Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(10):1917-1926. doi:10.7150/jca.18040 This issue Cite
Research Paper
Fluorofenidone Inhibits the Proliferation of Lung Adenocarcinoma Cells
Zheng-hao Deng1,2, Jie Meng3, Juan Tang4, Gao-yun Hu5, Li-jian Tao4,6 ![]()
1. Department of pathology, Xiangya Hospital, Central South University, Changsha, Hunan, 410008, China
2. Department of pathology, School of basic medicine, Central South University, Changsha, Hunan, 410078, China
3. Department of pulmonary Medicine, XiangYa Hospital, Central South University, Changsha, Hunan, 410008, China
4. Department of Nephropathy, XiangYa Hospital, Central South University, Changsha, Hunan, 410008, China
5. Faculty of Pharmaceutical Sciences, Central South University, Changsha Hunan 410013, China
6. State Key Laboratory of Medical Genetics of China, Changsha, Hunan 410078, China
Received 2016-10-23; Accepted 2017-4-8; Published 2017-7-4
Abstract
Background: Lung carcinoma is the leading cause of malignant tumor related mortality in China in recent decades, and the development of new and effective therapies for patients with advanced lung carcinoma is needed. We recently found that fluorofenidone (FD), a newly developed pyridine compound, reduced the activation of Stat3 (Signal transducer and activator of transcription 3) in fibroblasts. Stat3 plays a crucial role in the development of lung cancer and may represent a new therapeutic target. In this study, we examined the effect of FD on human lung adenocarcinoma cells in vivo and in vitro.
Methods: The effect of FD on the growth of lung cancer cells was measured with a CCK-8 assay, colony formation assay and xenograft tumor model. A flow cytometry analysis was performed to study cell cycle arrest and apoptosis. Western blotting and immunohistochemistry were used to observe the expression of Stat3. Changes in the expression of RNA induced by FD were assessed using gene chip and real-time RT-PCR assays.
Results: In vitro, FD inhibited the growth of lung adenocarcinoma A549 and SPC-A1 cells in a dose-dependent manner. After treatment with FD, the A549 and SPC-A1 cells were arrested in the G1 phase, and apoptosis was induced. In vivo, this compound significantly inhibited the growth of tumors that were subcutaneously implanted in mice. Moreover, FD decreased Stat3 activity in lung cancer cells and xenograft tumor tissue, and microarray chip results showed that FD altered the gene expression profile of lung cancer cells. Specifically, NUPR1, which plays a significant role in cancer development, was down-regulated by FD in lung cancer cells.
Conclusion: Our study supports the clinical evaluation of FD as a potential lung adenocarcinoma therapy.
Keywords: Fluorofenidone, lung adenocarcinoma, Stat3, NUPR1.
Introduction
Malignant tumor is a major public health problem worldwide, and lung carcinoma is the most frequently diagnosed malignant tumor and it is responsible for over 1.6 million deaths each year worldwide [1]. In China, approximately 705,000 new cases of lung carcinoma are diagnosed per year [2], and it is often found at an advanced stage, which results in a poor prognosis. Non-small-cell lung cancer (NSCLC) is the most common histologic type of lung carcinoma [3], and the treatment for NSCLC based on platinum chemotherapy. But, clinical interventions have only minimally reduced deaths from lung cancer in the past four decades [4-5]. Despite recent improvements in early diagnosis and the evolution of novel therapeutic strategies, the prognosis for all stages of NSCLC continues to be poor, and the 5-year survival rate for all patients remains at less than 15% [1-2,6]. Thus, novel therapeutic approaches are needed for the treatment of lung cancer.
Fluorofenidone [1-(3-fluorophenyl)-5-methyl-2-(1H)-pyridone, FD] is a new pyridine compound developed by our group. Our previous studies have reported that FD can attenuate fibrosis in the lung [7], kidney [8] and liver [9] via anti-inflammatory [10], antioxidative [11], and antifibrotic [12] activities. Recently, we found that FD reduced the activation of Stat3 (Signal transducer and activator of transcription 3) in fibroblast cells [13]. Notably, Stat3 plays a concernful role in carcinogenesis by regulating genes involved in anti-apoptosis, proliferation and angiogenesis [14]. Furthermore, More and more evidence indicates that Stat3 is constitutively activated in nearly 70% of tumors [15], and blocking Stat3 signaling in tumor cells inhibits tumor growth, angiogenesis, and metastasis [16]. Thus, Stat3 is recognized as a target for cancer therapy [17], but the ability of FD to reduce the activity of Stat3 and inhibit the growth of cancer cells remains unknown.
Based on the important role of Stat3 in cancer pathogenesis and the effect of FD on the activity of Stat3 in fibroblasts, we investigated the ability of FD to inhibit the growth of lung cancer cells. Specifically, we herein examine the effects of FD on the growth and tumorigenicity of lung cancer cells.
Materials and Methods
Reagents and antibodies
FD (lot no.070501) was purchased from Sunshine Lake Pharma (Guangzhou, GD, CN). Cell culture reagents including RPMI1640, fetal bovine serum (FBS), and penicillin/streptomycin were obtained from Invitrogen (Carlsbad, CA, U.S.). Antibodies against cyclin D1, p-Stat3 (Y705) and NUPR1 were obtained from CST (Danvers, MA, USA). Anti β-actin antibody was purchased from Sigma-Aldrich Ltd (St. Louis, MO, USA). Horseradish peroxide (HRP)-conjugated secondary antibodies for Western blotting were procured from Jackson (West Grove, PA, USA), and secondary antibodies for immunohistochemistry were obtained from GBI (Mukilteo, WA, USA). The ECL kit was purchased from GE Healthcare (Buckinghamshire, U.K.), and the CCK-8 assay kit was obtained from Dojindo (Rockville, USA).
Cell culture
The human lung adenocarcinoma cell lines A549 and SPC-A1 were obtained from the cell bank of the Type Culture Collection Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate at 37°C and 5% CO2. Confluent cells were passaged with 0.25% trypsin.
CCK-8 Assay
A CCK-8 assay was performed as described previously [18]. Briefly, cells were plated in 96-well plates at a density of 2000 cells in a final volume of 100 μl/well. Samples of subconfluent cells were treated with various concentrations of FD (0, 2, and 4 mM) for 24, 48 and 72 h, respectively. Cell growth was the assessed with a Cell Counting Kit-8 assay according to the manufacturer's protocol. All experiments were performed in triplicate.
Colony Formation Assay
A colony formation assay was performed as described previously [19]. Briefly, cells were trypsinized, and 1000 viable cells were subcultured in triplicate in six-well plates. The cells were treated with FD (0, 2, and 4 mM) for 72 h and then allowed to adhere and colonize for 14 days without the drug. The medium was refreshed every 3 days. The cells were washed with PBS and fixed with 4% paraformaldehyde. The fixed cells were stained with freshly prepared Giemsa stain for 20 min and colonies were counted under a microscope. All experiments were performed in triplicate.
Flow Cytometry Analysis
Flow cytometry was used to analyze the cell cycle and apoptosis as previously described [20]. In brief, cells were treated with FD (0, 2, and 4 mM) for 72 h and then harvested and fixed in 70% ethanol at 4°C overnight. After staining with annexin V to analyze apoptosis or propidium iodide to analyze the cell cycle, the cells were examined with a BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA).
Western Blot Analysis
Whole-cell lysates were prepared for the western blot analysis as previously described [21]. Cell lysates containing 30 μg of protein were separated on 10% SDS-polyacrylamide gels and transferred to PVDF membranes. The membranes were blocked with TBST buffer (10 mM Tris-HCl, pH 8.0, 0.15 M NaCl, and 0.05% Tween-20) containing 5% FBS and incubated with a rabbit anti-human p-stat3 antibody (1:2000), cyclin D1 antibody (1:1000) or β-actin antibody (1:5000) at room temperature for 2 h, followed by the addition of horseradish peroxidase-linked anti-goat IgG (1:2000). Blotted proteins were detected and quantified using an enhanced chemiluminescence detection system.
Animal Studies
The experimental protocol and the care of animals were under supervision of ethics committee of Central South University. Fifteen female BALB/c nude mice aged 4 to 6 weeks were purchased from Slac Laboratory Animal Co. Ltd. (Shanghai, China) and housed in individually ventilated cages. The mice were randomly divided into 3 groups (5 mice/group). Cultured A549 cells were harvested and resuspended in PBS at 1x107 cells/0.2 ml. The suspension was subcutaneously injected into the left upper extremity of each nude mouse, and treatment with FD was initiated 7 days after inoculation with the cells. The mice were treated daily with intragastrically administered 0.5% carboxymethyl cellulose sodium or fluorofenidone (500 mg/kg/day or 1000 mg/kg/day) for the next 4 weeks until they were euthanized. The tumor volume (TV) was calculated according to the following formula: TV (mm3) = d2×D/2, where d and D are the shortest and longest diameters, respectively. The mice were sacrificed 5 weeks after cell implantation, and the tumors were extracted, weighed, fixed in 10% formalin, embedded in paraffin, and cut into 4 µm-thick sections for further study.
Immunohistochemistry
The immunohistochemistry assay was performed as described previously [22]. Briefly, sections were incubated at 60℃ for 2 h in turpentine and deparaffinized, dried, and rehydrated in a decreasing ethanol: distilled water gradient series. The sections were then incubated for 30 min in 0.3% hydrogen peroxide in 0.01 M phosphate-buffered saline (PBS) to inactivate endogenous peroxidases. Subsequently, the sections were incubated for 20 min in citrate buffer (pH 6.0) at 95℃ for antigen retrieval, blocked with 5% normal goat serum in 0.01 M PBS for 15 min at 37℃, and incubated with primary antibodies overnight at 4℃ in a humidified chamber. The following primary antibodies were used: a rabbit anti-p-Stat3 (Y705) antibody (1:500) and a rabbit anti-cyclind1 (1:500). After washing, the samples were incubated with a biotinylated secondary antibody and a streptavidin-horseradish peroxidase (HRP) avidin working solution. Immunolabeling was visualized using a DAB substrate kit according to the manufacturer's recommendations.
Gene Microarray
The gene expression patterns of lung cancer cells treated with FD (4 mM, 72 h) were detected with a PrimeViewTM Human Gene Expression Array (Affymetrix, CA, USA). The cells were lysed in TRIzol (Invitrogen, CA, USA) to extract RNA, and microarray assays were then performed by CapitalBio Corporation (Beijing, China).
RNA extraction and real-time RT-PCR
RNA extraction and real-time RT-PCR were performed as previously described [23]. The primers were synthesized by Bio Basic Inc (Shanghai, China). The primer against NUPR1 was 5'-CTGTGTTTACTGGGCTGGATT-3' (forward) and 5'-AAGGCTGTGTCTCCTCCTCA-3' (reverse). The primer against β-actin was 5'-CCTTCCTGGGCATGGAGTCCT-3' (forward) and 5'-GGAGCAATGATCTTGATCTT-3' (reverse). Real-time RT-PCR was performed on an ABI Model 7500 Sequence Detector (Applied Biosystems, Foster City, CA) with a TaKaRa SYBR green real-time PCR kit (Takara, Dalian, China). The quantity of mRNA was calculated based on the cycle threshold (CT) values, and the relative abundance of NUPR1 mRNA was standardized to that of β-actin mRNA.
Statistical analysis
The SPSS 17.0 statistical software package (SPSS Inc., Chicago, IL, USA) was used to perform statistical analyses. Student's t-test was used to perform comparisons between two groups. Pearson's correlation coefficients were used to calculate bivariate correlations between study variables. The data represent the mean±S.D. A P-value <0.05 was considered significant.
Results
FD inhibited growth of lung adenocarcinoma cells
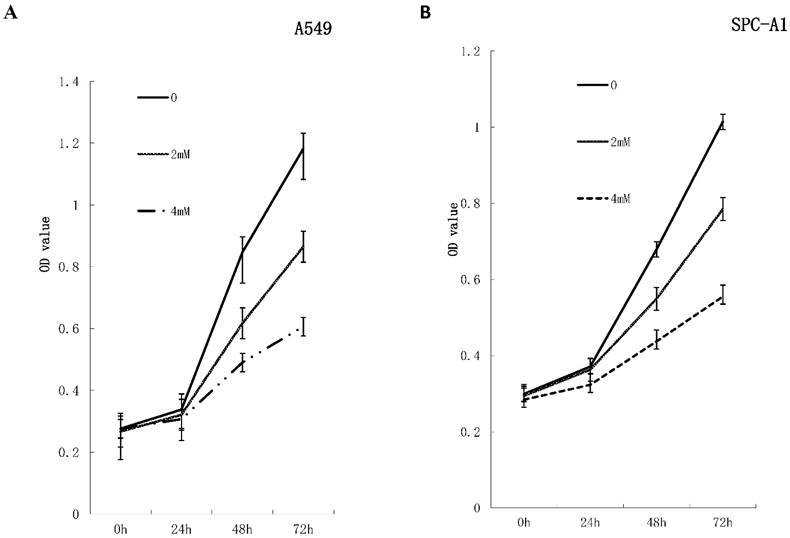
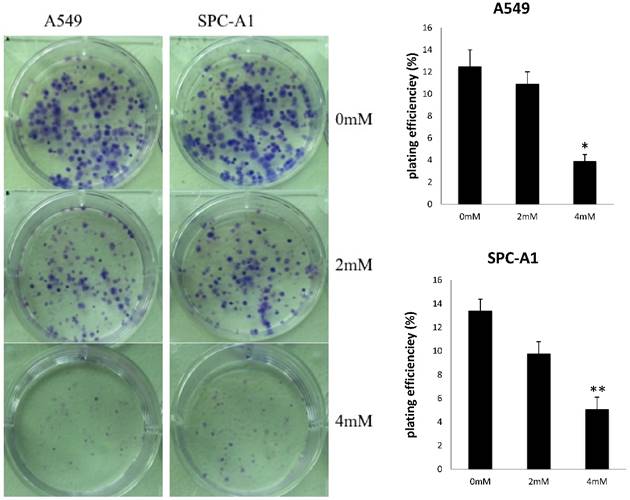
To investigate the effects of FD on the growth of human lung adenocarcinoma cells, a CCK-8 assay and colony formation assay were performed. FD inhibited the growth of A549 and SPC-A1 cells over 48 and 72 h of continuous exposure, as assessed by the CCK-8 assay (Figure 1). Specifically, the suppression rates were 42.14±0.17% at 48 h and 48.73±0.32% at 72 h for A549 cells treated with 4 µm FD, and the suppression rates were 35.64±0.48% at 48 h and 45.27±0.19% at 72 h for SPC-A1 cells treated with 4 µm FD. We analyzed the ability of plated A549 and SPC-A1 cells to form colonies because anchorage-independent growth often correlates with tumorigenicity. The respective plating efficiencies of the cells treated with 0, 2, and 4 mM FD were 12.5±1.5%, 10.9±2.1% and 3.9±0.6% for the A549 cells and 13.4±2.5%, 9.8±3.3% and 5.1±0.9% for the SPC-A1 cells (Fig. 2). FD decreased colony formation only at a dose of 4 mM compared with the other two groups(P<0.05). Therefore, it inhibited lung adenocarcinoma cell growth.
FD inhibits the growth of lung adenocarcinoma cells in vitro. A549 cells and SPC-A1cells were treated with FD at the indicated concentration for 72 h. Cell viability was measured with a CCK-8 assay. FD inhibited the growth of A549 (Fig. 1A) and SPC-A1 (Fig. 1B) cells over 48 and 72 h of continuous exposure. Experiments were performed in triplicate.
FD decreased the colonies formed by A549 and SPC-A1 cells in a dose-dependent. Experiments were performed in triplicate. (Compared with the control group,*, **, P<0.05)
FD induced lung adenocarcinoma cell G1 arrest and apoptosis
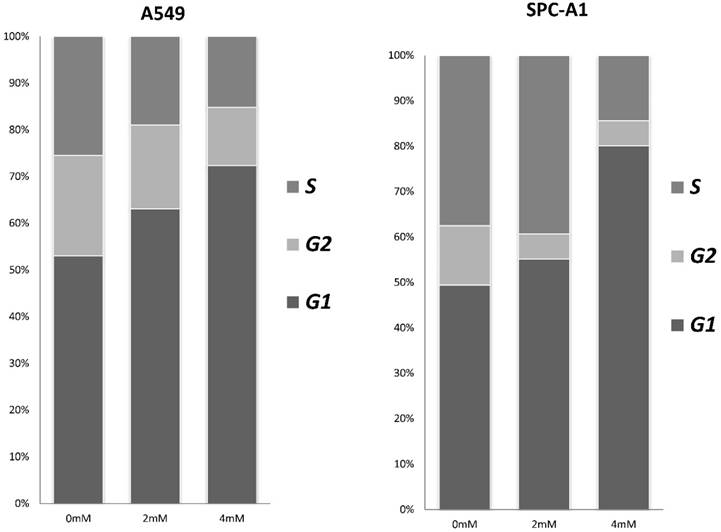
To investigate the effects of FD on the cell cycle and apoptosis of human lung adenocarcinoma cells, cells were assessed by flow cytometry after treatment with different concentrations of FD (0, 2, and 4 mM) for 72 h. As shown in Fig. 3, FD induced cell cycle transition to the G1 phase(P<0.05). The apoptosis rate of the cells treated with 0, 2, and 4 mM FD were 1.4±0.1%, 2.9±0.2% and 4.0±0.3% for the A549 cells and 3.4±0.6%, 6.8±0.3% and 10.7±1% for the SPC-A1 cells. Hence, FD increased the number of apoptotic cells compared with the respective controls(P<0.05).
FD induces G1 arrest in lung adenocarcinoma cells. Cells were stained with annexin-V-FITC (20 mg/ml) and PI (20 mg/l) following treatment with or without FD for 72 h. The cell cycle was analyzed using flow cytometry. Experiments were performed in triplicate.
FD inhibited lung adenocarcinoma cell growth in vivo
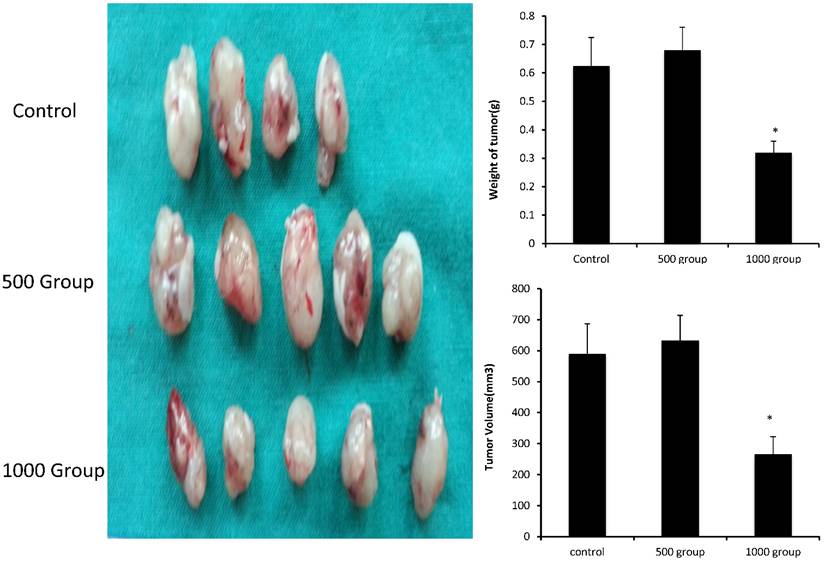
We evaluated the effects of FD in vivo in a xenograft tumor model by subcutaneously inoculating nude mice with A549 cells. Seven days after tumor cell injection, the mice were administered daily intragastric injections of FD (500 and 1000 mg/kg body weight) for 4 weeks. The tumors appeared at nearly the same time in the three groups. At the end of the study, all tumors were removed and dissociated, and their weights and volumes were measured. The mean tumor weights of the three groups (treated with FD 0,500 and 1000 mg/kg body weight) were 0.625±0.1 g, 0.68±0.08 g and 0.32±0.04 g, respectively. The mean tumor volumes of the three groups (treated with FD 0,500 and 1000 mg/kg body weight) were 589.6±97.7 mm3, 632.6±56.7 mm3 and 265.9±56.7 mm3, respectively. Consistent with the in vitro results, FD decreased the growth of A549 cells. FD administration was effective in inhibiting tumor growth in vivo throughout the course of treatment, resulting in decreases in tumor size and weight at a dose of 1000 mg/kg body weight(P<0.05) (Figure 4). It indicated that FD reduced tumor volume and the growth of A549 cells in vivo.
FD inhibits lung adenocarcinoma A549 cell tumor growth in vivo. Dissected tumors were photographed, and the weights and volumes of tumors from FD-treated mice (0, 500 mg/kg/day and 1000 mg/kg/day) were measured. (Compared with the control group, *, **, P<0.05)
FD decreased Stat3 activity in lung adenocarcinoma cells
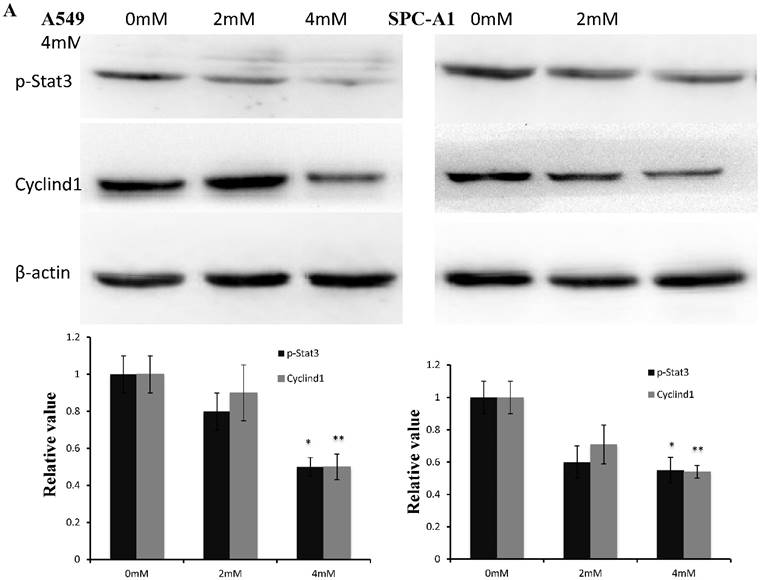
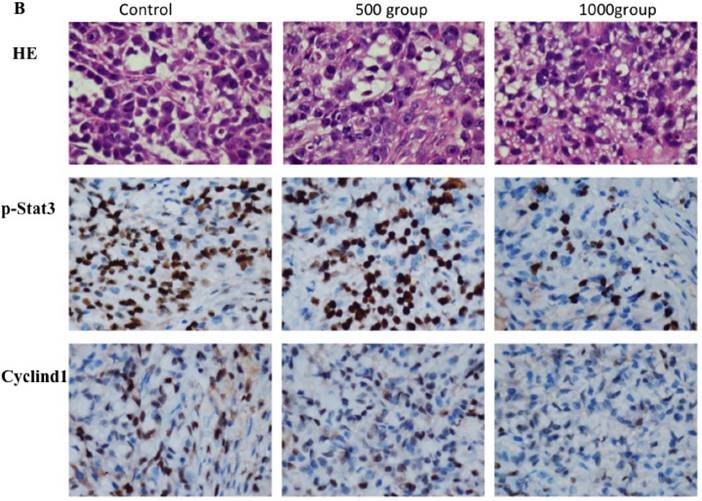
Western blotting data showed that p-stat3 and cyclin D1 expression were significantly inhibited in A549 and SPC-A1 cells treated with 4 mM FD compared with the control group and cells treated with 2 mM FD. Furthermore, immunohistochemistry assay showed that both p-Stat3 and cyclind1 expression were inhibited in xenograft tumors treated with 1000 mg/kg body weight compared with the other two groups(P<0.05) (Fig. 5).
FD decreased Stat3 activity. (1) Western blotting results showed that FD treatment repressed the expression of p-Stat3 and cyclin D1 in lung adenocarcinoma A549 and SPC-A1 cells (Fig. 5A). (2) Representative images of the immunohistochemistry analysis of p-Stat3 and cyclin D1 in xenograft tumors (×400) (Fig. 5B). (Compared with the control group, *, **, P<0.05)
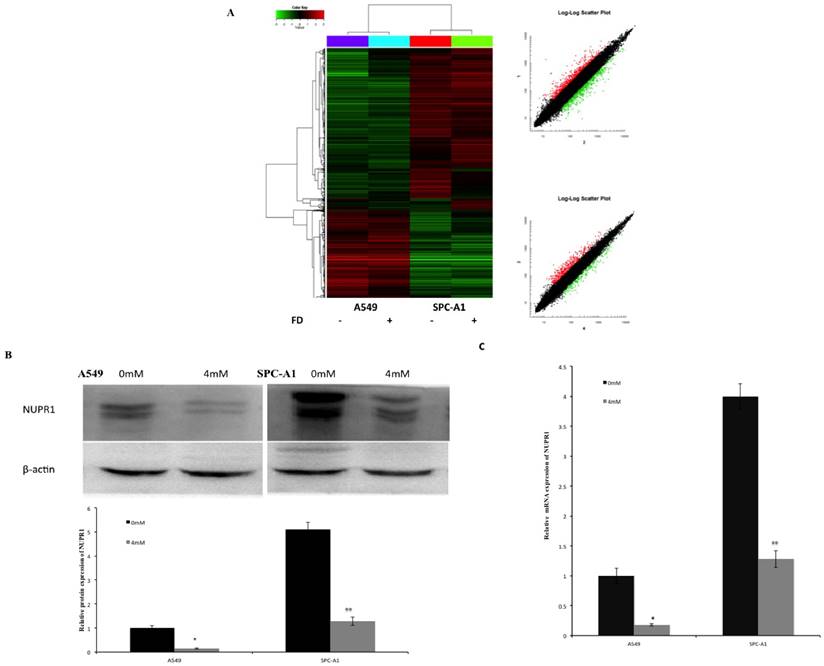
Effects of FD on gene expression pattern on lung cancer cells
The effect of FD on gene expression in A549 and SPC-A1 cells was assessed using Affymetrix gene microarrays. Gene expression profile was examined in cells treated with 0 or 4 mM FD for 72 h. The differentially expressed genes were identified including up-regulated 36 genes and down-regulated 40 genes in both A549 and SPC-A1 cells (compared with the control group, expression ratio showing a greater than 2-fold or a less than 0.5-fold was identified). These genes are involved in Wnt signaling, PPAR signaling, MAPK signaling and ErbB signaling (data not shown). Therefore, FD may have its anti-tumor effect on lung carcinoma cells by changing the expression of these genes. Among these differentially expressed genes, NUPR1 expression was increased more than 5-fold in A549 and SPC-A1 cells. Western blotting and real time-PCR results showed that the mRNA and protein expression levels of NUPR1 were markedly reduced in FD-treated lung carcinoma cells, which is consistent with the gene array analysis (Fig. 6).
Effects of FD on the gene expression pattern of lung cancer cells. (1) Cluster map and scatter plot of changes in gene expression levels in lung cancer cells (Fig. 6A). (2) FD decreased the protein expression of NUPR1 in lung cancer cells, as determined by Western blotting (Fig. 6B). (3) FD decreased the mRNA expression of NUPR1 in lung cancer cells, as determined by real-time qPCR (Fig. 6C). (Compared with the control group, *, **, P<0.05)
Discussion
In this study, we first described the effects of FD on lung cancer cells in vitro and in vivo. Specifically, we found that FD inhibited the growth of lung adenocarcinoma A549 and SPC-A1 cells using a CCK-8 assay and colony formation assay. Moreover, treatment with FD arrested these cells in the G1-phase and induced apoptosis. Furthermore, FD significantly inhibited the growth of tumors subcutaneously implanted in mice. We also found that FD suppressed the expression of p-STAT3 in lung adenocarcinoma A549 and SPC-A1 cells, and FD have markedly affected the mRNA expression pattern of these cells.
The same effects have been reported in malignant glioma [24] and pancreatic cancer cells [25] for pirfenidone, whose chemical structure is similar to that of FD. However, pirfenidone reportedly inhibited pancreatic cancer growth by suppressing desmoplasia via the regulation of pancreatic stellate cells [25]. In our study, the mice were orally administered 500 and 1000 mg/kg/day FD. Growth inhibition was only observed in the high-dose group without obvious side effects. Thus, FD may inhibit the growth of lung adenocarcinoma cells in a dose-dependent manner.
In this study, we also found that FD inhibited the expression of p-STAT3 in lung cancer cells. STAT3 is a well-known member of the STAT family of signal responsive transcription factors, which consists of seven members encoded by distinct genes [26]. The phosphorylation of a critical tyrosine residue (Tyr705) activates STAT3 [27], and phosphorylated STAT3 forms a homodimer by binding at the SH2 domain and is translocated to the nucleus to promote the transcription of target genes [28]. Specifically, STAT3 regulates cyclin D1, c-Myc, Bcl-XL, and p53, thereby mediating cellular proliferation and survival [29]. Moreover, STAT3 is persistently activated in 22~65% of non-small cell lung cancers (NSCLCs) and plays an important role as a signaling mediator in malignant diseases [30-32]. An increasing body of evidence has shown that STAT3 inhibitors exert anticancer and antiangiogenic effects in vitro and in vivo [33]. These results indicate that FD may serve as an anti-cancer agent by inhibiting the phosphorylation of STAT3.
Our previous studies indicated that FD is a multifunctional small molecule with anti-inflammatory, antioxidative and anti-apoptotic effects, and it can inhibit the activation and proliferation of myofibroblasts, promote the degradation of extracellular matrix and regulate the cellular signal transmission [34]. However, the exact mechanisms underlying these effects remain unknown. To further explore the mechanism of action of FD in lung cancer cells, we observed the changes in gene expression by A549 and SPC-A1 after treatment with FD. Our results showed that FD markedly changed the gene expression patterns of these cells. Specifically, FD significantly decreased the expression of NUPR1 (Nuclear protein 1) in both A549 and SPC-A1 cells.
NUPR1, also named P8 or com1 (candidate of metastasis-1), is a small, highly basic and loosely folded protein. Structurally, Nupr1 is related to the high-mobility group of transcriptional regulators [35], and its expression can be induced by several stressors, including LPS [36] and CCL4 [37]. Many studies have shown that NUPR1, which is frequently upregulated in several cancers, plays an important role in cancer development and progression [38]. For example, NUPR1 is essential for the survival of pancreatic ductal adenocarcinoma cells exposed to stress [39]. Similar to our results, X. Guo reported that the knockdown of NUPR1 significantly inhibited the proliferation and colony formation of lung cancer H1299 cells and suppressed tumor growth in vivo [40]. Moreover, Roxane reported that the expression of NUPR1 was activated by TGFβ at the transcriptional level [41], and our previous studies showed that FD attenuated TGF-β1-related signaling [13]. Thus, FD may have decreased the expression of NUPR1 by lung cancer cells via TGF-β signaling, but this mechanism requires further investigation.
Overall, the present study revealed that FD inhibits the growth of lung adenocarcinoma A549 and SPC-A1 cells in vitro and in vivo. Thus, FD may represent a new treatment strategy for lung cancer, but additional studies are needed to clarify the mechanisms of action of FD in cancer cells.
Abbreviations
FD: Fluorofenidone; Stat3: Signal transducer and activator of transcription 3; NSCLC: Non-small-cell lung cancer; CCK-8: Cell Counting Kit-8.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81470255 and 81273575).
Authors' contributions
Zhenghao Deng, Jie Meng, and Juan Tang collected and analyzed the data, drafted the manuscript, and conceived the main idea of the study. Gao-yun Hu and Lijian Tao participated in the acquisition of data and the design and intellectual conception of the study. All authors performed the statistical analysis and approved the final version of the manuscript.
Ethics approval and consent to participate
The experimental protocol and the care of animals accorded with the licenses held by the Central South University.
Competing Interests
The authors declare no conflicts of interest.
References
1. Ferlay J, Shin HR, Bray F. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-2917
2. Lindsey A. Torre, Freddie Bray et al. Global Cancer Statistics, 2012, CA CANCER J CLIN. 2015;65:87-108
3. Aisner DL, Marshall CB. Molecular pathology of non-small cell lung cancer: a practical guide. Am J Clin Pathol. 2012;138:332-346
4. Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350:379-392
5. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Eagl J Med. 2008;359:1367-1380
6. Zarogoulidis P, Darwiche K, Browning RF. et al. Molecular Targeted Drugs and Biomarkers in NSCLC, the Evolving Role of Individualized Therapy. J Cancer. 2013;4:736-754
7. Meng J, Zou Y, Hu C. et al. Fluorofenidone attenuates bleomycin-induced pulmonary inflammation and fibrosis in mice via restoring caveolin 1 expression and inhibiting mitogen-activated protein kinase signaling pathway. Shock. 2012;38:567-73
8. Yuan Q, Wang R, Peng Y. et al. Fluorofenidone attenuates tubulointerstitial fibrosis by inhibiting TGF-β(1)-induced fibroblast activation. Am J Nephrol. 2011;34:181-94
9. Peng Y, Yang H, Wang N. et al. Fluorofenidone attenuates hepatic fibrosis by suppressing the proliferation and activation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2014;306:253-63
10. Huang L, Zhang F, Tang Y. et al. Fluorofenidone attenuates inflammation by inhibiting the NF-кB pathway. Am J Med Sci. 2014;348:75-80
11. Qin J, Xie YY, Huang L. et al. Fluorofenidone inhibits nicotinamide adeninedinucleotide phosphate oxidase via PI3K/Akt pathway in the pathogenesis of renal interstitial fibrosis. Nephrology (Carlton). 2013;18:690-9
12. Peng Y, Yang H, Zhu T. et al. The antihepatic fibrotic effects of fluorofenidone via MAPK signalling pathways. Eur J Clin Invest. 2013;43:358-68
13. Tang J, Liu CY, Lu MM. et al. Fluorofenidone protects against renal fibrosis by inhibiting STAT3 tyrosine phosphorylation. Mol Cell Biochem. 2015;407(1-2):77-87
14. Wake MS, Watson CJ. STAT3 the oncogene - still eluding therapy? FEBS J. 2015;282(14):2600-11
15. Siveen KS, Sikka S, Surana R. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136-54
16. Wang X, Crowe PJ, Goldstein D. et al. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers. Int J Oncol. 2012;41(4):1181-91
17. Muhammad Furqan, Akintunde Akinleye, Nikhil Mukhi. STAT inhibitors for cancer therapy. Journal of Hematology & Oncology. 2013;6:90
18. Wang LL, Hu RC, Dai AG. et al. Bevacizumab induces A549 cell apoptosis through the mechanism of endoplasmic reticulum stress in vitro. Int J Clin Exp Pathol. 2015;8(5):5291-5299
19. Li Q, Lu XH, Wang CD. et al. Antiproliferative and apoptosis-inducing activity of schisandrin B against human glioma cells. Cancer Cell Int. 2015;15(1):12
20. Kim MO, Lee MH, Oi N. et al. [6]-shogaol inhibits growth and induces apoptosis of non-small cell lung cancer cells by directly regulating Akt1/2. Carcinogenesis. 2014;35(3):683-91
21. Liu J, Song C, Xiao Q. et al. Fluorofenidone attenuates TGF-β1-induced lung fibroblast activation via restoring the expression of caveolin-1. Shock. 2015;43(2):201-207
22. Hu C, Lv H, Pan G. et al. The expression of ADAM23 and its correlation with promoter methylation in non-small-cell lung carcinoma. Int J Exp Pathol. 2011;92(5):333-9
23. Xiong X, Mei W, Xie Y. et al. Fluorofenidone offers improved renoprotection at early interventions during the course of diabetic nephropathy in db/db mice via multiple pathways. PLoS One. 2014;9(10):e111242
24. Burghardt I, Tritschler F, Opitz CA. et al. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem Biophys Res Commun. 2007;354:542-7
25. Kozono S, Ohuchida K, Eguchi D. et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013;73:2345-56
26. Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets. 2004;8(5):409-22
27. Dutta P, Sabri N, Li J. et al. Role of STAT3 in lung cancer. JAKSTAT. 2015;3(4):e999503
28. Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert Opin Ther Targets. 2012;16(9):889-901
29. Hai-Feng Zhang, Raymond Lai. STAT3 in Cancer—Friend or Foe? Cancers. 2014;6:1408-1440
30. Zimmer S, Kahl P, Buhl TM. et al. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer Res Clin Oncol. 2009;135:723-30
31. Looyenga BD, Hutchings D, Cherni I. et al. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One. 2012;7:e30820
32. Jiang R, Jin Z, Liu Z. et al. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol.Diagn.Ther. 2011;15:347-52
33. Daijiro Harada, Nagio Takigawa, Katsuyuki Kiura. The Role of STAT3 in Non-small cell lung cancer. Cancer. 2014;6:708-22
34. MA Hong, PENG Zhangzhe, HU Gaoyun. et al. Effects of fluorofenidone on organ fibrosis and the mechanisms. Journal of Central South University. Medical Science. 2015;40(2):208-213
35. Sandro Goruppi, Juan Lucio Iovanna. Stress-inducible Protein p8 Is Involved in Several Physiological and Pathological Processes. J. Biol. Chem. 2010;285:1577-1581
36. Vasseur S, Hoffmeister A, Garcia-Montero A. et al. Mice with targeted disruption of p8 gene show increased sensitivity to lipopolysaccharide and DNA microarray analysis of livers reveals an aberrant gene expression response. BMC Gastroenterol. 2003;3:25
37. Taïeb D, Malicet C, Garcia S. et al. Inactivation of stress protein p8 increases murine carbon tetrachloride hepatotoxicity via preserved CYP2E1 activity. Hepatology. 2005;42(1):176-82
38. Cano CE, Hamidi T, Sandi MJ. et al. Nupr1: the Swiss-knife of cancer. J Cell Physiol. 2011;226(6):1439-43
39. Hamidi T, Algül H, Cano CE. et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J Clin Invest. 2012;122(6):2092-103
40. Guo X, Wang W, Hu J. et al. Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat Rec (Hoboken). 2012;295(12):2114-21
41. Pommier RM, Gout J, Vincent DF. et al. The human NUPR1/P8 gene is transcriptionally activated by transforming growth factor β via the SMAD signalling pathway. Biochem J. 2012;445(2):285-93
Author contact
![]() Corresponding author: Professor LiJian Tao, Email: taoljedu.cn
Corresponding author: Professor LiJian Tao, Email: taoljedu.cn