Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2456-2470. doi:10.7150/jca.19383 This issue Cite
Research Paper
Sulforaphane Induced Apoptosis via Promotion of Mitochondrial Fusion and ERK1/2-Mediated 26S Proteasome Degradation of Novel Pro-survival Bim and Upregulation of Bax in Human Non-Small Cell Lung Cancer Cells
Yang Geng1, Yan Zhou1, Sai Wu1, Yabin Hu1, Kai Lin1, Yalin Wang1, Zhongnan Zheng1, Wei Wu1, 2 ![]()
1. Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing, China;
2. Institute of Brain Tumor, Beijing Institute for Brain Disorders, Capital Medical University, Beijing, China.
Received 2017-1-28; Accepted 2017-5-29; Published 2017-8-2
Abstract
Previous studies in our laboratory showed that sulforaphane (SFN) induced apoptosis by sustained activation of extracellular regulated protein kinases 1/2 (ERK1/2). However, the underlying mechanisms associated with SFN-induced apoptosis and downstream cascades which are modulated by ERK1/2 were not elucidated. Herein we demonstrated for the first time that alteration of mitochondrial dynamics contributed to SFN-induced apoptosis in human non-small cell lung cancer (NSCLC) cells. Reports showed that protein Bim not only induced apoptosis but also promoted proliferation under certain circumstances. We found that Bim was related to cell growth in NSCLC cells. Pro-survival Bim downregulation was shown to induce apoptosis in response to SFN. Further, Using the ERK1/2 inhibitor, PD98059, we found that SFN upregulated Bax and downregulated Bim through the ERK1/2-dependent signaling pathway. Furthermore, SFN activated ERK1/2 to increase 26S proteasome activity to degrade Bim, while the proteasome inhibitor MG132 reversed this effect. Therefore, SFN phosphorylated ERK1/2 and activated the proteasome system leading to the degradation of Bim, which contributed to apoptosis in NSCLC cells. These findings provided a novel insight into SFN-related therapeutics in cancer treatment.
Keywords: Sulforaphane, Bim, ERK1/2, Mitochondrial Fusion, Apoptosis, Non-Small Cell Lung Cancer.
Introduction
There were approximately 221,200 new cases and 158,040 deaths due to lung cancer globally in 2015; nearly 80% of these cases belong to non-small cell lung cancer (NSCLC) [1]. Surgery, chemotherapy, and radiotherapy are the currently used ways to treat NSCLC [2]. However, radiotherapy or chemotherapy often causes side effects on normal tissues. Novel phytochemicals have been introduced to improve traditional chemotherapy and surgery [3]. Among phytochemicals, sulforaphane (SFN) is a low-toxicity isothiocyanate of organosulfur compounds [4-6].
Sulforaphane (1-isothiocyanato-[4R]-[methylsulfinyl]-butane) is generally found in broccoli sprouts and in most cruciferous vegetables [7] and thought to be effective in cancer treatment [8-13]. It has been found that SFN modulates the Bax/Bcl-2 ratio, enhances proteasomal activity, and activates mitogen activated protein kinase (MAPK) [14]. Our previous data have shown that sustained activation of ERK1/2 with SFN treatment of tumor cells for 24h inhibited invasion in both human glioblastoma and prostate cancer cells [15, 16]. However, whether SFN induces apoptosis through persistent activation of ERK1/2 in NSCLC cells and what are the underlying mechanisms are unclear.
The mitochondrial-dependent apoptotic pathway is the primary signal pathway by which SFN activates ERK1/2 to induce apoptosis and inhibit invasion [16-18]. Many studies have reported that SFN induces changes in mitochondrial morphology, leading to a release of cytochrome c and apoptosis inducing factor (AIF) [19]. Mitochondria are dynamic organelles that are regulated by fusion and fission events that occur constantly to maintain mitochondrial morphology [20, 21]. Recently, it has been reported that upregulated mitochondrial fission is a prerequisite for promoting hepatocellular carcinoma (HCC) cell proliferation [22]. Despite the fact that the excessive mitochondrial fission was associated with proliferation, whether mitochondrial fusion was an opposite process of fission leading to cancer cell deaths has not been identified.
Bcl-2 family members are the regulatory factors that are associated with the mitochondrial membrane and regulate apoptotic signals. Recently, many studies have shown that multiple proapoptotic proteins, such as Bax/Bak, Bid, Bad, Noxa, and Puma, possess non-apoptotic/pro-survival functions [23]. It has been reported that Bim, a BH3-only protein, was upregulated in prostate cancer and breast cancer cells [23]. Therefore, it is important to explore whether Bim downregulation was associated with apoptosis in NSCLC cells.
The ERK1/2 signaling pathway is the major pathway known so far to be involved in Bim ubiquitination and consequent proteasomal degradation [24, 25]. The ubiquitin-proteasome system (UPS) was activated to degrade target proteins into small peptides in eukaryotic cells [26, 27]. The 26S proteasome is a cylindrical particle that consists of two distinct sub-complexes, a 20S core particle (CP) and a 19S regulatory particle (RP) [26]. The 26S proteasome is a target for many post-translational modifications (PTMs), including phosphorylation, N-acetylation, ubiquitination, SUMOylation, and O-glycosylation [26, 27]. Therefore, we will investigate the novel apoptotic mechanism by which ERK1/2, activated by SFN, mediates proteasomal activity, causing pro-survival Bim degradation and apoptosis in NSCLC cells.
In the present study, we aimed to find the signaling pathway which SFN induced apoptosis in NSCLC cells. These findings will give some new data to enable targeting of the research on the use of SFN in clinical trials.
Materials and Methods
Cell Culture
SK-1 cells and A549 cells from Cell Resource Center, Peking Union Medical College (CRC/PUMC) were cultured in DMEM/F-12 medium (Lifetechnologies, Shanghai, China) supplemented with 10% fetal bovine serum(Invitrogen), and 100 U/ml penicillin/streptomycin (Solarbio, Beijing, China) at 37℃in 5% CO2, maintaining cells logarithmic growth. Cells were washed with PBS three times and incubated in DMEM/F-12 medium supplemented with 10% FBS and 100U/ml penicillin/streptomycin and SFN (Sigma) for 24h. MG132 (Sigma) and PD98059 (Cell Signaling Technologies) had been added before SFN was added into F-12 medium according to PD98059 product specification.
Cell Proliferation Assay
SK-1 and A549 cells (4-6×103) were plated in 96-well plates. When cells grew up to 80% density, SFN at the different concentrations at time gradients was added to 96-well plates for 24h. MTS (Promega, Madison, USA) working solution (20μl) was injected to each of the pool and the 96-well plates were incubated at 37℃ for 2h. The OD values were detected at 490 nm wavelength via a BioTekH microplate reader (Winooski, VT, USA).
Electron Microscopy Observation
NSCLC cells were prepared for EM observation. Two cell lines were classified into control groups (0μM) and SFN (15μM) groups. Cell pellets were collected by centrifugation for 3 min. Then cell pellet was fixed with cold glutaraldehyde (2%) for 2h, the fixed cells were postfixed with 1% osmic acid and further the cell pellet was embedded in epoxy resin and sectioned for EM observation.
Morphological Observation
According to a series of concentrations and times, SK-1 and A549 cells were treated by SFN and incubated in 6-well plates. Cell morphological modification was observed with a phase-contrast microscope (Leica, Germany). Then digital camera (Olympus, Japan) was used to take photograph at different time and concentration points.
Apoptosis Detection
Apoptosis was determined by flow cytometry (GenStar, Beijing, China) analysis. Following cell cultivation, cells were treated with SFN at dose-dependent and time-dependent manners. Adhesive cells were collected and washed twice in cold PBS and binding buffer. Solution was centrifuged at 1000 g for 7 min and stained with FITC reagent for 30min as well as PI reagent for 5min. Cells were analyzed by the flow cytometer (BD Biosciences, Rutherford, NJ).
Immunofluorescence Assay
The NSCLC cells (4×104) were cultivated in glass coverslips for 12h at 37℃. 15μM SFN was added into Petri dishes for 24h. Then cells were fixed with 4% paraformaldehyde for 15min and permeabilized with 0.2% Tween 20 for 10min. Cells were treated with DPBS (containing 3% FBS and 0.5% Tween 20) for 1h. The cells were incubated by primary antibody (anti-Bim, 1:100) (Abcam) for 12h at 4℃ and fluorescence-labeled secondary antibody for 1h. Finally, the glass coverslips were stained with DAPI and detected on confocal laser-scanning microscope (Olympus FV1000, Tokyo, Japan). To observe the mitochondrial fusion, the cells were treated with or without SFN at 15μM and 0μM for 24h, then were stained with Mito-Tracker Green (Beyotime, Shanghai, China) for 45min and observed under confocal laser-scanning microscope.
Western blot
Cell lysates were prepared with RIPA lysis buffer (Thermo Fisher Scientific). and protein concentrations were determined by BCA protein assay kit (Invitrogen). Protein was loaded and run through 12% or 15% SDS-PAGE gels and transferred to nitrocellulose membranes. The blots were probed with Caspase 3 (Santa Cruz, 1:500), 26S Proteasome S5 (Abcam, 1:500) and Bim (Abcam, 1:500), ERK1/2 and phosphorylated ERK1/2 (Cell Signaling Technologies, 1:1000), Bax (Sangon Biotech, Shanghai, China, 1:400) antibodies, and fluorescence‑labeled secondary antibody (LI-COR Biosciences, Lincoln, NE, USA) and protein bands were detected by Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Statistical Analysis
The results were expressed as the mean ± SD, and analyzed by SPSS software. The differences were considered statistically significant at p<0.05.
Results
Determination of the optimal SFN concentration to induce apoptosis
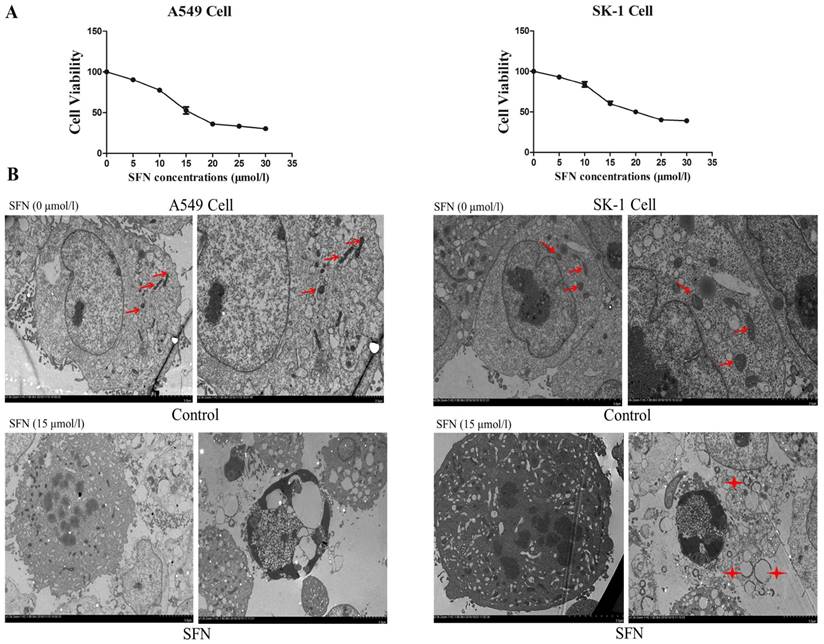
We previously found in vitro experiments that the optimal time at which SFN inhibited cell growth was 24 h. In order to determine the optimal SFN concentration that induced cell death in NSCLC cells, we selected different SFN concentrations (0, 5, 10, 15, 20, 25, and 30 μM) to treat SK-1 and A549 cells for 24 h, and then performed the MTS assay. As shown in Figure 1A, the IC50 values, calculated by statistical analysis, of SK-1 cells and A549 cells were 15.29μM and 14.45μM. We found that 15μM SFN reduced cell viability efficiently in NSCLC cells (Figure 1A). Further work demonstrated that SFN induced cell apoptosis. Electron microscopy was used to detect apoptosis, characterized by cytoplasmic shrinkage, nuclear fragmentation and apoptotic bodies in the 15μM SFN group (Figure 1B). Therefore, 15μM SFN caused apoptosis in NSCLC cells. Surprisingly, we found many circular and flattened mitochondria around the apoptotic bodies in SK-1 cell. We considered that morphological changes of mitochondria might be associated with apoptosis.
Determination of the optimal SFN concentration to induce apoptosis. A. SFN inhibited cell growth. The SK-1 and A549 cells were treated with increasing concentrations of SFN for 24h. Cell viability was determined by the MTS assay. Data are presented as the percentage of control. The results represent three independent experiments. B. Electron microscopy was used to observe the cell membrane and nuclei of normal cells in the control group. In the SFN group, it showed chromatin accumulation, nuclear fragments, and apoptotic bodies. Red arrows point to normal mitochondria; Asterisks indicated circle mitochondria.
SFN-induced apoptosis was associated with mitochondrial fusion
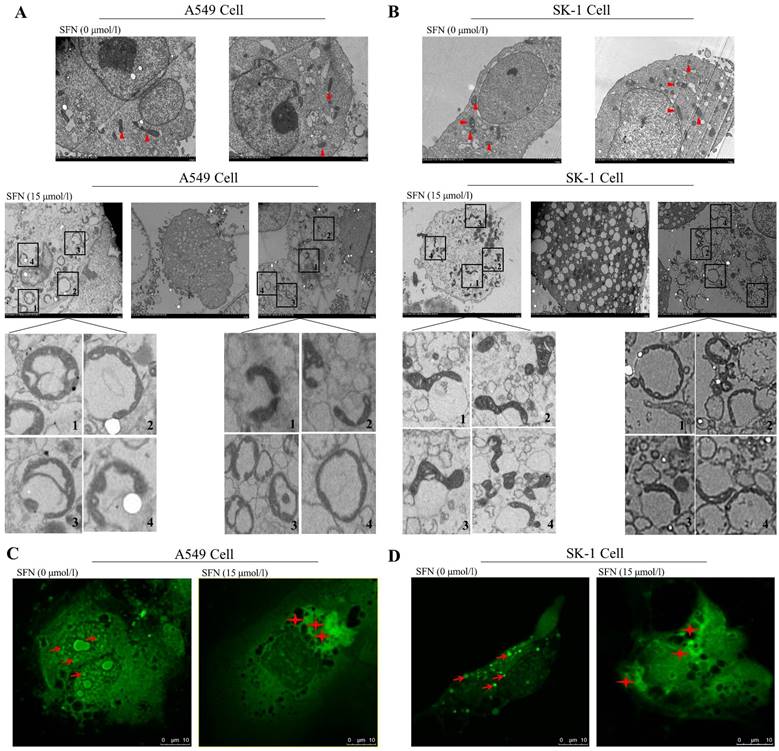
Mitochondrial behaviors mainly affect cell apoptosis by changing mitochondrial morphology, such as fission and fusion. We therefore observed alteration of mitochondrial ultrastructure with electron microscopy with or without SFN treatment in NSCLC cells. We found that granular mitochondria gathered and formed a circular structure in the SFN-treated cells. Each circle was composed of many small mitochondria. The cell nuclei lost their original conformation or were broken into pieces. The mitochondria forming the circles were more flattened and smaller when compared with the control cells. Irregularly shaped mitochondria gathered together. Eventually, the circular fused mitochondria became vacuolated, inducing cell apoptosis (Figure 2A, 2B). Confocal microscopy indicated that mitochondrial particles were spread throughout the cytoplasm in the control cells. However, mitochondrial particles were clustered and fused around cells that had lost their original structure in the SFN-treated cells (Figure 2C, 2D).
SFN-induced apoptosis was associated with mitochondrial fusion. A-B. SK-1 and A549 cells were treated with 15μM SFN for 24h; the control-untreated cells were also incubated for 24h. The mitochondrial morphology was observed under electron microscopy. Triangles represent normal mitochondria; Rectangles point to mitochondrial fusion (The mitochondrial fusion marked with 1, 2, 3, 4 in the A, B diagrams were further magnified). C-D. Mitochondrial morphologic changes were shown via confocal laser scanning microscopy after staining with Mito-Tracker Green in SK-1 cells and A549 cells. Red arrows point to mitochondria; Asterisks indicate mitochondrial clusters.
SFN induced apoptosis in a dose-dependent manner
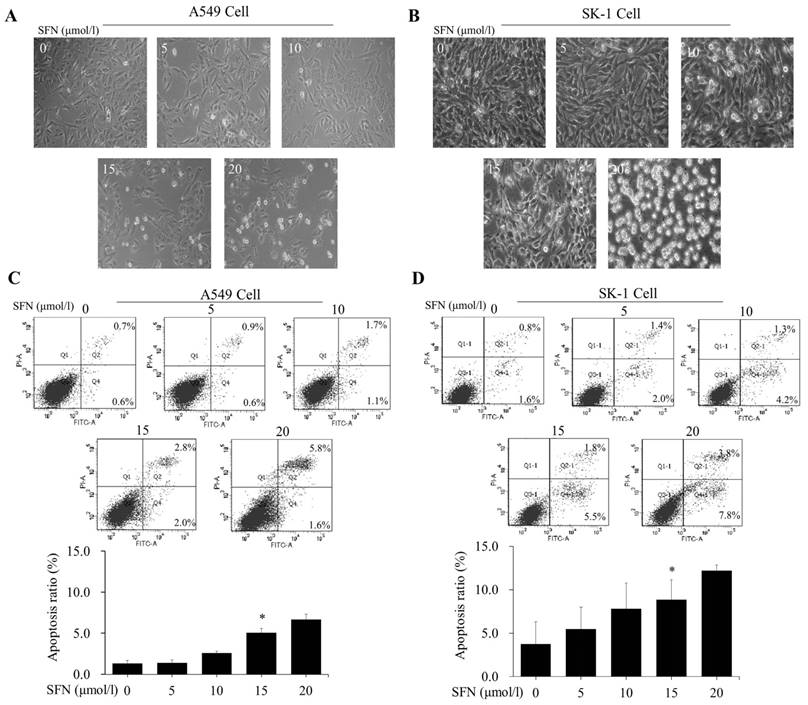
After NSCLC cells had been treated with SFN (0, 5, 10, 15, and 20μM) for 24 h, morphological observation and flow cytometry were performed. The apoptosis rates were elevated versus control groups (0μM) in a dose-dependent manner (Figure 3A, 3B, 3C, 3D). Although 20μM SFN also induced higher apoptosis rates than 15μM, the morphological structures in the 20μM SFN-treated samples were completely destroyed (Figure 3A, 3B). Thus, the optimal concentration of 15μM was established for SFN-induced apoptosis.
SFN induced apoptosis in a dose-dependent manner. A-B. The SK-1 and A549 cells treated with 0, 5, 10, 15, or 20μM SFN for 24 h exhibited morphological alterations. Cell morphology was observed via a Leica DMIRB Microscope at×100 magnification. C-D. Apoptosis ratios were determined by Annexin V-FITC/PI staining. The percentage of apoptotic cells was detected by flow cytometry. The diagram (ratio [mean± SD]) under each flow cytometry image showed the apoptosis percentage. p<0.05 versus 0 μM, n=3.
SFN induced apoptosis in a time-dependent manner
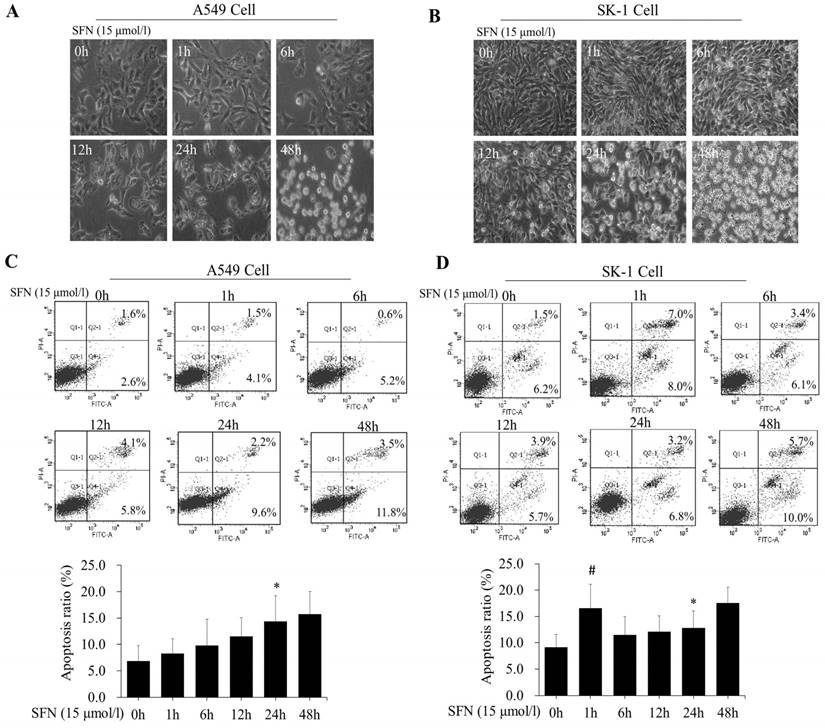
We further determined that the degree of apoptosis varied by treatment times. At different time points (0, 1, 6, 12, 24, and 48h), morphological observation and flow cytometry were used to detect apoptosis rates, which increased gradually in both cell lines (Figure 4A, 4B, 4C, 4D). Apoptosis rate was reached a peak at 48 h. Apoptosis rates in A549 cells were elevated, but those in SK-1 cells were clearly higher at 1 h and 48 h, respectively. The morphological structures at 48 h were completely altered, which was unfavorable for apoptosis signal research (Figure 4A, 4B). Therefore, the optimal time point was found to be 24 h for SFN-induced apoptotic analysis.
SFN induced apoptosis in a time-dependent manner. A-B. SK-1cells and A549 cells were treated with 15μM SFN for 0, 1, 6, 12, 24, or 48h, and cell morphology was observed using a Leica DMIRB Microscope at×100 magnification. C-D. The apoptosis ratios were determined by Annexin V-FITC/PI staining in SK-1 and A549 cells. The percentage of apoptotic cells was detected by flow cytometry. The diagram (ratio [mean± SD]) under each flow cytometry image showed the apoptosis percentage. * p<0.05 versus 0 h, # p<0.05 versus 24 h, n=3.
SFN induced apoptosis by upregulating apoptosis-related proteins and downregulating pro-survival Bim in a dose-dependent manner
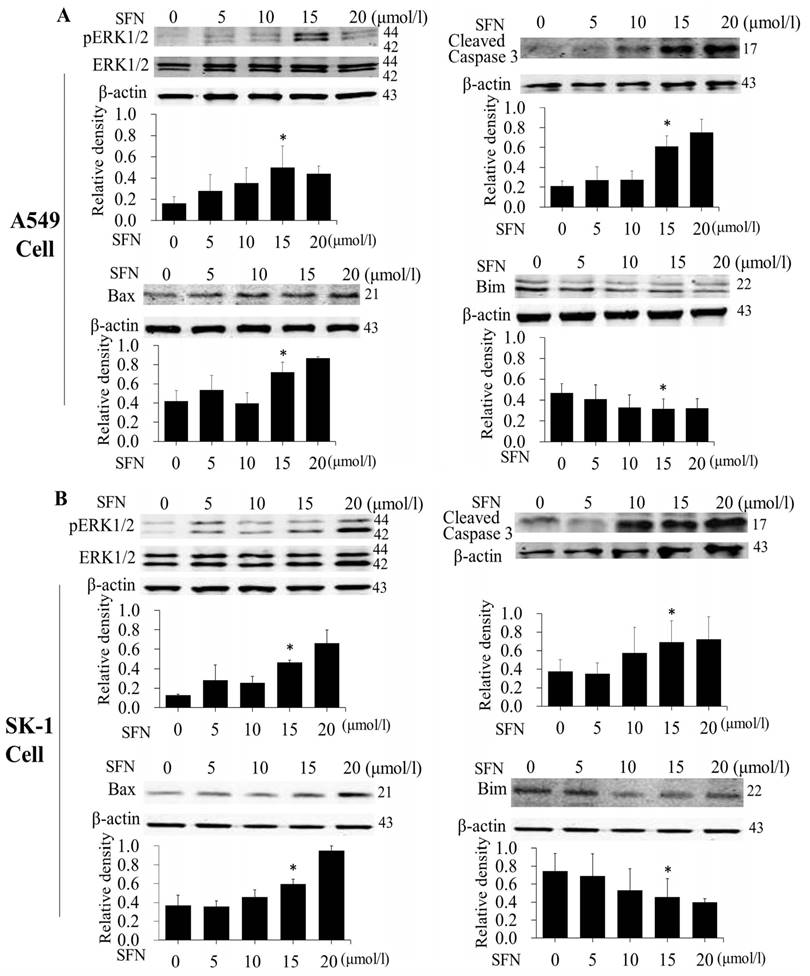
After NSCLC cells had been treated with SFN (0, 5, 10, 15, and 20μM) for 24 h, and Western blot showed that SFN upregulated phosphorylated ERK1/2, Bax and cleaved Caspase 3 (Figure 5A, 5B). Similar to the results of flow cytometry, these data also followed a dose-dependent manner. In contrast to upregulated phosphorylated ERK1/2, Bax, and cleave Caspase 3 proteins, SFN downregulated Bim leading to apoptosis in NSCLC cells.
SFN induced apoptosis by upregulating apoptosis-related proteins and downregulating pro-survival Bim in a dose-dependent manner. A-B. The level of protein expression in NSCLC cells, when the cells were treated in a dose-dependent manner. Western blot was used to measure phosphorylated ERK1/2, ERK1/2, Bim, Bax and cleaved Caspase3 protein expression. SFN (0μM) was used as the negative control. Phospho-ERK1/2 was normalized to total proteins, and β-actin was used as a loading control. Densitometric analysis of the bands was done via ImageJ analysis software. The quantification of band intensity was presented as a mean ± SD after normalization to β-actin or total proteins. The diagram showed these protein density ratios at different drug doses. Each bar reflects the average protein density ratio; * p <0.05, n=3.
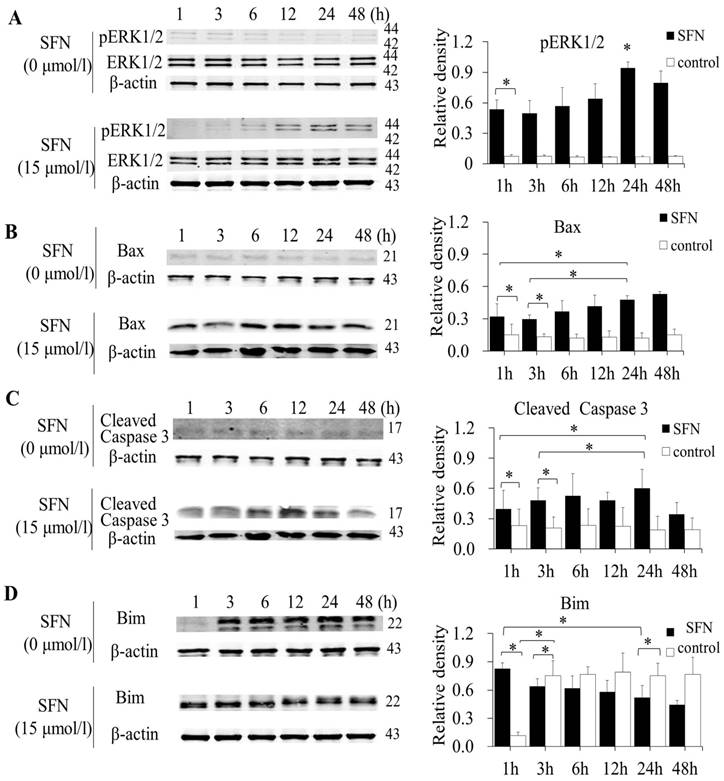
SFN induced apoptosis by upregulating apoptosis-related proteins and downregulating pro-survival Bim in a time-dependent manner
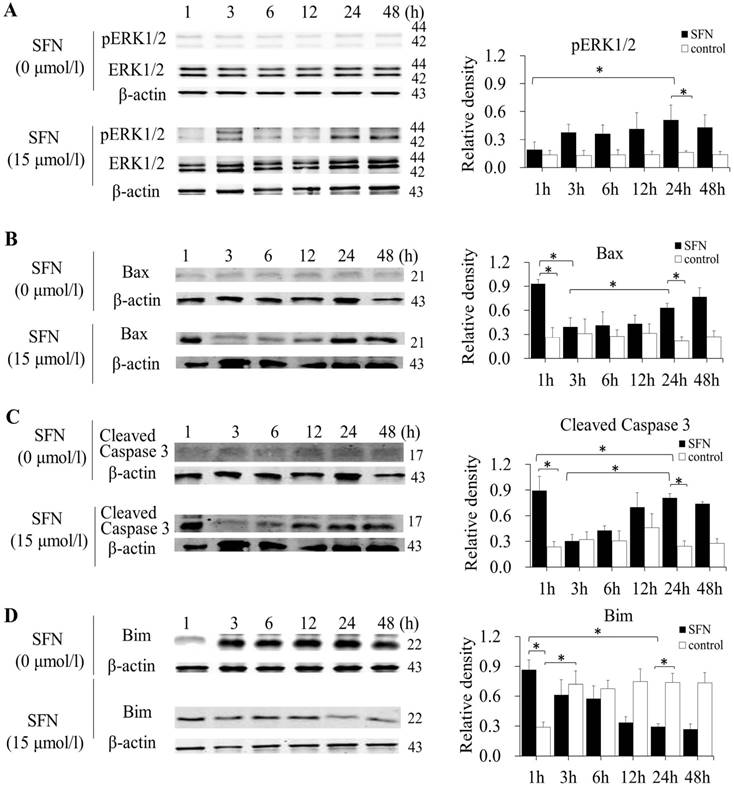
SK-1 and A549 cells were treated with 15μM or 0μM SFN for 1, 3, 6, 12, 24, or 48h, and Western blot indicated that SFN upregulated phosphorylated ERK1/2 (Figure 6A, Figure 7A), Bax (Figure 6B, Figure 7B) and cleaved Caspase 3 (Figure 6C, Figure 7C). In the control cells, Bim (Figure 6D, Figure 7D) was expressed extensively when cells were cultured for more than 1 h, and ERK1/2, Bax, and cleaved Caspase 3 were not upregulated. After treatment with SFN, Bim was decreased significantly, but phosphorylated ERK1/2, Bax and cleaved Caspase 3 were increased significantly as the treatment time increased.
SFN induced apoptosis by upregulating apoptosis-related proteins and downregulating pro-survival Bim by a time-dependent manner in A549 cells. A-D. Cells were treated with 15μM or 0μM SFN for 1, 3, 6, 12, 24, or 48 h, then phosphorylated ERK1/2, Bax, cleaved Caspase 3 and Bim protein level were measured by Western blot. SFN (0 μM) was used as the negative control. Phospho-ERK1/2 was normalized to total proteins, and β-actin was used as a loading control. Densitometric analysis of the bands was done using ImageJ analysis software. The quantification of band intensity was presented as a mean ± SD after normalization to actin or total proteins. The diagram showed these protein density ratios at different time points. Each bar reflects the average protein density ratio; * p <0.05, n=3.
SFN induced apoptosis by upregulating apoptosis-related proteins and downregulating pro-survival Bim by a time-dependent manner in SK-1 cells. A-D. Cells were treated with 15μM or 0μM SFN for 1, 3, 6, 12, 24, or 48 h, then phosphorylated ERK1/2, Bax, cleaved Caspase 3 and Bim protein level were measured by Western blot. SFN (0 μM) was used as the negative control. Phospho-ERK1/2 was normalized to total proteins, and β-actin was used as a loading control. Densitometric analysis of the bands was done using ImageJ analysis software. The quantification of band intensity was presented as a mean ± SD after normalization to actin or total proteins. The diagram showed these protein density ratios at different time points. Each bar reflects the average protein density ratio; * p <0.05, n=3.
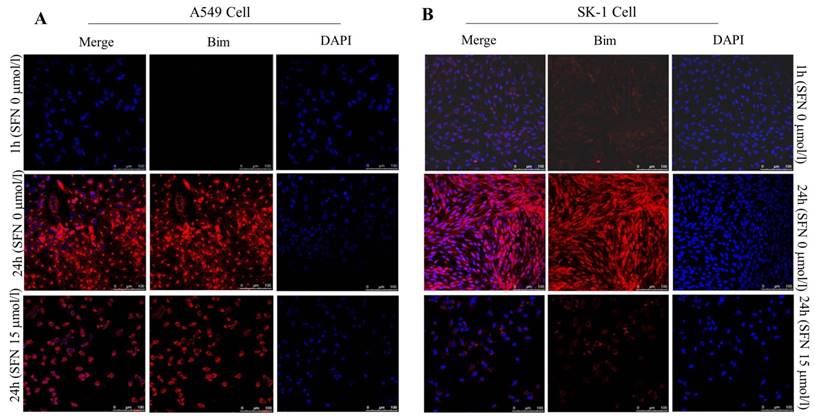
SFN downregulated Bim expression in NSCLC cells
Similar results of Bim expression were obtained with fluorescence confocal microscope. The immunofluorescence results also showed that, in A549 and SK-1 cells without treatment of SFN for 1h and 24h, respectively, Bim expression was increased remarkably at 24h versus 1h treatment. But cells were incubated with 15μM SFN for 24h, highly expressed Bim was decreased dramatically. DAPI staining showed that the cell nucleus was out of shape following SFN treatment, when compared with the nucleus integrity in the control (0μM SFN, 24h) (Figure 8A, 8B).
SFN downregulated Bim expression in NSCLC cells. A-B. Fluorescence confocal microscopy was used to detect the distribution of Bim in SK-1 and A549 cell lines when 0 μM SFN was applied for 1 h and 24 h; cells were incubated with 15 μM SFN for 24 h. Fluorescence images of cell nuclei were stained with DAPI; the red stain showed Bim protein. Merged images indicated that Bim is localized in the nucleus and cytoplasm.
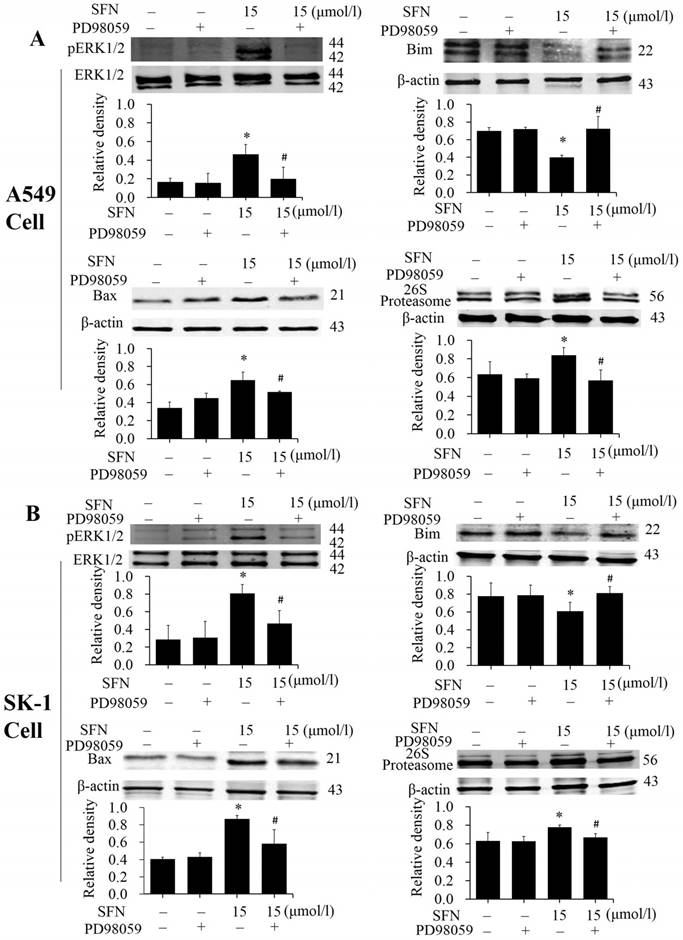
SFN activated ERK1/2 to upregulate Bax, 26S proteasome, and downregulate Bim
The phosphorylated ERK1/2 inhibitor PD98059 was used to treat NSCLC cells for 24 h. Western blots showed that expression of phosphorylated ERK1/2 and Bax was inhibited in presence of SFN and PD98059 (Figure 9A, 9B), indicating that SFN eventually increased Bax by activating ERK1/2 signal pathway. It is possible that the proteasome was regulated by phosphorylated ERK1/2. More, we examined 26S proteasome expression and found that SFN raised the 26S proteasome. With the ERK1/2 inhibitor and SFN, Western blot showed that PD98059 reversed this effect. (Figure 9A, 9B). Thus, the 26S proteasome could be regulated by phosphorylated ERK1/2. In order to demonstrate that downregulation of Bim also resulted from the ERK1/2-dependent pathway cells were treated with SFN for 24 h. We found that SFN downregulated Bim and phosphorylated ERK1/2; when SFN combined with PD98059 were used to treat the cells, the downregulation of Bim expression was reversed (Figure 9A, 9B). These data suggested that Bim was downregulated by an ERK1/2-dependent pathway.
SFN activated ERK1/2 to upregulate Bax, 26S proteasome, and downregulate Bim. A-B. PD98059 was added to the cell medium 30 min before SFN treatment. SK-1 and A549 cells were incubated with 15 μM SFN for 24 h. Western blot was used to analyze the protein level of phosphorylated ERK1/2, Bax, Bim, and 26S proteasome; Phospho-ERK1/2 was normalized to total protein, β-actin was used as a loading control. Densitometric analysis of the bands was done via ImageJ analysis software. The quantification of band intensity was presented as a mean ± SD after normalization to β-actin or total protein. The diagram under each blot showed these protein density ratios, * p<0.05 versus 0 μM, # p<0.05 versus SFN-only group, n=3.
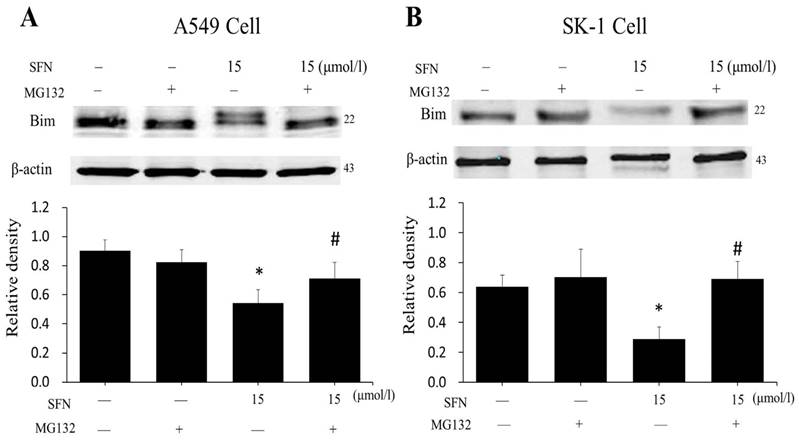
SFN degraded Bim via the proteasome pathway
In order to demonstrate that downregulation of Bim resulted from proteasome activated, the proteasome inhibitor MG132 was applied. After cells had been treated with SFN for 24 h, we found that SFN downregulated Bim; when both SFN and MG132 were used to treat cells for 24h, the downregulation of Bim was diminished (Figure 10A, 10B). The data suggested that Bim is degraded by the proteasome pathway. In addition, in all western blot results (Figure 5A-B, 6D, 7D, 9A-B, 10A-B), only one 22kDa band was detected with the Bim antibody in SK-1 cells, whereas in A549 cells, two bands were detected, one band located at 22 kDa and the other at about 24 kDa, showing one of the Bim isoforms in NSCLC cells.
SFN degraded Bim via the proteasome pathway. A-B. After treatment with MG132 for 30 min, SK-1 and A549 cells were incubated with 15 μM SFN for 24 h. Western blot showed that Bim level was upregulated dramatically compared with the SFN-only group; β-actin was used as a loading control. Densitometric analysis of the bands was done via ImageJ analysis software. The quantification of band intensity was presented as a mean ± SD after normalization to β-actin. The diagram under each blot showed the protein density ratio, * p<0.05 versus 0 μM, # p<0.05 versus SFN-only group, n=3.
Discussion
Increasing evidence showed that SFN induced apoptosis and reduced the incidence of cancer; however, the underlying mechanisms had not been investigated. Herein we found some novel apoptotic features induced by SFN, which might play a key role in cancer inhibition and cell death. SFN is a drug with low toxicity and the optimal plasma concentration of SFN is about 20μM in animal experiments [14]. In this study we verified that 15μM SFN inhibited cell proliferation in NSCLC cells. Analysis of cell apoptosis and morphological observation indicated that 15μM SFN effectively induced apoptosis. Further, electron microscopy observation revealed that 15μM SFN caused a chromatin staining pattern, nuclear fragmentation and generation of apoptotic bodies. These results indicate that SFN has the potential to inhibit cell proliferation and induce apoptosis at a lower dose in NSCLC cells.
Many studies have shown that SFN induces apoptosis through the regulation of intrinsic apoptotic pathways (mitochondrial pathway) in various cancer cells [10, 12, 14, 28]. Mitochondrial outer membrane permeabilization (MOMP) and mitochondrial swelling are the prominent characteristics of mitochondrial morphology during apoptosis [29]. The apoptotic signal ultimately activates ERK1/2 and Bax to promote MOMP, resulting in the release of cytochrome c and Caspase 3 activation [29, 30]. Western blot results indicated that SFN persistently activated ERK1/2, upregulated Bax and Caspase 3 in a dose-dependent and time-dependent manner. Consistently, electron microscopy observation showed that SFN induced mitochondrial swelling and vacuolated structures, suggesting that SFN induced apoptosis via sustained activation of ERK1/2, Bax, Caspase 3 and alternations of mitochondrial morphology in NSCLC cells.
Recently, it has been reported that benzyl isothiocyanate (BITC) derived from cruciferous vegetables induces apoptosis by disruption of the mitochondrial fission-fusion balance in breast cancer cells [19]. Whether SFN induced changes of mitochondrial morphology by disruption of the mitochondrial fission-fusion balance was not been reported. Electron microscopy observation revealed that SFN treatment caused circular structures that were formed by the fusion of many mitochondria. Mitochondria with diverse morphological features along the borderline of the vacuoles formed a circle, which might contribute to mitochondrial vacuolization. These circular mitochondria clustered together in a specific location. Particularly, we observed many fused mitochondria clustered around apoptotic bodies in SK-1 cells. Immunofluorescence also indicated that SFN-induced mitochondria gathered around cells that had lost their original structure, in comparison with the dispersed mitochondria in NSCLC cells without SFN treatment. We proposed that increased mitochondrial fusion represents a critical event in SFN-induced apoptosis in NSCLC cells.
It has been reported that Bcl-2 family proteins play a critical role in regulation of mitochondrial morphology [21]. The anti-apoptotic Bcl-2 proteins are responsible for maintaining the integrity of the mitochondrial membrane (OMM), whereas the pro-apoptotic protein Bax mediates mitochondrial fusion-fission, resulting in the loss of mitochondrial membrane potential and release of cytochrome c [19]. These proteins are all not activated in cytoplasm. The BH3-only protein is required for binding to Bcl-2 or Bax to activate them. The BH3 peptide of Bim binds with high affinity to all pro-survival relatives and weakly to Bax, so that Bim is more easily regulated by survival stimulation in tumor cells [31]. Overexpressed Bim interacted with microtubules, Bcl-xL, and Mcl-1 to induce cell growth and attenuated the release of cytochrome c [23, 32, 33]. Several studies have shown that overexpressed Bim is localized to the mitochondrial membrane, inhibiting interaction with Bax, performing pro-survival functions [21, 23]. We found that when NSCLC cells were cultured for 24h with SFN treatment, Bim level was downregulated, but Bax and cleaved Caspase 3 were upregulated. Immunofluorescence indicated that NSCLC cells without SFN proliferated most dramatically, and Bim level was highest at 24h, indicating that Bim might be an apoptosis inhibitor. With SFN treatment for 24h, cell morphology and the cell nucleus were modified, and Bim level was also decreased. Thus, Bim plays a pro-proliferative role in NSCLC cells.
Degradation of Bim protein via proteasome is a rapid measure which perturbs the high-affinity association of Bim with pro-survival proteins [31, 34]. The 26S proteasome comprises dozens of subunits and might be a target for many post-translational modifications. However, the mechanisms by which the different modifications exert their effects on proteasomal function are not understood. As we know, proteasome activity is regulated by phosphorylation of proteasome subunits. c-Abl kinase, PKA and CKII phosphorylate subunits of the mammalian 20S CP, ASK-1, DYRK2, PKA and CaMKII phosphorylates 19S RP, affecting the association between the 19S RP and 20S CP, substrate recognition, gate opening of 20S CP, and assembly of 19S RP [27]. However, the means by which ERK1/2 exert an effect on 26S proteasome is unknown. Here, we found that 26S proteasome level was upregulated following SFN treatment of NSCLC cells, while upregulated 26S proteasome activation was blocked by addition of the ERK1/2 inhibitor PD98059. Thus, SFN activates ERK1/2 persistently to modify specific 26S proteasomal subunits, leading to degradation of target protein.
It has been demonstrated that the poly-ubiquitination of Bim is degraded by 26S proteasome [35]. The downregulation of Bim was suppressed in the cells treated with SFN plus the proteasome inhibitor MG132. The proteasomal degradation of protein requires a preconditioning protein modification. The Ser69 residue of Bim phosphorylated by ERK1/2 regulates Bim ubiquitination and proteasomal degradation [35, 36]. Using the ERK1/2 inhibitor PD98059, we found that ERK1/2 activated by SFN not only stimulated 26S proteasome activity but also downregulated Bim and upregulated Bax level. Thus, SFN might induce Bim ubiquitination, contributing to recognition of 26S proteasome activated by ERK1/2 and Bim degradation.
Interestingly, Western blot showed that the Bim antibody (22kDa) detected two protein bands: one was about 24 kDa and abundant in A549 cells. Bim exists as three common isoforms, BimS (15kDa), BimL (19kDa), and BimEL (23kDa). The molecular weights of the Bim isoforms are less than 22 kDa, with the exception of BimEL (23 kDa). Many studies have showed that BimEL is the major form of Bim that interacts with pro-survival Bcl-2 protein [34]. BimEL is overexpressed in certain prostate cancer cell lines [23, 33]. Thus, this band might represent the Bim isoform BimEL, judging from the molecular weight; further study is on the way.
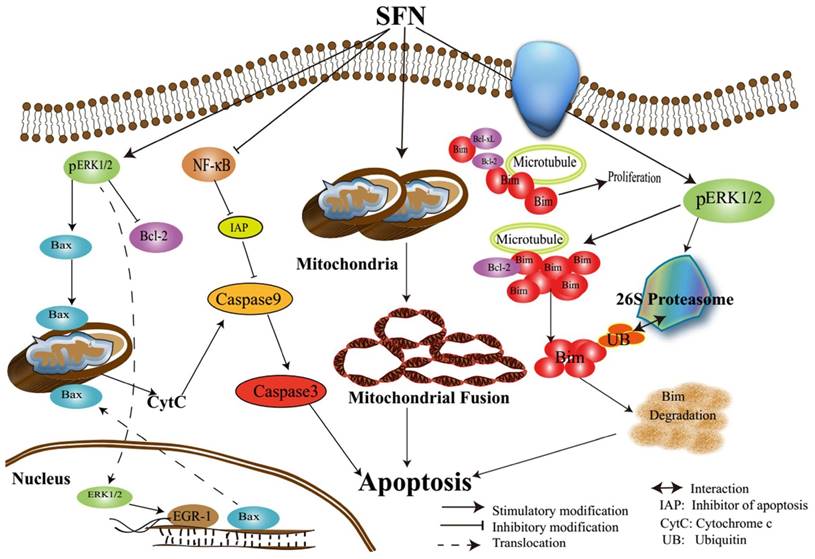
In conclusion, we characterized the underlying mechanisms by which SFN induces apoptosis in NSCLC cells (Figure 11). The characterization of the inhibitory mechanisms may provide a new insight into cancer cell death and therapy. The results might produce some new ideas for targeting pro-survival Bim in the treatment of NSCLC.
Assumed signaling map that SFN induces apoptosis via mediating ERK1/2 and Bim.
Abbreviations
SFN: Sulforaphane
NSCLC: Non-small cell lung cancer
AIF: Apoptosis inducing factor
HCC: Hepatocellular carcinoma
MOMP: Mitochondrial outer membrane permeabilization
OMM: Outer mitochondrial membrane
PTMs: Post-translational modifications
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant numbers 81272843, 81601993).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ansari J, Shackelford RE, El-Osta H. Epigenetics in non-small cell lung cancer: from basics to therapeutics. Translational Lung Cancer Research. 2016;5:155-71
2. Johnson DH, Schiller JH, Bunn PA Jr. Recent clinical advances in lung cancer management. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2014;32:973-82
3. Tan XL, Spivack SD. Dietary chemoprevention strategies for induction of phase II xenobiotic-metabolizing enzymes in lung carcinogenesis. Lung Cancer. 2009;65:129-37
4. Jo GH, Kim GY, Kim WJ, Park KY, Choi YH. Sulforaphane induces apoptosis in T24 human urinary bladder cancer cells through a reactive oxygen species-mediated mitochondrial pathway: The involvement of endoplasmic reticulum stress and the Nrf2 signaling pathway. Int J Oncol. 2014;45:1497-506
5. Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. The AAPS Journal. 2010;12:87-97
6. Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Letters. 2008;269:291-304
7. Ziaei A, Schmedt T, Chen Y, Jurkunas UV. Sulforaphane decreases endothelial cell apoptosis in fuchs endothelial corneal dystrophy: a novel treatment. Invest Ophth Vis Sci. 2013;54:6724-34
8. Cipolla BG, Mandron E, Lefort JM, Coadou Y, Della Negra E, Corbel L. et al. Effect of sulforaphane in men with biochemical recurrence after radical prostatectomy. Cancer Prev Res (Phila). 2015;8:712-9
9. Huang TY, Chang WC, Wang MY, Yang YR, Hsu YC. Effect of sulforaphane on growth inhibition in human brain malignant glioma GBM 8401 cells by means of mitochondrial- and MEK/ERK-mediated apoptosis pathway. Cell Biochem Biophy. 2012;63:247-59
10. Li M, Wang Q, Lin W, Wang B. Regulation of ovarian cancer cell adhesion and invasion by chloride channels. International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society. 2009;19:526-30
11. Kim MG, Lee HS. Growth-inhibiting activities of phenethyl isothiocyanate and its derivatives against intestinal bacteria. Journal of Food Science. 2009;74:M467-71
12. D'Agostini F, Mastracci L, Izzotti A, Balansky R, Pennisi TM, Steele VE. et al. Modulation by phenethyl isothiocyanate and budesonide of molecular and histopathologic alterations induced by environmental cigarette smoke in mice. Cancer Prev Res (Phila). 2009;2:546-56
13. Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224-36
14. Sestili P, Fimognari C. Cytotoxic and antitumor activity of sulforaphane: the role of reactive oxygen species. Biomed Research International. 2015;2015:402386
15. Peng X, Zhou Y, Tian H, Yang G, Li C, Geng Y. et al. Sulforaphane inhibits invasion by phosphorylating ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer DU145 cells. Oncol Rep. 2015;34:1565-72
16. Lee JW, Li C, Zhou Y, Peng X, Du L, Tian H. et al. Sulforaphane inhibits invasion via activating ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS ONE. 2014;9:e90520
17. Galkin A, Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem. 2007;282:37448-53
18. Tang L, Zhang Y. Mitochondria are the primary target in isothiocyanate-induced apoptosis in human bladder cancer cells. Mol Cancer Ther. 2005;4:1250-9
19. Sehrawat A, Croix CS, Baty CJ, Watkins S, Tailor D, Singh RP. et al. Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion. 2016;30:67-77
20. Rao SG. Mitochondrial changes in cancer. Handbook of Experimental Pharmacology. 2016;338:1-17
21. Brooks C, Dong Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle. 2007;6:3043-7
22. Zhan L, Cao H, Wang G, Lyu Y, Sun X, An J. et al. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget. 2016;7:65001-65011
23. Gogada R, Yadav N, Liu J, Tang S, Zhang D, Schneider A. et al. Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J Biol Chem. 2013;288:368-81
24. Pontes MS, Van Waesberghe C, Nauwynck H, Verhasselt B, Favoreel HW. Pseudorabies virus glycoprotein gE triggers ERK1/2 phosphorylation and degradation of the pro-apoptotic protein Bim in epithelial cells. Virus Res. 2016;213:214-8
25. Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G. et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785-93
26. Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cancer Res. 2016;26:869-85
27. Im E, Chung KC. Precise assembly and regulation of 26S proteasome and correlation between proteasome dysfunction and neurodegenerative diseases. BMB Rep. 2016;49:459-73
28. Choi WY, Choi BT, Lee WH, Choi YH. Sulforaphane generates reactive oxygen species leading to mitochondrial perturbation for apoptosis in human leukemia U937 cells. Biomedicine & Parmacotherapy. 2008;62:637-44
29. Thornton C, Hagberg H. Role of mitochondria in apoptotic and necroptotic cell death in the developing brain. Clinica chimica acta; International Journal of Clinical Chemistry. 2015;451:35-8
30. Seo HJ, Choi SJ, Lee JH. Paraquat induces apoptosis through cytochrome C release and ERK activation. Biomol Ther. 2014;22:503-9
31. Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA. et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186:355-62
32. Luna-Vargas MP, Chipuk JE. Physiological and pharmacological control of BAK, BAX, and beyond. Trends Cell Biol. 2016;26:906-17
33. Liu JW, Chandra D, Tang SH, Chopra D, Tang DG. Identification and characterization of Bimgamma, a novel proapoptotic BH3-only splice variant of Bim. Cancer Res. 2002;62:2976-81
34. Ewings KE, Wiggins CM, Cook SJ. Bim and the pro-survival Bcl-2 proteins: opposites attract, ERK repels. CellCycle. 2007;6:2236-40
35. Ordonez AN, Jessick VJ, Clayton CE, Ashley MD, Thompson SJ, Simon RP. et al. Rapid ischemic tolerance induced by adenosine preconditioning results in Bcl-2 interacting mediator of cell death (Bim) degradation by the proteasome. International Journal of Physiology, Pathophysiology and Pharmacology. 2010;2:36-44
36. Abutin RM, Chen J, Lung TK, Lloyd JA, Sawyer ST, Harada H. Erythropoietin-induced phosphorylation/degradation of BIM contributes to survival of erythroid cells. Exp Hematol. 2009;37:151-8
Author contact
![]() Corresponding author: Wei Wu, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, No. 10, Xitoutiao, You An Men Wai Ave, Feng Tai District Beijing 100069, China Tel: 011-8610-8391-1823 Fax: 011-8610-8391-1823 Email Address: weiwu207edu.cn
Corresponding author: Wei Wu, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, No. 10, Xitoutiao, You An Men Wai Ave, Feng Tai District Beijing 100069, China Tel: 011-8610-8391-1823 Fax: 011-8610-8391-1823 Email Address: weiwu207edu.cn