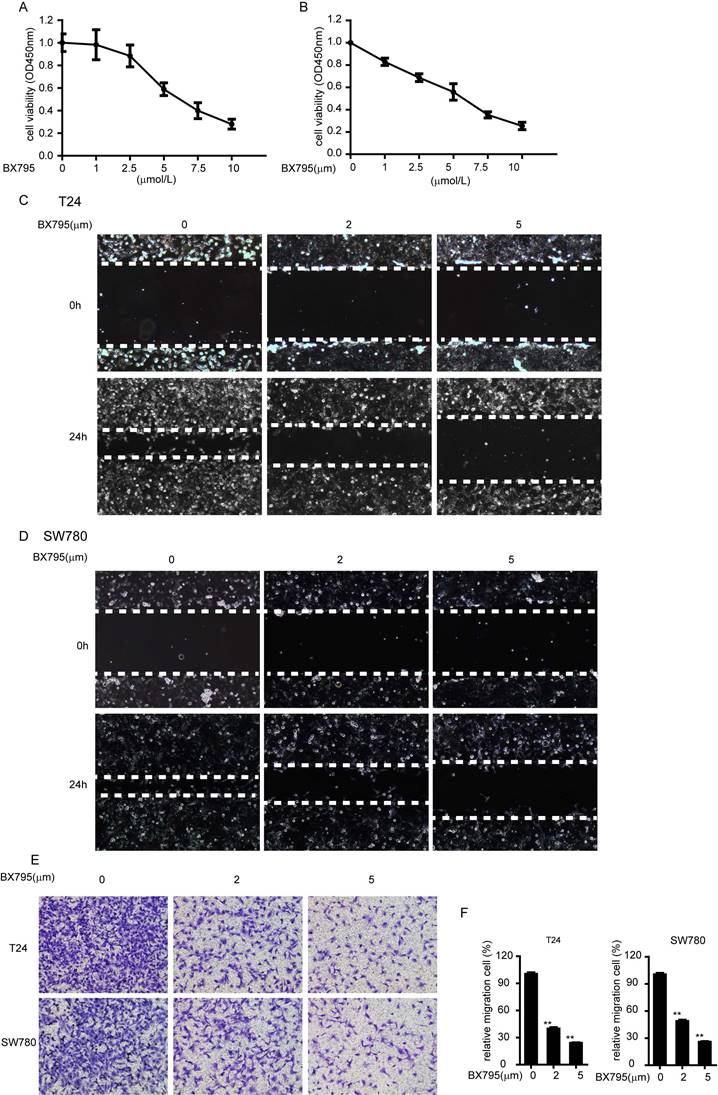
BX795, an inhibitor of TBK1, inhibit the proliferation and migration of bladder cancer cell
Furthermore, we demonstrated that whether the role of TBK1 in bladder tumorigenesis depends on its kinase activity. We choose BX795 as a TBK1 inhibitor, which inhibits the catalytic activity of TBK1 by blocking its phosphorylation [13]. BX795 treatment could dramatically inhibit the proliferation and migrations of bladder cancer cell T24 and SW780 (Fig. 5A-5F), which indicated that BX795 is a useful therapeutic agent to target bladder cancer cell line T24 and SW780. These data demonstrated the function of TBK1 in bladder tumorigenesis is dependent on its kinase activity.
BX795 inhibit the proliferation and migration of bladder cancer. (A and B). T24 (A) or SW780 (B) cells were treated with BX795 or DMSO, and cell viability was assessed by CCK8 assays. (C to E). T24 or SW780 cells were exposed to BX795 at the indicated concentrations for 24 h. The cell migration was detected by Wound healing assay (C and D) and transwell assay (E). (F) The relative cell number of migration (E) in each well was statistically analyzed.
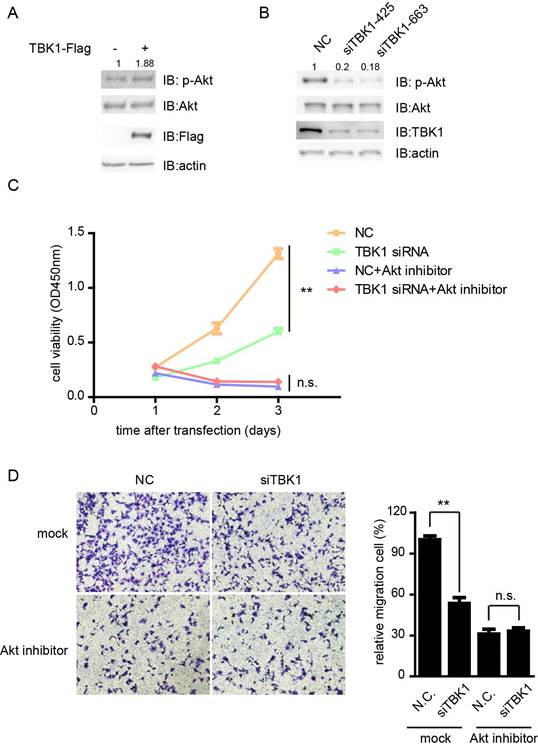
TBK1 promotes proliferation and metastasis via the Akt signaling
Previous study showed that TBK1 directly phosphorylates AKT to inhibit apoptosis and support oncogenic transformation [14, 15]. Next we tested whether TBK1 promote the proliferation and metastasis of bladder cancer cells via the Akt signaling. The phosphorylation of Akt was enhanced in the presence of TBK1, whereas knockdown of TBK1 dramatically inhibited the phosphorylation level of Akt (Fig. 6A and 6B). Furthermore, the attenuation of the proliferation and migration ability of T24 cells by silencing of TBK1 was markedly eliminated in response to perifosine inhibition of Akt pathway (Fig. 6C and 6D). These data suggested that TBK1 promoted the proliferative and metastasis phenotype of bladder cancer cells by Akt signaling.
TBK1 promote bladder tumorigenesis via Akt signaling. (A). T24 cells were transfected with Flag-tagged TBK1. Twenty-four hours later, cell extracts were analyzed for Akt phosphorylation. (B). The nonspecific control (NC) or TBK1 siRNA were transfected into T24 cells. Fourty-eight hours later, cell extracts were analyzed for Akt phosphorylation. (C and D). Cells knocking down TBK1 were treated with perifosine, a PI3K/AKT inhibitor. The cell proliferation and migration were determined by CCK-8 (C) and transwell assay (D).
Discussion
Besides its canonical function as an essential component of innate immune response, the vital role of TBK1 in oncogenesis has attracted attention [10, 11]. In this study, we demonstrated that TBK1 is up-regulated in both bladder cancer cells and bladder cancer tissue. In addition, knockdown of TBK1 inhibited the proliferation and migration of bladder cancer, and this action is dependent on its kinase activity.
Akt is established unambiguously as a key signaling scaffold and downstream effector of the PI3K pathway [16, 17]. Akt plays a vital role in cell growth, proliferation, energy metabolism and survival [18]. Under physiological circumstances, Akt activity is subjected to stringent regulations spatially and temporally, whereas aberrant activation of Akt has been identified in a wide range of cancers [19, 20]. Numerous research showed that aberrant activation of the PI3K/Akt signaling pathway was critical to the tumorigenesis and progression of bladder cancer and could enhance the malignant phenotypes of bladder cancer [21]. Our results suggested overexpression of TBK1 promotes the phosphorylation of Akt, and silencing of TBK1 inhibits the phosphorylation level.
Chemical inhibitors of TBK1 will be useful for further defining the therapeutic value of this kinase target. As a tool compound, BX795 was found to be effective in controlling the proliferation and migration of bladder cancer cells, which indicated that BX795 have great potential in bladder cancer therapy. We predict that further study will uncover the side effect of BX795 in bladder cancer treatment.
In summary, this study demonstrates that up-regulated TBK1 functioned as an oncogene by modulating the Akt signaling pathway. TBK1 plays a vital role in the progress of bladder cancer and maybe a potential therapy target in bladder cancer.
Supplementary Material
Fig. S1.
Acknowledgements
This work was supported by the National Key Basic Research Program of China (973 Program) (2014CB745201), National Natural Science Foundation of China (81602224), Natural Science Foundation of Guangdong Province, China (2016A030310031), China Postdoctoral Foundation (2016M592576), Shenzhen Municipal Government of China (ZDSYS201504301722174, JCYJ20150330102720130, GJHZ20150316154912494, JCYJ20160425100840929), Special Support Funds of Shenzhen for Introduced High-Level Medical Team, and Shenzhen High-Level Medical Discipline Development Program (2016031638).
Competing Interests
The authors have declared that no competing interests exist.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62:10-29
2. Guillaume L, Guy L. [Epidemiology of and risk factors for bladder cancer and for urothelial tumors]. La Revue du praticien. 2014;64:1372-4 8-80
3. Jacobs BL, Lee CT, Montie JE. Bladder cancer in 2010: how far have we come? CA: a cancer journal for clinicians. 2010;60:244-72
4. van Kessel KE, Zuiverloon TC, Alberts AR, Boormans JL, Zwarthoff EC. Targeted therapies in bladder cancer: an overview of in vivo research. Nature reviews Urology. 2015;12:681-94
5. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148-51
6. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT. et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491-6
7. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631-41
8. Korherr C, Gille H, Schafer R, Koenig-Hoffmann K, Dixelius J, Egland KA. et al. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4240-5
9. Wei C, Cao Y, Yang X, Zheng Z, Guan K, Wang Q. et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E601-10
10. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL. et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157-70
11. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12
12. Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE. et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15:1064-74
13. Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. The Journal of biological chemistry. 2009;284:14136-46
14. Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T. et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Molecular cell. 2011;41:458-70
15. Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G. et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6474-9
16. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7
17. Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer cell. 2007;12:104-7
18. Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene. 2005;24:7391-3
19. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer cell. 2003;4:257-62
20. Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Current opinion in genetics & development. 2009;19:12-7
21. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:1075-83
Author contact
![]() Corresponding authors: E-mail: pony8980com; caizhiming2000com.
Corresponding authors: E-mail: pony8980com; caizhiming2000com.