Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2384-2393. doi:10.7150/jca.19486 This issue Cite
Research Paper
Interferon-α Promotes the Expression of Cancer Stem Cell Markers in Oral Squamous Cell Carcinoma
Hailong Ma1#, Shufang Jin1,2#, Wenyi Yang1, Zhuowei Tian1, Shuli Liu1, Yang Wang1, Ge Zhou3, Mei Zhao3, Shalva Gvetadze4, Zhiyuan Zhang1, ![]() , Jingzhou Hu1,
, Jingzhou Hu1, ![]()
1. Department of Oral Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, Shanghai 200011, China
2. Department of Oral and Maxillofacial Surgery, The Second Affiliated Hospital Zhejiang University School of Medicine, Hangzhou 310009, China
3. Department of Head and Neck Surgery, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA
4. Central Research Institute of Dentistry and Maxillofacial Surgery, Congenital Maxillofacial Defects and Deformations, Timura Frunze 16, Moscow 119034, Russia
#Hailong Ma and Shufang Jin contributed equally to this study.
Received 2017-2-4; Accepted 2017-5-1; Published 2017-7-22
Abstract
Objectives: IFNα can stimulate an antitumor immune response and has a direct inhibition on cancer cells. This study is to test whether IFNα can activate dormant cancer stem cell (CSC) in oral squamous cell carcinoma (OSCC) to facilitate their elimination by chemotherapy.
Materials and methods: Nude mouse transplantation tumor model was established and administrated with IFNα and saline. The influence on CD44 and ALDH1A1 expression under IFNα treatment was detected by in vivo experiments. Flow cytometry, western blot, and immunofluorescence were used to detect the expression of CD44 and ALDH1A1 after INFa treatment in OSCC cell lines. Tumorsphere formation assay was conducted under incubation with IFNα for 2 weeks. Chromatin immunoprecipitation (ChIP) assays was used to examine the IFNα-induced transcriptional regulation of CD44 and ALDH1A1 expression. That IFNα-primed enhanced killing effect of chemotherapy was evaluated by MTT and western blot.
Results: IFNα transcriptionally activated the expression of CD44 and ALDH1A1 expression both in vivo and in vitro. IFNα-primed enhanced the cytotoxic inhibition effect of CDDP, erlotinib and nimotuzumab on OSCC cells.
Conclusion: These results suggest that IFNα could be administrated to patients prior to chemotherapeutic drugs, which will facilitate the killing of cancer stem cells in OSCC.
Keywords: Interferon alpha, CD44, ALDH1A1, Oral squamous cell carcinoma
Introduction
Type I interferons (IFNs) mainly comprise IFNα and IFNβ proteins. IFNα has a major role not only in antiviral immune responses but also in the natural and the therapy-induced immunological control of virus-unrelated malignancies [1]. IFNα displays direct cytotoxicity on tumor cells in cell culture and preclinical studies [2]. IFNα has been reported to affect tumor cell proliferation, tumor lymph/angiogenesis, and tumor metastasis [3]. IFNα induced by plasma dendritic cells (pDC) leads to tumor regression of melanoma, basal cell carcinoma, and T cell lymphoma [4]. Type I IFNs intervene all the phases of cancer immunoediting including elimination, equilibrium and escape [5]. IFNα can activate proliferation of DC and NK cells, enhance the cytotoxicity and survival of NK cells and CD4+ T cell, and promote tumor antigen expression of tumor cells [6-9]. Trials of IFNα therapies in solid malignancies such as melanoma, renal cell carcinoma and Kaposi sarcoma have achieved varied success [10]. Given its pleiotropic actions, IFNα is expected to not only stimulate an antitumor immune response, but also directly impact on cancer cell proliferation and survival, which together can achieve maximum therapeutic effect on cancer cells.
CSC has emerged as a critical factor for cancer chemoresistance, relapse and metastasis which often lead to treatment failure. CSC can undergo self-renewal and differentiation to promote the initiation and maintenance of tumor. CD44 and ALDH1A1 were considered as classical surface markers of CSC in head and neck squamous cell carcinoma (HNSCC) [11]. CD44 is a type I transmembrane glycoprotein that mediates cell-cell and cell-matrix interaction through affinity for hyaluronic acid. It is the one of the first markers to identify HNSCC CSCs [12]. OSCC CSCs can be also isolated on the basis of functional activities of ALDH1A1, which contributes to chemotherapy resistance [13]. Recently, more and more therapies were investigated to target the CSC population. However, CSCs usually lie dormant to avoid killing by chemotherapeutic drugs [14]. So how to promote the exit of CSCs out of the dormant stage is particularly important. Pre-treated/primed with IFNα can awaken dormant haematopoietic stem cells in vivo [15]. Whether IFNα can also activate dormant CSCs in OSCC to facilitate CSC's elimination by chemotherapy remains unclear.
In our current study we demonstrated that IFNα promoted the expression of CSC markers CD44 and ALDH1A1 in vitro and in vivo models of OSCC. Moreover, we showed that primed-IFNα can enhance the tumor-killing effect of chemotherapy. Our results strongly suggest that IFNα could be given to patients prior to chemotherapy in order to activate maximal therapeutic effects on CSC.
Materials and methods
Materials
Human recombinant IFNα was purchased from PeproTech company (Rocky Hill, NJ). A mouse monoclonal antibody against CD44 (CST) and a rabbit monoclonal antibody against ALDH1A1 (Abcam) were applied in this study. GAPDH and FITC-labeled secondary antibody were purchased from Proteintech (Rosemont, IL).
In vivo experiments
The nude mouse transplantation tumors were established with Cal27, a OSCC cell line, at 1×106 cells/point. IFNα was administrated 20 000 IU/day by subcutaneous injection after tumor formation, while the controls with normal saline (5 mice/group). Three weeks after administration, all the mice were sacrificed and the tumor tissues were preserved in liquid nitrogen and embedded in paraffin. Protein was extracted from the fresh tissue. Paraffin-cut sections were prepared for immunohistochemistry. The animal experiment was approved by Ethics Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine.
RNA isolation and qPCR analysis
Total RNA was extracted by TRIzol reagent (Invitrogen, Life Technologies) according to the manufacturer's instructions, and cDNA was generated using a Reverse Transcriptase Kit. Real-time quantitative PCR was performed in triplicate with Power PCR SYBR Green Master Mix (Takara Biotechnology, Dalian, China) using the ABI PRIAM Step-One Real-time PCR System (Applied Biosystems, Carlsbad, CA, USA) with results normalized to GAPDH expression. The relative expression was calculated using the2-ΔΔCT method. CD44 primers: Forward: 5'- GGACCTTCATCCCAGTGACC -3', Reverse: 5'- GCTCCACCTTCTTGACTCCC - 3'. ALDH1A1 primers: Forward: 5'- ATCAAAGAAGCTGCCGGGAA -3', Reverse: 5'- GCATTGTCCAAGTCGGCATC -3'. GAPDH primers: Forward: 5'- CCTCTGACTTCAACAGCGAC -3', Reverse: 5'- TCCTCTTGTGCTCTTGCTGG -3'.
Immunohistochemistry and immunofluorescence
Xenograft tumors of nude mice were formalin-fixed and paraffin-embedded. Sections were heated by water bath at 100 °C with citrate buffer solution (pH 6.0) for 20 min to retrieve antigen. CD44 at 1:50 dilution and ALDH1A1 at 1:400 dilution were incubated overnight at 4 ℃. Goat secondary antibody against mouse/rabbit immunoglobulin was incubated for 1 hour at room temperature. 3, 3`-diaminobenzidine (DAB) detection was developed under light microscope. Every step of the wash was used phosphate buffered saline solution (PBS) for 5 min three times. The immunoreaction staining score was calculated by multiplying the percentage of positive cells and the staining intensity. For immunofluorescence, FITC-labeled secondary antibodies against mouse and rabbit were incubated for 1 hour in the cassette. The percentage of positive cells was counted under fluorescence microscope. Image-pro plus 6.0 software was used to measure mean optical density for immunohistochemistry analysis.
Cell cultures
The OSCC cell lines HN4 and HN30 were kindly presented by the by the University of Maryland Dental School (USA). These cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin-streptomycin. All cells were cultured in a humidified atmosphere of 5% CO2 at 37 ℃.
Flow cytometry analysis of CSC markers
Flow cytometry was performed as described previously [16]. Briefly, HN4 and HN30 were treated with 0, 2, 20 ng/ml IFNα in 12 well-plate for 72 h. The cells were collected, washed and incubated with CD44 and ALDH1 antibodies at 1:100 dilution. After 1 hour incubation, cells were washed and incubated with FITC-labeled secondary antibody for 30 min. Then cells were washed twice and resuspended in 100 μl of FACS buffer. The stained cells were analyzed on BD FORTASA flow cytometer. The final results were operated using FlowJo software (Tree Star).
Western blot
Western blot assay was performed as briefly described [17]. Cells were lysed and quantified. Protein samples (30 μg/lane) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then electrophoretically transferred onto PVDF membranes using a wet transfer system (Bio-Rad, USA). The membranes were incubated overnight with the primary antibody of rabbit anti-ALDH1A1 monoclonal antibody (1:1 000) and mouse anti-CD44 monoclonal antibody (1:1 000). Antibodies against PARP, p-STAT1 (Tyr701), p-Akt (Ser473), p53, p-ERK1/2 (Thr202/Tyr204), and p-P38 (Thr180/Tyr182) were purchased from CST company and incubated at 1:1 000 dilution overnight. After incubating with fluorescent-based anti-rabbit/mouse IgG second antibody (Fermentas, Vilnius, Lithuania) at 1:10 000 dilution for 1 h in a dark place, immunoreactive bands were scanned and analyzed using Odyssey Infrared Imaging System (LICOR Biosciences, Lincoln, NE). GAPDH, β-actin, α-tubulin was used as internal control.
Tumorsphere formation
The sphere formation analysis was performed as described elsewhere [18]. Briefly, HN4 and HN30 cells were cultivated in the serum free RPMI-1640 medium containing 20 ng/ml of fibroblast growth factor, 20 ng/ml epithelial growth factor, and 1% B27, with 0, 2, 20 ng/ml of IFNα in low adhesion plate. After two weeks, all the spheres were filtered through a 70 μm-filter membrane. Only the spheres greater than 70 μm were counted. The sphere formation efficiency was calculated.
Chromatin Immunoprecipitation (ChIP)
ChIP was strictly operated according the protocol of SimpleChIP Enzymatic Chromatin IP kit (purchased from CST). After incubation with 20 ng/ml IFNα for 48 h, HN4 was fixed with 1% formaldehyde for 10 min and quenched with glycine for 5 min. Cells were washed twice in PBS and re-suspended in lysis buffer. Then the lysis was digested by micrococcal nuclease at 37°C for 20 min to length of approximately 150-900 bp. Digestion was terminated by the addition of 0.5 M EDTA. Chromatin was sonicated at 30% output for 6×10 sec. Clarify lysates by centrifugation at 10 000 rpm for 10 min at 4 °C. The supernatant was diluted with ChIP buffer (1:9). Add the immunoprecipitating antibodies into 500 μl of the diluted chromatin. For the positive control Histone H3 rabbit mAb, add 10 μl to the IP sample. For the negative control normal rabbit IgG, add 5 μl to the IP sample. 5 μl p-STAT1 (Try 701) (CST) was added into the sample. Incubate IP samples overnight at 4 °C with rotation. Add 30 μl of ChIP Grade Protein G Magnetic beads and incubate for 2 h at 4 °C with rotation. Wash the immunoprecipitated chromatin with three times low salt wash and once high salt wash. Eluted chromatin was reversed cross-links by adding 6 μl 5M NaCl and 2 μl proteinase K into every samples overnight at 65 °C. Reverse cross-linked DNA was purified by spin columns. Quantitative polymerase chain reaction analysis of purified ChIP DNA (ChIP-qPCR) was performed to calculate the enrichment percentage of promoter region using 2-ΔΔCT formula. The CD44 promoter region primers: Forward: 5'- ATCTAGGTGTTCTAGCTCCTGAATC -3', Reverse: 5'- CTACCATTCCTAGAGAAGGGAGTC -3'. The ALDH1A1 promoter region primers: Forward: 5'- CTTGGTCCCTAGGTTCTCACAA -3', Reverse: 5'- CACTTAAGCAGATCTTTTCTGCC -3'.
Cell viability assay
HN4 and HN30 was incubated with 0, 2 ng/ml IFNα for 48 h. After INFα primed, 3 000 cells per well were seeded in the 96-well plates. They were then treated with the indicated concentration of drugs in 10% FBS DMEM containing CDDP, erlotinib, nimotuzumab for 72 h. The total cell numbers were assessed after incubation with 20 μl of MTT for 4 h. The optical density (OD) of each well was measured using a microplate reader at 490 nm, and the OD values are presented as the means ± SD.
Statistical analysis
Statistical analysis was performed using the software of GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA, USA). All values are expressed as mean ± SD, and Student's t test was performed to assess the statistical significance of difference. P < 0.05 was considered statistically significant.
Results
IFNα induces the expression of CSC markers CD44 and ALDH1A1 in vivo
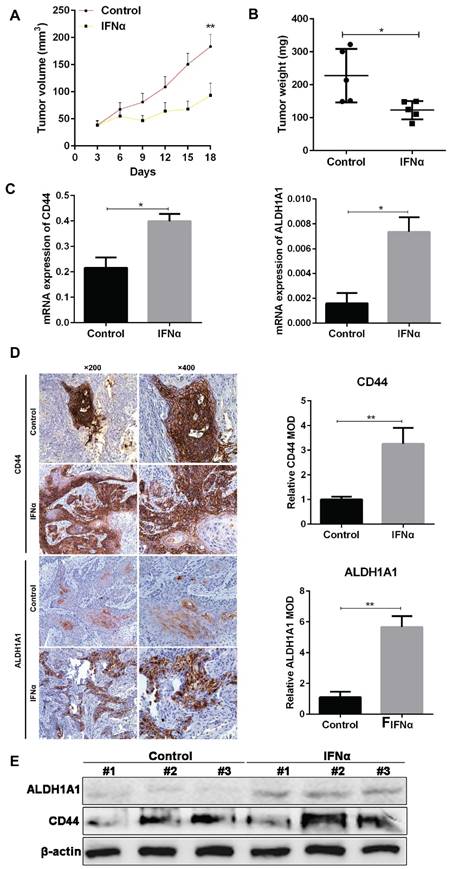
From in vivo experiment, we have confirmed the tumor inhibitory effect of IFNα in OSCC xenograft in nude mice. The tumor volume in IFNα group was much smaller than control (104.9 ± 22.1 vs 60.84 ± 7.88, P < 0.01. Figure 1A). The tumor weight in treatment group also decreased compared to control (227.5 ± 36.4 vs 122.9 ± 12.5, P = 0.02. Figure 1B). PCR assay demonstrated that mRNA expression of CD44 and ALDH1A1 was also significantly elevated in treatment group (Figure 1C). Immunohistochemistry (IHC) assay indicated that CD44 localizes on cell membrane, while ALDH1A1 is in the cytoplasm (Figure 1D). The relative mean optical density (MOD) of CD44 that represents the IHC score for CD44 expression was significantly higher in IFNα-treatment tumors than that in the control (5.667 ± 0.409 vs 1.100 ± 0.208, P < 0.01). Similarly, MOD of ALDH1A1 was also much higher in IFNα treatment group. Our western blot showed that CD44 and ALDH1A1 proteins was elevated in tumors of random three mice of the treatment group compared to the untreated control (Figure 1E).
IFNα induces the expression of CSC markers CD44 and ALDH1A1 in vivo. (A) The tumor volume was measured every three days with vernier caliper and calculated as a×b2/2 (a: length, b: width). (B) The tumor weight in each group was measured after administration. (C) Real-time PCR analyses of CD44 and ALDH1A1 mRNA in tumor samples from OSCC mouse xenograft with and without IFNα treatment. (D) IHC staining of CSC markers in OSCC xenograft tumor samples with and without IFNα treatment. Relative MOD of CD44 and ALDH1A1 in IHC staining was analyzed. (E) Western blot of tumor samples from OSCC mouse xenograft with and without IFNα treatment. Magnification: ×200, ×400.
IFNα upregulates the expression of CD44 and ALDH1A1 and promotes tumorsphere formation in vitro
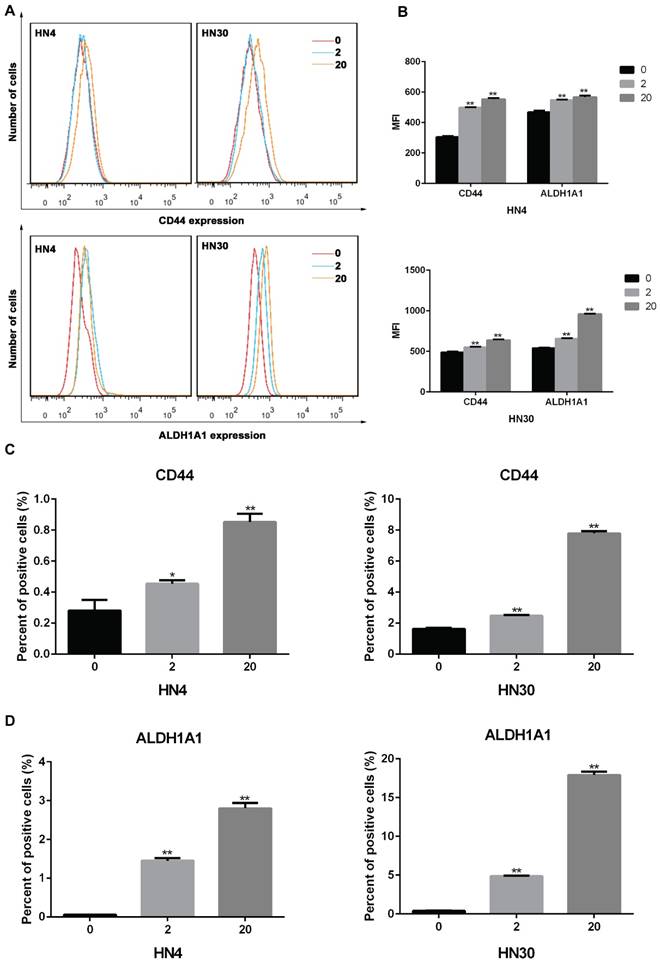
To investigate the impact of INFα on expression of markers in HNSCC cells, we treated HN4 and HN30 cells with IFNα and examined the expression CD44 and ALDH1A1 using flow cytometry. As shown in Figures 2A-B, after 72 h of treatment the mean fluorescence intensity (MFI) of CD44 and ALDH1A1 were increased in an INFα-dependent manner. Similarly, the percentage of CD44 and ALDH1A1 positive cells were significantly elevated after treatment (Figures 2C-D), suggesting that IFNα upregulated CD44 and ALDH1A1 expression in both HN4 and HN30 cells.
IFNα upregulates the expression of CD44 and ALDH1A1 in vitro. (A) Flow cytometry analyses of CD44 and ALDH1A1 expression in HN4 and HN30 cells in the absence (0) and presence of INFα treatment (2ng/ml; 20ng/ml) for 48h. (B) Mean fluorescence intensity (MFI) of CD44 and ALDH1A1 in the absence (0) and presence of INFα treatment (2ng/ml; 20ng/ml) for 48 h. (C-D) Percent of positive CD44 and (C) ALDH1A1 (D) cells in HN4 and HN30 before and after INFα treatment.
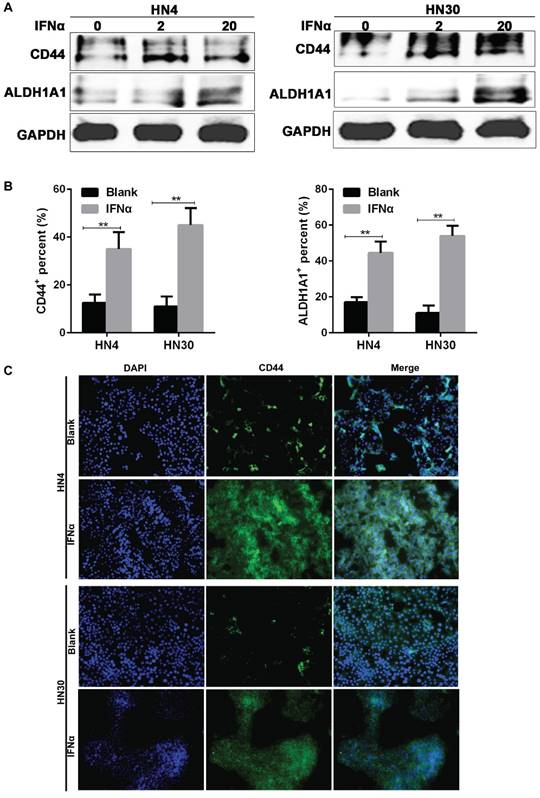
Consistent with the results from flow cytometry, western blot also indicated that the CD44 and ALDH1A1 expression were increased after IFNα incubation (Figure 3A), while immunofluorescence assay showed that CD44 and ALDH1A1 positive cells were enriched after 20 ng/ml IFNα treatment (Figure 3B). CD44 located on the cell membrane (Figure 3C), whereas ALDH1A1 located in the cytoplasm of HN4 and HN30 cells (Supplementary Figure 1). Together, our results demonstrated that IFNα upregulated CD44 and ALDH1A1 expression in both HN4 and HN30 cells in a dose-dependent manner.
IFNα upregulates the expression of CD44 and ALDH1A1 in OSCC cells. (A) Western blot of CD44 and ALDH1A1 in HN4 and HN30 cells before and after IFNα treatment (2ng/ml; 20ng/ml) for 48h. (B) Immunofluorescent (IF) assay of CSC markers in HN4 and HN30 cells before and after IFNα treatment. (C) Representative images of IF staining of CD44 in HN4 and HN30 in the presence and absence of IFNα treatment. Magnification: ×200.
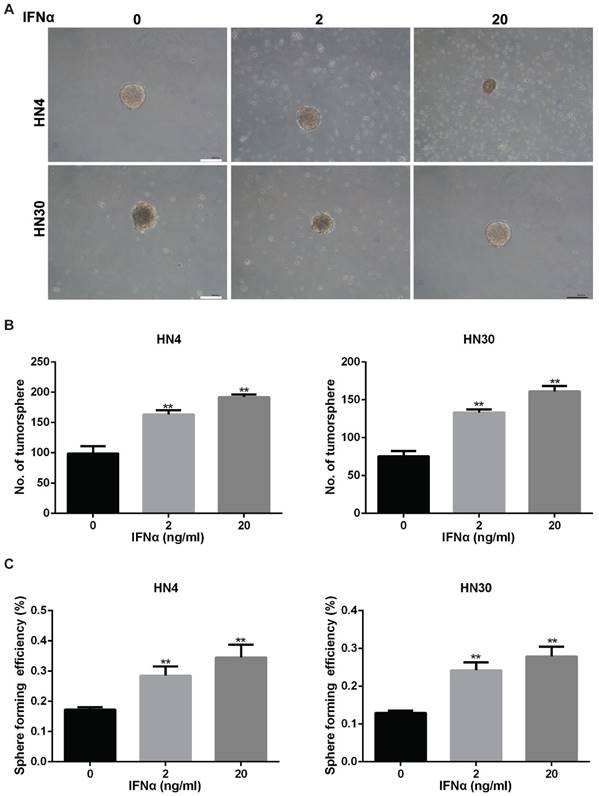
Sphere-forming capacity of tumor cells correlates directly with CSC phenotype and thus tumorigenic efficiency of the cancer cells can be determined based on the number of spheres that originate from specific number of seeded cells [19]. HN4 and HN30 were examined for their ability to form tumorspheres in vitro (Figure 4A). The number of tumorsphere increased after IFNα stimulation for two weeks (Figure 4B). Furthermore, sphere forming efficiency also increased in HN4 and HN30 with the treatment of IFNα (Figure 4C), suggesting that IFNα promoted tumorsphere formation of HN4 and HN30 cells.
IFNα enhances tumorsphere formation ability in HN4 and HN30 cells. (A) Representative images of tumorsphere formation in conditioned medium with indicated concentration of IFNα (2ng/ml; 20ng/ml) for 2 weeks. Scar bar: 1mm. (B) Quantitation of the number of tumorsphere formation in HN4 and HN30 cells in the presence and absence of IFNα incubation. (C) Sphere forming efficiency of HN4 and HN30 cells in the presence and absence of IFNα incubation.
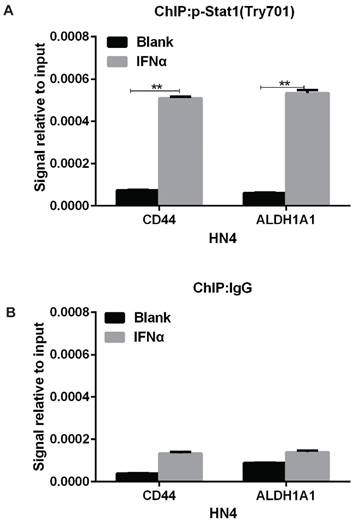
IFNα transcriptionally activates CD44 and ALDH1A1 expression through inducing the binding of phosphorylated (activated) signal transducer and activator of transcription 1 (STAT1) to CD44 and ALDH1A1 promoters
In response to IFNα stimulation, STAT1 forms heterodimers with STAT2 that bind the Interferon-Stimulated Response Element (IRSE) promoter element [20]. So we speculated that IFNα transcriptionally activated CD44 and ALDH1A1 expression through similar mechanism. In support of this, ChIP-PCR assay indicated that Tyr-701 phosphorylated STAT1 bound to the promoter region of CD44 and ALDH1A1 under IFNα stimulation in HN4 cells (Figure 5A), whereas no obvious binding was detected in normal IgG control (Figure 5B). This result strongly suggests that IFNα can induce phosphorylation (activation) of STAT1, leading its binding to the upstream promoters of CD44 and ALDH1A1 and transcriptionally activate the expression of CD44 and ALDH1A1 in HN4 cells.
IFNα transcriptionally activates CD44 and ALDH1A1 expression through inducing the binding of phosphorylated STAT1 to CD44 and ALDH1A1 promoters. (A) ChIP assay of p-STAT1 (pTry701) in promoter regions of CD44 and ALDH1A1 were under IFNα treatment for 48 h. (B) ChIP assay of control IgG in promoter regions of CD44 and ALDH1A1 were under IFNα treatment.
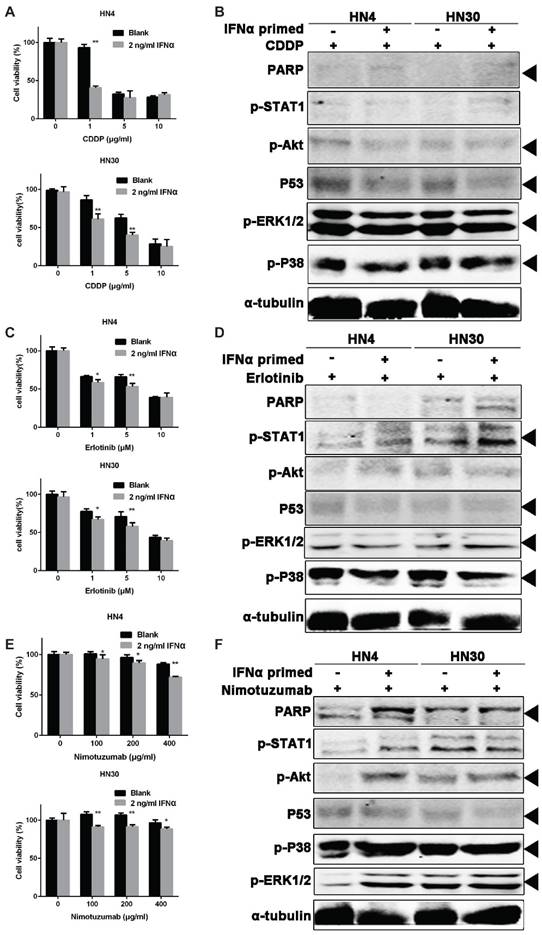
Primed-IFNα can enhance the tumor-killing effect of CDDP, Erlotinib and Nimotuzumab
From the above results, we confirmed the activation of CSC markers by IFNα in OSCC cells. We hypothesized that the tumor cells would be more sensitive to chemotherapy after activation of CSC. To test this, HN4 and HN30 were treated with 20 ng/ml IFNα for 48 hour, and then seeded in 96-well plates and further treated with CDDP (a classic cytotoxic drug for OSCC), Erlotinib and Nimotuzumab (epidermal growth factor receptor inhibitors), respectively. Nimotuzumab binds to the epidermal growth factor receptor (EGFR) and Erlotinib is a tyrosine kinase inhibitor of EGFR in HNSCC [21]. Our result showed that IFNα sensitized the cells to the tumor inhibitory effect of CDDP (Figure 6A), Erlotinib (Figure 6C), and Nimotuzumab (Figure 6E), respectively. Combination of primed-INFα treatment with 1 μg/ml CDDP, PARP, p-ERK and p-P38 were elevated while p-Akt and p53 decreased (Figure 6B). Combination of primed-INFα treatment with 5 μM Erlotinib, p53 decreased while p-STAT1, p-ERK and p-P38 increased (Figure 6D). In addition, PARP, p-Akt, p-P38 and p-ERK were increased and only p53 decreased under 200 μg/ml Nimotuzumab (Figure 6F). Taken together, our results demonstrated that OSCC cells primed with IFNα can be efficiently eliminated by treatment of CDDP, Erlotinib and Nimotuzumab in vitro.
IFNα pretreatment enhances the sensitivity of OSCC cells to chemotherapy. (A) Cell viability of OSCC cells pretreated with 2 ng/ml IFNα, followed by CDDP treatment at different concentration. (B) Western blot of OSCC cells pretreated with 2 ng/ml IFNα, followed by CDDP treatment (1μg/ml). (C) Cell viability of OSCC cells pretreated with 2 ng/ml IFNα, followed by erlotinib at different concenrtartion. (D) Western blot of OSCC cells pretreated with 2 ng/ml IFNα, followed by 5 μM erlotinib incubation. (E) Cell viability of OSCC cells pretreated with 2 ng/ml IFNα, followed by nimotuzumab treatment at different concentration. (F) Western blot of OSCC cells pretreated with 2 ng/ml IFNα, followed by 200 ng/ml nimotuzumab incubation for 48 h.
Discussion
In this study, we demonstrated that IFNα can upregulated CSC markers of oral cancer cells and primed-IFNα sensitized the therapeutic effect of CDDP, Erlotinib and Nimotuzumab. CSCs have stem features such as self-renewal, high migration capacity, drug resistance, high ability of tumorigenicity. In OSCC, CSCs have been increasingly shown to have an integral role in tumor initiation, disease progression, metastasis and treatment resistance [22]. So targeting for the eradication of CSCs was critical. In haematopoietic stem cells, three factors-granulocyte colony-stimulating factor, IFNα and arsenic trioxide - have been shown to efficiently activate dormant stem cells and thereby could break their resistance to anti-proliferative chemotherapy [14]. IFNα up-regulated the expression of the CSC markers CD24, CD44 and CD133 in in vitro and in vivo models of pancreatic ductal adenocarcinoma [23], which was consistent with our results. Our results suggested that IFNα promoted the expression of CSC markers through transcription activation in HN4 cells (Figure 5). However, we failed to detect IFNα-induced binding of STAT1 to the promoter of CSC markers in HN30 cells (data not shown). The difference may come from cellular heterogeneity, such as wild-type p53 expression in HN30 [24]. IFNα may promote the expression of ALDH1A1A in HN30 through other STAT members, which perhaps can bind the promoter region of ALDH1A1. Furthermore, IFNα may promote it through post-transcriptional regulation, posttranslational regulation and the modulation by interacting proteins. A number of studies have demonstrated that IFNα had a direct cytotoxic effect on various tumors and acted as a chemo-sensitizing agent [25, 26]. So IFNα can enrich CSCs in tumors while directly kill the bulk of the tumor cells. This distinctive feature of IFNα can provide a new method for CSC-targeted therapy in OSCC.
While CD44 and ALDH1A1 were used as biomarkers for CSC in OSCC in our study, BMI-1 is another stem cell-related biomarker involved in the carcinogenesis in HNSCC [12]. CD133 antigen, known as prominin-1 has been also identified as a CSC marker in several cancers and particularly in the laryngeal cancer [27]. However, the combined use of ALDH1 and CD44 is more relevant for identifying CSC-like populations as it is more selective than any other marker used alone [28]. ALDH expression was seen in only 1%-7.8% of HNSCC cells, and were able to form tumors with injections of as few as 500 cells into mice. While, HNSCC tumors shown a proportion of CD44+ cells ranging from 0.1% to 41.72% of the tumor population [27]. Combination of CD44 and ALDH1A1 can identify almost all the CSC population in HNSCC, which had strong capacity of tumorigensis. So two generally accepted biomarkers - CD44 and ALDH1A1, were adopted in our investigation.
Accumulating evidences have demonstrated that IFNα possessed chemo-sensitizing characteristics under different drugs. Radical surgery followed by IFNα and 5-FU combination chemotherapy offers possibility of long-term survival despite massive hepatocellular carcinoma with multiple intrahepatic metastases [29, 30]. IFNα pre-treatment followed by a combination of IFNα plus Erlotinib significantly enhanced the sensitivity of colon cancer cell lines [31]. IFNα enhanced the sensitivity to an anti-epidermal growth factor receptor monoclonal antibody (Nimotuzumab) in a human lung cancer cell line with intermediate expression of the receptor [32]. Low dose of IFNα intensified antineoplastic effect of a histone deacetylase -inhibitor, valproic acid, and the mammalian target of rapamycin inhibitor everolimus in prostate cancer cells [33]. It should be noted that in these studies IFNα was administrated together with chemoradiotherapy. Patients can benefit from unspecific activation of the immune system through IFNα to obtain better outcomes. While the future of enrichment of CSCs was implied with IFNα-primed in our study, only CDDP, Erlotinib and Nimotuzumab can be sensitized with pretreated with low dose IFNα in HN4 and HN30 (Figure 6). This synergistic effect should be further explored in in vitro study. Our results showed that p38 and ERK1/2 were activated after IFNα pretreatment, while p53 decreased in OSCC cells (Figure 6B, D, F). The activation of Akt depend the chemotherapies. p38 could be strongly activated by a wide variety of environmental and cellular stresses or by inflammatory cytokines [34]. Under certain conditions, ERK1/2 can also have pro-apoptotic functions which has lead this pathway to receive significant attention for development of cancer therapies [35]. Poly (ADP-ribose) polymerase (PARP) can be activated in cells experiencing stress and/or DNA damage. Our results showed that PARP was also elevated and activated under IFNα-primed and chemotherapy treatment. It should be noted that STAT1 that would be directly activated by IFNα was not significantly activated because of IFNα withdrawal under IFNα-primed and chemotherapy treatment. Taken together, IFNα could be given prior to chemotherapy to enrich the CSC and followed with concurrent with drugs to activate immune system. This hypothesis should be confirmed in future preclinical studies.
In summary, our studies indicate that IFNα can activate dormant CSCs in OSCC to facilitate the killing of CDDP, Erlotinib and Nimotuzumab. Our results provide experimental evidences and new treatment strategy for application of IFNα in OSCC.
Supplementary Material
Figure S1.
Acknowledgements
This study was supported by grants from Project of National Natural Science Foundation of China (Grant No. 31140007, 81472516), Natural Science Foundation of Shanghai (No. 14ZR1424200), Shanghai Leading Academic Discipline Project (No. S30206), and Doctoral Innovation Fund of Shanghai Jiao Tong University School of Medicine (BXJ201728).
Competing Interests
The authors declare no conflicts of interest.
References
1. Zitvogel L, Galluzzi L, Kepp O. et al. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405-14
2. Iacopino F, Ferrandina G, Scambia G. et al. Interferons inhibit EGF-stimulated cell growth and reduce EGF binding in human breast cancer cells. Anticancer Res. 1996;16:1919-24
3. Demoulin S, Herfs M, Delvenne P. et al. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: insight into the molecular mechanisms. J Leukoc Biol. 2013;93:343-52
4. Urosevic M, Dummer R, Conrad C. et al. Disease-independent skin recruitment and activation of plasmacytoid predendritic cells following imiquimod treatment. J Natl Cancer Inst. 2005;97:1143-53
5. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565-70
6. Greiner JW, Hand PH, Noguchi P. et al. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984;44:3208-14
7. Schiavoni G, Mattei F, Gabriele L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front Immunol. 2013;4:483
8. Brinkmann V, Geiger T, Alkan S. et al. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993;178:1655-63
9. Biron CA, Nguyen KB, Pien GC. et al. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189-220
10. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131-44
11. Dionne LK, Driver ER, Wang XJ. Head and Neck Cancer Stem Cells: From Identification to Tumor Immune Network. J Dent Res. 2015;94:1524-31
12. Prince ME, Sivanandan R, Kaczorowski A. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973-8
13. Wei Y, Wu S, Xu W. et al. Depleted aldehyde dehydrogenase 1A1 (ALDH1A1) reverses cisplatin resistance of human lung adenocarcinoma cell A549/DDP. Thorac Cancer. 2016
14. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. 2010;10:201-9
15. Essers MA, Offner S, Blanco-Bose WE. et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904-8
16. Fang S, Huang Y, Wang S. et al. IL-17A Exacerbates Fibrosis by Promoting the Proinflammatory and Profibrotic Function of Orbital Fibroblasts in TAO. J Clin Endocrinol Metab. 2016;101:2955-65
17. Ma HL, Jin SF, Tao WJ. et al. Overexpression of stathmin/oncoprotein 18 correlates with poorer prognosis and interacts with p53 in oral squamous cell carcinoma. J Craniomaxillofac Surg. 2016;44:1725-32
18. Yan M, Yang X, Wang L. et al. Plasma membrane proteomics of tumor spheres identify CD166 as a novel marker for cancer stem-like cells in head and neck squamous cell carcinoma. Mol Cell Proteomics. 2013;12:3271-84
19. Wu J, Mu Q, Thiviyanathan V. et al. Cancer stem cells are enriched in Fanconi anemia head and neck squamous cell carcinomas. Int J Oncol. 2014;45:2365-72
20. Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675-87
21. Zibelman M, Mehra R. Overview of Current Treatment Options and Investigational Targeted Therapies for Locally Advanced Squamous Cell Carcinoma of the Head and Neck. Am J Clin Oncol. 2016;39:396-406
22. Mery B, Guy JB, Espenel S. et al. Targeting head and neck tumoral stem cells: From biological aspects to therapeutic perspectives. World J Stem Cells. 2016;8:13-21
23. Zhu Y, Karakhanova S, Huang X. et al. Influence of interferon-alpha on the expression of the cancer stem cell markers in pancreatic carcinoma cells. Exp Cell Res. 2014;324:146-56
24. Song X, Xia R, Li J. et al. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin Cancer Res. 2014;20:701-10
25. Vitale G, van Eijck CH, van Koetsveld Ing PM. et al. Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann Surg. 2007;246:259-68
26. Kang Y, Perry RR. Effect of alpha-interferon on P-glycoprotein expression and function and on verapamil modulation of doxorubicin resistance. Cancer Res. 1994;54:2952-8
27. Wei XD, Zhou L, Cheng L. et al. In vivo investigation of CD133 as a putative marker of cancer stem cells in Hep-2 cell line. Head Neck. 2009;31:94-101
28. Krishnamurthy S, Dong Z, Vodopyanov D. et al. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 2010;70:9969-78
29. Tanaka K, Yabushita Y, Nakagawa K. et al. Debulking surgery followed by intraarterial 5-fluorouracil chemotherapy plus subcutaneous interferon alfa for massive hepatocellular carcinoma with multiple intrahepatic metastases: a pilot study. Eur J Surg Oncol. 2013;39:1364-70
30. Yamashita T, Arai K, Sunagozaka H. et al. Randomized, phase II study comparing interferon combined 5-fluorouracil plus cisplatin hepatic arterial infusion with interferon combined 5-fluorouracil hepatic arterial infusion in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2009;27:4588
31. Yang JL, Qu XJ, Russell PJ. et al. Interferon-alpha promotes the anti-proliferative effect of Erlotinib (OSI-774) on human colon cancer cell lines. Cancer Lett. 2005;225:61-74
32. Diaz A, Batista AE, Montero E. Interferon-alpha conditioned sensitivity to an anti-epidermal growth factor receptor monoclonal antibody in a human lung cancer cell line with intermediate expression of the receptor. J Interferon Cytokine Res. 2009;29:433-40
33. Tsaur I, Hudak L, Makarevic J. et al. Intensified antineoplastic effect by combining an HDAC-inhibitor, an mTOR-inhibitor and low dosed interferon alpha in prostate cancer cells. J Cell Mol Med. 2015;19:1795-804
34. Zou X, Blank M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 2017;384:19-26
35. Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105-43
Author contact
![]() Corresponding authors: Jingzhou Hu, Department of Oral Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, No 639, Zhizaoju Rd, Shanghai 200011, China. Tel: +86 021 23271699 5160; Fax: +86 021 63136856; E-mail: huyayiedu.cn and Zhiyuan Zhang, Department of Oral Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, No 639, Zhizaoju Rd, Shanghai 200011, China. Tel: +86 021 23271699 5160; Fax: +86 021 63136856; E-mail: zhzhy0502com
Corresponding authors: Jingzhou Hu, Department of Oral Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, No 639, Zhizaoju Rd, Shanghai 200011, China. Tel: +86 021 23271699 5160; Fax: +86 021 63136856; E-mail: huyayiedu.cn and Zhiyuan Zhang, Department of Oral Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, No 639, Zhizaoju Rd, Shanghai 200011, China. Tel: +86 021 23271699 5160; Fax: +86 021 63136856; E-mail: zhzhy0502com