Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(11):1952-1958. doi:10.7150/jca.19029 This issue Cite
Research Paper
A Role for The ATP7A Copper Transporter in Tumorigenesis and Cisplatin Resistance
Sha Zhu1,3,*, Vinit Shanbhag1,3,*, Yanfang Wang2,3, Jaekwon Lee4, Michael Petris1,2,3, ![]()
1. Department of Biochemistry, University of Missouri, Columbia, MO, 65211
2. Nutrition and Exercise Physiology, University of Missouri, Columbia, MO, 65211
3. The Christopher S. Bond Life Science Center, University of Missouri, Columbia, MO, 65211
4. Redox Biology Center, Department of Biochemistry, University of Nebraska, Lincoln, Nebraska, 68588
*These authors contributed equally to this study.
Received 2017-1-3; Accepted 2017-4-25; Published 2017-7-5
Abstract
The ATP7A protein is a ubiquitously expressed copper-translocating P-type ATPase that controls cytoplasmic copper concentrations by mediating cellular copper egress. In vitro studies have previously demonstrated that ATP7A abundance in various tumor cell lines is correlated with increased resistance to cisplatin, a widely-used chemotherapy agent. However, to date no studies have examined a role for ATP7A in tumor growth or cisplatin sensitivity in vivo. In this study, we deleted ATP7A in H-RAS transformed tumorigenic mouse embryonic fibroblasts (MEFRAS7A-). Interestingly, loss of ATP7A was found to markedly suppress tumorigenesis in MEFRAS7A- cells relative to wild type parental cells. This was associated with hyperaccumulation of copper and sensitivity to reactive oxygen species and hypoxia. Tumor grafts lacking ATP7A were markedly more sensitive to cisplatin chemotherapy compared to ATP7A-expressing control tumors. These findings identify ATP7A at the nexus between tumorigenesis and cisplatin resistance pathways, underscoring its potential as a therapeutic target for regulating both tumor growth and the efficacy of cisplatin treatment.
Keywords: ATP7A, Copper transporter, Cisplatin resistance, Tumorigenesis
Introduction
Copper is an enzymatic cofactor that in mammals is necessary for cellular respiration, iron homeostasis, melanogenesis, neurotransmitter biosynthesis, and connective tissue formation. The essentiality of copper lies in its ability to cycle between Cu1+ and Cu2+ oxidation states, which confers upon cuproenzymes the ability to catalyze redox reactions. However, this same redox property also underlies its potential toxicity if copper concentrations surpass the capacity of cellular detoxification mechanisms. The principle regulators of copper homeostasis in virtually all cell types include CTR1, a plasma membrane copper importer, and ATP7A, a copper exporting P-type ATPase that regulates cytoplasmic copper concentrations. The intracellular roles of ATP7A include the delivery of copper to essential cuproenzymes of the secretory pathway, and copper excretion across the plasma membrane [1-3]. Accordingly, loss of ATP7A in cultured cells results in the accumulation of copper in the cytosol, and hypoactivity of various copper-dependent enzymes that require copper to be transported into the secretory pathway [4-7].
Previous studies have shown that copper transporters play key roles in the cellular accumulation of the chemotherapy agent cisplatin [8-12], a front-line treatment against several types of cancer [13]. As with many chemotherapy agents, acquired cisplatin resistance becomes problematic upon repeated exposure [14, 15]. The expression of CTR1 in cultured cells is inversely proportional to cisplatin accumulation and resistance, suggesting that CTR1 is responsible for cisplatin uptake across the plasma membrane. In contrast, ATP7A abundance is positively correlated with increased resistance to cisplatin [16-18]. Elevated expression of ATP7A is associated with worse outcomes in patients with ovarian or non-small cell lung cancer who are treated with platinum- containing drugs [19-21]. Similar correlations have been made between cisplatin resistance and a closely related copper ATPase, ATP7B [22]. Although the mechanisms by which ATP7A/B contribute to cisplatin resistance are not well understood, several hypotheses have been proposed including direct transport and sequestration of the drug into intracellular vesicles, or chelation through interactions between cisplatin and metal-binding domains in these proteins [23-25]. In addition to playing roles in cisplatin resistance, pathways of copper homeostasis also appear to regulate tumor growth, although the underlying mechanisms are poorly understood. Changes in systemic copper metabolism are known to accompany various types of cancer. Serum copper levels are elevated in cancer patients, which correlate with disease outcome and response to therapies [26, 27]. Copper-chelating drugs such as tetrathiomolybdate or penicillamine are known to have anti-angiogenic and anti-neoplastic activity, indicating that copper is rate limiting for tumor growth [28-31]. Copper chelation has been shown to reduce ATP levels and suppress pancreatic tumors in mice, in part due to restricting the activity of copper-dependent mitochondrial cytochrome c oxidase [32]. Other studies suggest that NFkappaB-dependent tumorigenesis is also impaired by copper chelation [30]. Copper also appears to be rate limiting for mitogenic signaling pathways necessary for tumor growth such as RAS/MAPK signaling, in part due to a direct role for copper ions in regulating MEK1 activity [33]. Studies have also shown that copper-dependent superoxide dismutase is necessary for receptor tyrosine kinase signaling by producing hydrogen peroxide, which blocks downstream inhibitory phosphatases [34]. These studies suggest that cancer cells are vulnerable to disturbances in copper homeostasis, a weakness that might be exploited by novel therapeutic strategies to restrict copper delivery to one or more key enzymes with roles in cancer biology.
The goal of the current study was to investigate the extent to which ATP7A is necessary for tumor growth and cisplatin resistance in vivo. Deletion of ATP7A in tumorigenic fibroblasts was found to significantly increase cellular accumulation of copper, sensitize these cells to both hypoxia and reactive oxygen species, reduce tumor formation in mice, and increase cisplatin sensitivity in vitro and in vivo. These data reveal a requirement for ATP7A in both tumor growth and cisplatin resistance, highlighting its potential as a novel target for anti-cancer therapies.
Results and Discussion
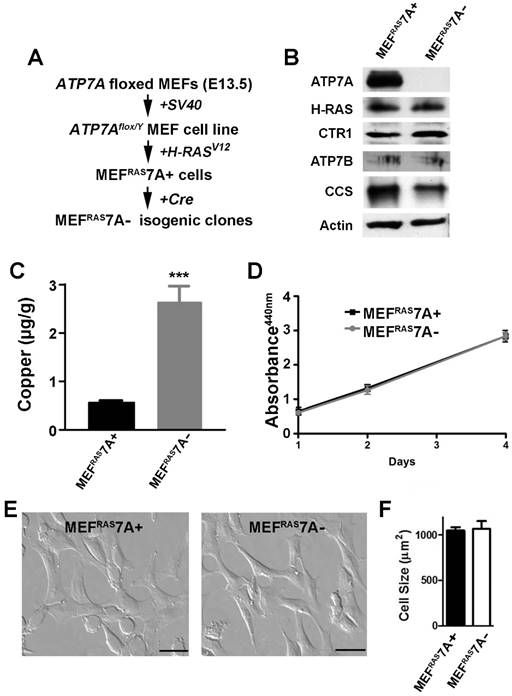
Generation of tumorigenic ATP7A knockout MEF cells. An initial goal of this study was to generate a tumorigenic mouse embryonic fibroblast cell line containing a floxed ATP7A allele that would enable subsequent ATP7A gene deletion using Cre recombinase. Previously, we generated a mouse embryonic fibroblast cell line carrying a floxed ATP7A allele (MEF7A fl/Y) that was immortalized by expression of the Simian virus 40 (SV40) large T-antigen [35] (Figure 1A). As transformation usually requires collaboration between at least two oncogenes in rodent cells, we transfected these cells with a plasmid carrying a mutant RAS oncogene (H-RASV12). Several independent clones were isolated and tested for tumorigenicity by subcutaneous injection into immunocompromised mice. A single clone was chosen for further study, hereafter called MEFRAS7A+, which gave rise to palpable tumors at 100% incidence with a latency period of approximately 10 days (Figure S1). To generate isogenic clones in which ATP7A was deleted, the MEFRAS7A+ cells were infected with adenovirus carrying the Cre recombinase gene, and single clones were isolated in which ATP7A expression was absent. One of these clones, MEFRAS7A- was chosen for further study (Figure 1A and 1B). Consistent with the known role of ATP7A in copper efflux, the MEFRAS7A- cells were found to hyperaccumulate copper relative to MEFRAS7A+ cells (Figure 1C). The MEFRAS7A- cells also exhibited lower levels of CCS (Figure 1B), a protein whose abundance is inversely proportional to cytoplasmic copper concentration [35, 36]. No differences were found in the levels of the ATP7B protein, which was weakly detected in both MEFRAS7A+ and MEFRAS7A- cells. A similar level of expression of the CTR1 copper importer was found in both cell lines (Figure 1B). Despite the hyperaccumulation of copper in MEFRAS7A- cells, these cells grew at a similar rate compared to control MEFRAS7A+ cells in regular growth medium (Figure 1D).
Characterization of MEFRAS7A+ and MEFRAS7A- cells. (A) MEFRAS7A+ cells were generated from a mouse embryonic fibroblast cell line carrying a floxed Atp7a allele and transformed by sequential transfection of SV40 large T-antigen and mutant H-RAS oncogene. Isogenic MEFRAS7A- cells were generated by infecting with adenovirus carrying the Cre recombinase. (B) Immunoblot analyses. Cell lysates were separated by SDS-PAGE and immunoblotted with the indicated antibodies. (C) Copper concentration determination within cultured MEFRAS7A+ and MEFRAS7A- cells by inductively coupled plasma mass spectrometry (mean ± S.E.M; ***P<0.001; n = 3). (D) Cell proliferation was evaluated using WST-1 colorimetric assay by measuring the absorbance at 440nm (mean ± S.E.M; n = 3). (E) Representative images of MEFRAS7A+ and MEFRAS7A- cells Scale bars: 50µM. (F) Cell size determination of MEFRAS7A+ and MEFRAS7A- cells (mean ± S.E.M; n = 30 cells in three fields).
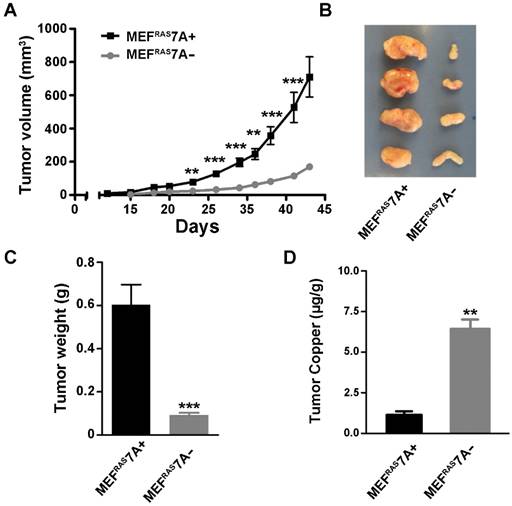
ATP7A deletion suppresses tumorigenesis. To investigate the effect of ATP7A gene deletion on tumor growth, the MEFRAS7A- and MEFRAS7A+ cells were subcutaneously injected into nude mice, and tumor volume was measured at regular intervals over 5 weeks. Tumors derived from MEFRAS7A- cells were found to be markedly smaller than those from MEFRAS7A+ cells (Figure 2A-C). This difference in tumor size was observed in multiple independent clones of MEFRAS7A- cells (data not shown). Histological analyses of the tumors did not reveal obvious differences in markers of vascular density (CD31) or proliferation (Ki67) (Figure S2). The findings suggest that loss of ATP7A is tumoristatic in RAS-transformed fibroblasts.
Deletion of Atp7a suppresses tumor growth of MEFRAS7A- cells Subcutaneous growth of MEFRAS7A+ and MEFRAS7A- cells in nude mice. Tumor volume (A) and tumor weight (C) are shown. B) Representative MEFRAS7A+ and MEFRAS7A- tumors at 5 weeks after cell implantation (n= 10-12 mice; mean ± S.E.M; **P<0.01; ***P<0.001). (D) Tumor copper concentrations measured by ICP-MS (mean ± S.E.M; **P<0.01).
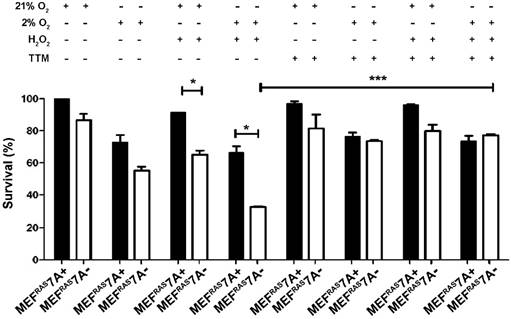
Loss of ATP7A causes sensitivity to ROS and hypoxia. The observation that MEFRAS7A- cells grew at a similar rate as MEFRAS7A+ cells in cell culture media, and yet showed reduced tumor growth in mice, suggests that the growth inhibition mediated by ATP7A deletion is specific to in vivo conditions. As the rapid growth of solid tumors is often associated with hypoxia and oxidative stress, we investigated whether MEFRAS7A- cells might be more sensitive to hypoxia and/or hydrogen peroxide compared to MEFRAS7A+ cells using an MTT assay of cell viabiility. Hydrogen peroxide alone, or in the presence of hypoxia, resulted in a significant inhibition of growth in MEFRAS7A- cells relative to MEFRAS7A+ cells (Figure 3). Importantly, this hypersensitivity was prevented by the addition of the membrane permeable copper chelator, tetrathiomolybdate (Figure 3). Similar results were obtained using a Crystal violet assay of cell proliferation (Figure S3). These findings are consistent with the hypothesis that loss of ATP7A renders MEFRAS7A- cells hypersensitive to ROS in a copper dependent manner.
Loss of ATP7A confers copper-dependent hypersensitivity to hydrogen peroxide. MEFRAS7A- and MEFRAS7A+ cells in a 24 well plate were exposed to 40 µM H2O2 in either normoxia (21% O2) or hypoxia (2% O2) in the presence of absence of the copper chelator tetrathiomolybdate (TTM; 4µM). Cell viability was then determined using the MTT assay after 24 hours of treatment (mean ± S.E.M., *P<0.05, **P<0.01, ***P<0.001).
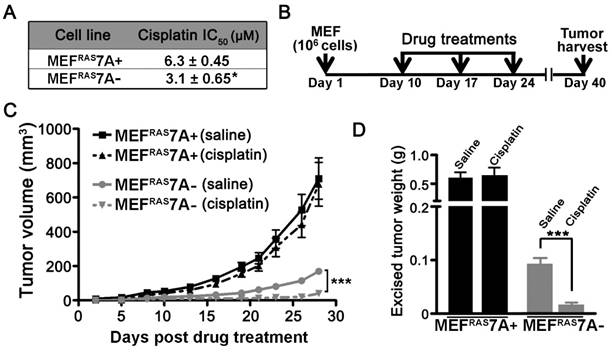
ATP7A contributes to cisplatin resistance in vitro and in vivo. While previous studies have demonstrated that ATP7A plays a role in cisplatin resistance in cultured cell lines, there have been no studies examining this requirement in vivo [16]. In support of previous studies, the MEFRAS7A- cells exhibited reduced survival compared to MEFRAS7A+ cells in cell culture medium containing cisplatin (Figure 4A). To test the in vivo requirement for ATP7A in cisplatin resistance, we compared the inhibitory effect of cisplatin chemotherapy on tumor growth of MEFRAS7A- and MEFRAS7A+ cells in mice. Tumor growth was initiated by subcutaneous injection of either MEFRAS7A- or MEFRAS7A+ cells in nude mice. After a 10-day initial growth period, cisplatin (4mg/kg) or saline control was administered via intraperitoneal injection at weekly intervals over a three-week period, and tumors were then harvested at day 40 (Figure 4B). Consistent with our earlier observations, in the saline-treated control animals the MEFRAS7A- tumors were markedly smaller than the MEFRAS7A+ tumors (Figure 4C and 4D). Importantly, cisplatin treatment significantly reduced the size of the MEFRAS7A-tumors compared to the saline treated controls, however, there was no significant inhibitory effect of cisplatin on the growth of MEFRAS7A+ tumors (Figure 4C and 4D).
Deletion of Atp7a increases cisplatin sensitivity in vitro and in vivo (A) IC50 values for cisplatin were calculated for MEFRAS7A+ and MEFRAS7A- cells exposed to increasing doses of cisplatin for 24 hours. Cell viability was measured using the Prestoblue assay. (B) Schematic illustration of the cisplatin treatment regimen used to assess the role of ATP7A in cisplatin sensitivity in tumor-bearing mice. MEFRAS7A+ and MEFRAS7A- cells were injected subcutaneously into flanks of nude mice. Cisplatin (4mg / kg) or an equal volume of saline control was intraperitoneally injected on days 10, 17 and 24. At day 40, tumors were harvested for tumor volume measurement (C) and weight (D) (n= 10-12 mice per group; mean ± S.E.M., ***P<0.001).
Taken together, the results of this study indicate that deletion of the ATP7A gene is rate limiting for tumor growth, and enhances the efficacy of cisplatin-mediated chemotherapy in a RAS- transformed tumor model. Loss of ATP7A protein attenuated the growth of MEFRAS7A- tumors in mice compared to wild type cells, but had little impact on their growth in cell culture. The tumoristatic effects of copper hyperaccumulation have been demonstrated in previous studies involving deletion of a related copper exporter, ATP7B [37, 38]. However, the tumoristatic effect of ATP7A deletion in our study was not solely attributable to copper hyperaccumulation per se, because the magnitude of the increased copper accumulation in MEFRAS7A- cells in vitro was similar to that in MEFRAS7A- tumors. These findings suggest that the loss of ATP7A was deleterious specifically within the tumor microenvironment. It is well documented that cancer cells produce increased levels of ROS relative to normal cells [39-42], and that copper ions are known to potentiate the toxicity of ROS, in part, by catalyzing Fenton-like chemistry [43]. Our finding that cultured MEFRAS7A- cells were more sensitive to hypoxia and / or hydrogen peroxide relative to wild type cells offers a possible model by which for the tumoristatic effect of ATP7A deletion by augmenting the deleterious effects of ROS generated within the hypoxic environment of the tumor. This model does not exclude the possibility that loss of ATP7A may perturb other pathways important for tumor growth. Indeed, because ATP7A functions to transport copper to nascent polypeptides within the secretory pathway, it is possible that the tumoristatic effect of ATP7A may be the result of reduced cuproenzyme activity. Studies have shown that copper deficiency by either deletion of the CTR1 copper importer, or the use of copper chelators, prevents tumor growth by restricting the MEK1 protein, which has been shown to require copper as a cofactor for ERK phosphorylation [29, 33, 44]. We found no differences in ERK phosphorylation in MEFRAS7A- cells relative to wild type cells (Figure S4), suggesting that the excess copper accumulation in MEFRAS7A- cells does not increase MEK1 activity. Thus, it would appear from these data that tumor inhibition via perturbation of copper homeostasis may be achieved by restricting copper-dependent pathways for tumor growth or by generating an unfavorable environment for tumor growth by over-accumulating this potentially toxic metal. Previous studies have shown that silencing of ATP7A or ATP7B increases the sensitivity of cultured cells to cisplatin treatment, whereas forced expression of these proteins increases cisplatin resistance [16, 17, 20, 22] [45, 46]. Our results expand on these observations by demonstrating that ATP7A deletion can enhance the efficacy of cisplatin treatment in vivo. These findings point to the possibility that inhibitors of ATP7A/B might be used in combination with cisplatin to increase efficacy of cisplatin-mediated killing or reverse the resistance to cisplatin that is acquired by increased ATP7A/B expression [16, 21], [47]. In this context it is interesting to note that the proton-pump inhibitor, omeprazole, which was recently shown to block ATP7A-dependent melanogenesis in B16 melanoma cells [48], has been shown to increase the sensitivity of solid tumors to cisplatin chemotherapy in nude mice [49]. While omeprazole is known to affect numerous pathways in cancer cells including extracellular- and lysosomal pH [50], it is tempting to speculate that the potentiation of cisplatin sensitivity by omeprazole may occur via blocking ATP7A activity. The results and strategies described in our study open new avenues of research into the potential use of ATP7A inhibitors in controlling both tumor growth and cisplatin sensitivity.
Supplementary Material
Supplementary figures and methods.
Abbreviations
MEF: Mouse embryonic fibroblasts; CTR1: copper transporter 1; CCS: copper chaperone for superoxide dismutase 1; ROS: Reactive oxygen species.
Acknowledgements
We thank all members of our laboratory for their support and helpful comments. The pBabe H-RAS 12V was a gift from Channing Der (Addgene plasmid # 12545).
Funding
This work was fully supported by National Institutes of Health grants DK093386 and CA190265 to MJP, and DK079209 to JL.
Ethics approval and consent to participate
Research Involving Animals - All animal husbandry and euthanasia procedures were performed in accordance with, and under the approval of, the Animal Care and Use Committee (ACUC) of the University of Missouri. No human subjects, materials or data have been used in this study.
Availability of supporting data
All data generated or analysed during this study are included in this article and its supplementary information files.
Author contributions
SZ, VS and YW performed experiments. SZ, VS, and MJP wrote the manuscript. JL performed metal measurements and provided technical advice. All authors read and approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests
References
1. Prohaska JR. Role of copper transporters in copper homeostasis. Am J Clin Nutr. 2008;88:826S-9S
2. La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149-67
3. Mercer JF, Llanos RM. Molecular and cellular aspects of copper transport in developing mammals. J Nutr. 2003;133:1481S-4S
4. Masson W, Hughes H, Papworth D, Boyd Y, Horn N. Abnormalities of copper accumulation in cell lines established from nine different alleles of mottled are the same as those found in Menkes disease. J Med Genet. 1997;34:729-32
5. Qian Y, Tiffany-Castiglioni E, Welsh J, Harris ED. Copper efflux from murine microvascular cells requires expression of the menkes disease Cu-ATPase. J Nutr. 1998;128:1276-82
6. La Fontaine SL, Firth SD, Camakaris J, Englezou A, Theophilos MB, Petris MJ. et al. Correction of the copper transport defect of Menkes patient fibroblasts by expression of the Menkes and Wilson ATPases. J Biol Chem. 1998;273:31375-80
7. Petris MJ, Strausak D, Mercer JF. The Menkes copper transporter is required for the activation of tyrosinase. Hum Mol Genet. 2000;9:2845-51
8. Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002;99:14298-302
9. Chen HH, Kuo MT. Overcoming platinum drug resistance with copper-lowering agents. Anticancer Res. 2013;33:4157-61
10. Dmitriev OY. Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem Cell Biol. 2011;89:138-47
11. Gupta A, Lutsenko S. Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem. 2009;1:1125-42
12. Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010;77:887-94
13. Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495-535
14. Rybak LP, Mukherjea D, Jajoo S, Ramkumar V. Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J Exp Med. 2009;219:177-86
15. Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115-24
16. Samimi G, Safaei R, Katano K, Holzer AK, Rochdi M, Tomioka M. et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10:4661-9
17. Inoue Y, Matsumoto H, Yamada S, Kawai K, Suemizu H, Gika M. et al. Association of ATP7A expression and in vitro sensitivity to cisplatin in non-small cell lung cancer. Oncol Lett. 2010;1:837-40
18. Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M. et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559-65
19. Samimi G, Varki NM, Wilczynski S, Safaei R, Alberts DS, Howell SB. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin Cancer Res. 2003;9:5853-9
20. Li ZH, Qiu MZ, Zeng ZL, Luo HY, Wu WJ, Wang F. et al. Copper-transporting P-type adenosine triphosphatase (ATP7A) is associated with platinum-resistance in non-small cell lung cancer (NSCLC). J Transl Med. 2012;10:21
21. Nakayama K, Kanzaki A, Terada K, Mutoh M, Ogawa K, Sugiyama T. et al. Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin Cancer Res. 2004;10:2804-11
22. Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T. et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60:1312-6
23. Katano K, Safaei R, Samimi G, Holzer A, Tomioka M, Goodman M. et al. Confocal microscopic analysis of the interaction between cisplatin and the copper transporter ATP7B in human ovarian carcinoma cells. Clin Cancer Res. 2004;10:4578-88
24. Dolgova NV, Olson D, Lutsenko S, Dmitriev OY. The soluble metal-binding domain of the copper transporter ATP7B binds and detoxifies cisplatin. Biochem J. 2009;419:51-6 3 p following 6
25. Safaei R, Adams PL, Maktabi MH, Mathews RA, Howell SB. The CXXC motifs in the metal binding domains are required for ATP7B to mediate resistance to cisplatin. J Inorg Biochem. 2012;110:8-17
26. Linder MC, Moor JR, Wright K. Ceruloplasmin assays in diagnosis and treatment of human lung, breast, and gastrointestinal cancers. J Natl Cancer Inst. 1981;67:263-75
27. Pagliardi E, Giangrandi E. Clinical significance of the blood copper in Hodgkin's disease. Acta Haematol. 1960;24:201-12
28. Brem SS, Zagzag D, Tsanaclis AM, Gately S, Elkouby MP, Brien SE. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am J Pathol. 1990;137:1121-42
29. Yoshii J, Yoshiji H, Kuriyama S, Ikenaka Y, Noguchi R, Okuda H. et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int J Cancer. 2001;94:768-73
30. Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C. et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62:4854-9
31. Cox C, Teknos TN, Barrios M, Brewer GJ, Dick RD, Merajver SD. The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope. 2001;111:696-701
32. Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci U S A. 2013;110:19507-12
33. Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM. et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32:1284-95
34. Tsai CY, Finley JC, Ali SS, Patel HH, Howell SB. Copper influx transporter 1 is required for FGF, PDGF and EGF-induced MAPK signaling. Biochem Pharmacol. 2012;84:1007-13
35. Wang Y, Zhu S, Weisman GA, Gitlin JD, Petris MJ. Conditional knockout of the Menkes disease copper transporter demonstrates its critical role in embryogenesis. PLoS One. 2012;7:e43039
36. Caruano-Yzermans AL, Bartnikas TB, Gitlin JD. Mechanisms of the copper-dependent turnover of the copper chaperone for superoxide dismutase. J Biol Chem. 2006;281:13581-7
37. Cater MA, Pearson HB, Wolyniec K, Klaver P, Bilandzic M, Paterson BM. et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem Biol. 2013;8:1621-31
38. Denoyer D, Pearson HB, Clatworthy SA, Smith ZM, Francis PS, Llanos RM. et al. Copper as a target for prostate cancer therapeutics: copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget. 2016;7:37064-80
39. Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14
40. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97-110
41. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875-84
42. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175-6
43. Aust SD, Morehouse LA, Thomas CE. Role of metals in oxygen radical reactions. J Free Radic Biol Med. 1985;1:3-25
44. Goodman VL, Brewer GJ, Merajver SD. Copper deficiency as an anti-cancer strategy. Endocr Relat Cancer. 2004;11:255-63
45. Matsuzaki S, Enomoto T, Serada S, Yoshino K, Nagamori S, Morimoto A. et al. Annexin A4-conferred platinum resistance is mediated by the copper transporter ATP7A. Int J Cancer. 2013
46. Song L, Li Y, Li W, Wu S, Li Z. MiR-495 enhances the sensitivity of non-small cell lung cancer cells to platinum by modulation of copper-transporting P-type adenosine triphosphatase A (ATP7A). J Cell Biochem. 2013
47. Aida T, Takebayashi Y, Shimizu T, Okamura C, Higasimoto M, Kanzaki A. et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a prognostic factor in human endometrial carcinoma. Gynecol Oncol. 2005;97:41-5
48. Matsui MS, Petris MJ, Niki Y, Karaman-Jurukovska N, Muizzuddin N, Ichihashi M. et al. Omeprazole, a Gastric Proton Pump Inhibitor, Inhibits Melanogenesis by Blocking ATP7A Trafficking. J Invest Dermatol. 2015;135:834-41
49. Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A. et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96:1702-13
50. Fais S. Evidence-based support for the use of proton pump inhibitors in cancer therapy. J Transl Med. 2015;13:368
Author contact
![]() Corresponding author: E-mail: petrismedu
Corresponding author: E-mail: petrismedu