Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2478-2486. doi:10.7150/jca.18648 This issue Cite
Research Paper
Uncarboxylated Osteocalcin Induces Antitumor Immunity against Mouse Melanoma Cell Growth
Yoshikazu Hayashi1, 2, Tomoyo Kawakubo-Yasukochi1, 3 ![]() , Akiko Mizokami1, 4, Mai Hazekawa3, Tomiko Yakura5, Munekazu Naito5, Hiroshi Takeuchi6, Seiji Nakamura2, Masato Hirata1, 7
, Akiko Mizokami1, 4, Mai Hazekawa3, Tomiko Yakura5, Munekazu Naito5, Hiroshi Takeuchi6, Seiji Nakamura2, Masato Hirata1, 7 ![]()
1. Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan;
2. Section of Oral and Maxillofacial Oncology, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan;
3. Department of Immunological and Molecular Pharmacology, Faculty of Pharmaceutical Science, Fukuoka University, Fukuoka 814-0180, Japan;
4. OBT Research Center, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan;
5. Department of Anatomy, Aichi Medical University, Aichi 480-1195, Japan;
6. Division of Applied Pharmacology, Kyushu Dental University, Kitakyushu 803-8580, Japan;
7. Fukuoka Dental College, Fukuoka 814-0193, Japan.
Received 2016-12-7; Accepted 2017-6-4; Published 2017-8-2
Abstract
Because of the poor response to chemotherapy and radiation therapy, new treatment approaches by immune-based therapy involving activated T cells are required for melanoma. We previously reported that the uncarboxylated form of osteocalcin (GluOC), derived from osteoblasts, potentially suppresses human prostate cancer cell proliferation by direct suppression of cell growth. However, the mechanisms in vivo have not been elucidated. In this study, we found that GluOC suppressed tumor growth of B16 mouse melanoma transplants in C57Bl/6N wild-type mice. Our data demonstrated that GluOC suppressed cell growth by downregulating phosphorylation levels of receptor tyrosine kinases and inducing apoptosis in vitro. Additionally, stimulation of primary mouse splenocytes with concanavalin A, a polyclonal T-cell mitogen, in the presence of GluOC increased T cell proliferation and their interferon-γ production. Taken together, we demonstrate that GluOC exerts multiple antitumor effects not only in vitro, but also in vivo through cellular immunostimulatory effects against B16 mouse melanoma cells.
Introduction
Melanoma is one of the most severe forms of malignant diseases (1). Although recent therapeutic agents for melanoma have prolonged patient survival, their prognosis remains poor. In recent years, some epochal strategies against melanoma have been proposed, focusing on immunostimulatory therapy (2), because cellular immunity, especially interferon (IFN)-γ released from cytotoxic T lymphocytes (CTLs) is essential to reject bulky melanoma tumors (3).
Serum osteocalcin (OC), a noncollagenous bone matrix protein secreted by osteoblasts, in serum has been correlated with bone remodeling under pathological conditions including cancer bone metastasis (4), and during normal bone turnover. OC in serum exits as two types, γ-carboxylated OC (GlaOC) and lower- (or un-)γ-carboxylated OC (GluOC) (5-8). We have reported that GluOC, but not GlaOC, improves glucose metabolism (5-8), which is consistent with a previous report (9). These results indicate that each OC type might play a different role in various pathophysiological conditions. In terms of cancer, our recent study revealed that GlaOC and GluOC have completely opposite effects on the growth of human prostate cancer cells in vitro (10), that is, GluOC suppresses cancer cell growth, whereas GlaOC accelerates it.
In the present study, we investigated anticancer mechanisms of exogenous GluOC using a mouse melanoma cell line, B16. There is an intimate crosstalk between bone lineage cells and immune cells (11), and OC has been reported to be regulated by proinflammatory cytokines (12) and decreases in a weakened immune system (13). Therefore, we investigated the immunological aspect of GluOC in cancer progression, as well as its direct antitumor effects.
Materials and Methods
Materials
Recombinant GluOC was prepared as described previously (6). GlaOC was purchased from AnaSpec (Fremont, CA, USA).
Cells
The mouse melanoma cell line, B16, was obtained from RIKEN BioResource Center. The cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), penicillin (100 U/ml), and streptomycin (0.1 mg/ml) at 37°C in humidified air containing 5% CO2.
Measurement of cell viability
Cell viability was determined with a WST8 assay (Cell Count Reagent SF, Nacalai Tesque, Kyoto, Japan) and a BrdU uptake assay (BrdU Cell Proliferation ELISA Kit, Exalpha Biologicals, Shirley, MA, USA), as reported previously (10). Each cell seeding density was optimized before experiments (GluOC: 1 × 104 cells/ well, GlaOC: 5 × 103 cells/ well).
Receptor tyrosine kinase (RTK) phosphorylation antibody array
The RTK phosphorylation antibody array was emploved a Mouse Phospho-RTK Array Kit (R&D Systems, Minneapolis, MN, USA), as reported previously (10). After precultured for 24 h, B16 cells were treated with GluOC (10 ng/ml) or GlaOC (10 ng/ml) for 6 h. Cells were lysed in lysis buffer 17 (R&D Systems) including 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin. Cell lysate (57 μg protein) was applied to the array.
Animals
Female C57Bl/6N mice (Charles River Laboratories, Yokohama, Japan) were maintained under specific pathogen-free conditions. All animal experiments and a part of experiments were approved by the Animal Ethics Committees of Kyushu University and Fukuoka University, respectively.
Determination of tumor growth in B16 transplants of C57Bl/6N mice
A micro-osmotic pump (ALZET, model 1004; DURECT, Cupertino, CA, USA) was used to administer saline, GluOC at a high dose (7.5 µg/mouse/day), or GluOC at a low dose (1.5 µg/mouse/day) by sustained release into the left flank. After 1 week, B16 cells (1×106 cells) in 0.1 ml of PBS were injected subcutaneously into the right flank of the 9-week-old female C57Bl/6N mice (n=8-10) a week later. Tumor size was measured with calipers every 3 or 4 days. Tumor volume was calculated as ab2/2, where a and b are the largest and smallest central cross-sectional dimensions, respectively. At 20 days after tumor transplantation, tumors were extracted after euthanasia.
Immunohistochemistry
Tissue was fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 4-6 μm. Following routine procedures including quenching of endogenous peroxidase and antigen retrieval, the sections were incubated with rabbit polyclonal antibodies against osteocalcin (1:100 dilution, ab93876; Abcam, Cambridge, UK), CD3 (1:100 dilution, 17A2; Biolegend, San Diego, CA, USA), or IFN-γ (1:100 dilution, bs-0480R; Bioss, Woburn, MA, USA). After washing, the sections were incubated for 30 min with biotin-conjugated goat anti-rabbit (1:500 dilution, BA-1000; Vector, Burlingame, CA, USA) or mouse IgG (1:500 dilution, BA-9200; Vector, Burlingame, CA, USA). Tissue sections were visualized using and avidin-horseradish peroxidase (HRP) and 3,3′-diaminobenzidine (DAB), followed by counterstaining with hematoxylin. Images were acquired under a microscope.
Primary splenocyte culture
Splenocytes were prepared as described previously (14). Briefly, spleens were disrupted in RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal calf serum (Life Technologies Japan, Tokyo, Japan). After erythrocytes were lysed by treatment with 0.83% ammonium chloride, the remaining splenocytes were washed twice in RPMI-1640 with 10% fetal calf serum. Splenocytes were seeded at a cell density of 1 × 106 cells/ well in a 96-well culture plate. The cells were stimulated with 5 μg/ml concanavalin A (ConA) (Sigma-Aldrich) in the presence or absence of GluOC or GlaOC. For mitogen-induced lymphocyte proliferation assays, we used Cell Count Reagent SF (Nacalai Tesque) was used. The culture supernatants of lymphocytes were collected at 15, 45, 65 h after stimulation to measure cytokine concentrations by ELISAs.
Enzyme-immuno assay (EIA) and enzyme-linked immunosorbent assays (ELISAs)
To measure IFN-γ and IL-6 concentrations in mouse serum or splenocyte culture supernatants, anti-IFN-γ antibody (Affymetrix, San Diego, CA, USA) or anti-IL-6 antibodies (Affymetrix) were pre-coated onto 96-well plates (Nunc-Immuno Module; Nunc A/S, Roskilde, Denmark) at 4°C overnight. After blocking with ELISA Diluent Solution (eBioscience, San Diego, CA, USA) for 1 h, samples were incubated in the wells at room temperature for 1 h. After washing three times, biotin-anti-mouse IFN-γ (Biolegend, San Diego, CA, USA) or biotin-anti-mouse IL-6 (Biolegend) antibodies were added, followed by incubation at room temperature for 1 h. HRP-streptavidin and tetramethylbenzidine substrate were applied each for 1 h each. Then, absorbance was measured using a microplate reader at 450 nm (655 nm reference). For measurement of serum GluOC concentrations, we used a mouse Glu-osteocalcin high sensitive EIA kit (Takara Bio, Shiga, Japan TAKARA BIO INC. TAKARA BIO INC. TAKARA BIO INC.) was used.
Immunoblot analysis
B16 cells were lysed in 20 mM phosphate buffer, containing 5 mM EDTA, 1% Triton X-100, pepstatin A (2.5 μg/ml), leupeptin (5 μg/ml), 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (25 μg/ml), and aprotinin (1.7 μg/ml). The lysates were centrifuged at 12,000 ×g for 30 min at 4 °C. The protein concentration of the resulting supernatants was determined with a Protein Assay Rapid Kit (Wako, Osaka, Japan). Protein samples (10 μg) were fractionated by polyacrylamide gel electrophoresis on 8-15% polyacrylamide gels containing 0.1% sodium dodecyl sulfate. The separated proteins were transferred to a polyvinylidene difluoride membrane (Merck-Millipore, Darmstadt, Germany), that was then exposed to Blocking One (Nacalai Tesque) at 4°C overnight. After incubation with anti-cleaved Caspase-3 (Cell Signaling Technology, Danvers, MA, USA) or anti-β-actin (Sigma-Aldrich) antibodies at 4 °C overnight, immune complexes were detected with HRP-conjugated secondary antibodies and a chemiluminescence substrate kit (GE Healthcare, Buckinghamshire, England).
Fluorimetric caspase 3/7 assay
An Amplite™ Fluorimetric Caspase 3/7 assay kit (AAT Bioquest®, Sunnyvale, CA, USA) was used to determine caspase 3/7 proteolytic activities according to the manufacturer's protocol. Precultured B16 cells for 24 h in 100 μl medium/well were treated with the vehicle, GluOC (10 ng/ml), or GlaOC (10 ng/ml) for 6 h in 96-well culture plates. Caspase assay solution (100 µl/well) was then applied at room temperature for 1 h in the presence or absence of Ac-DEVD-CHO, a caspase 3/7 inhibitor. For measurement, the fluorescence increase at 350/450 nm (Ex/Em) was monitored.
Statistical analysis
The student's t-test or Dunnett's test were performed for statistical analysis as appropriate. P values of less than 0.05 were considered to be statistically significant. Results are expressed as the mean ± standard error of the mean (SEM).
Results and Discussion
Antitumor effects of GluOC in vivo
We first investigated the effect of GluOC on B16 melanoma isografts in vivo using C57Bl/6N wild-type mice (Fig. 1A). As a result, B16 tumor growth was significantly attenuated by GluOC administration (7.5 μg/mouse/day) (Fig. 1B-C). GluOC administrated by a micro-osmotic pump was detected in the tumor mass and tumor-bearing mouse serum in a dose-dependent manner (Fig. 1D-E). These results indicated that GluOC administrated through a subcutaneous micro-osmotic pump was hematogenously delivered to the isografts to directly exert antitumor effects on B16 melanoma cells.
GluOC attenuates B16 tumor growth in syngenic C57Bl/6N mice in vivo. (A) Experimental protocol. (B) Tumor volume in wild-type female mice administrated saline (closed circle), GluOC at 7.5 µg/mouse/day (open square), or GluOC at 1.5 µg/mouse/day (open circle). (C) Tumor weight at the end point. (D) Immunohistochemical analysis of OC (brown) in B16 transplants. (E) EIA of serum GluOC. (F) ELISA of serum IFN-γ. Immunohistochemical analysis of IFN-γ (G) and CD3 (H) (brown) and in B16 transplants. All IHC sections were counterstained with hematoxylin. Data represent the mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the control (n=8-10).
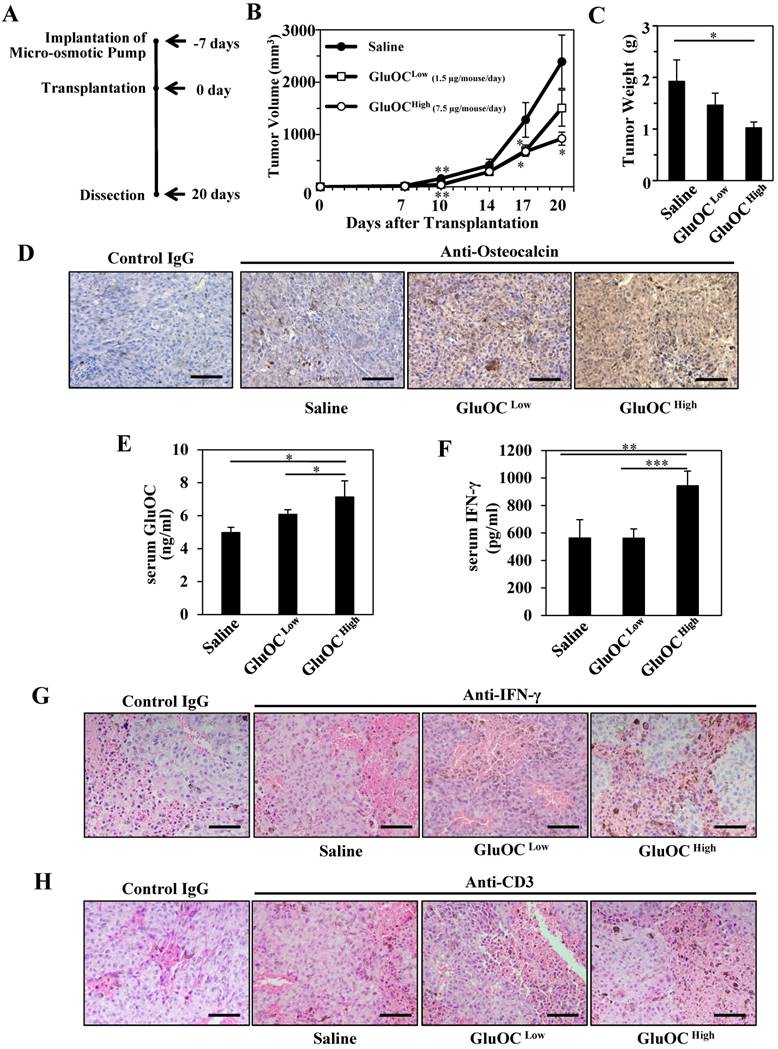
We next examined serum IFN-γ levels in serum and tumors and T cell population in tumors, because recent studies focusing on biological therapy of malignant melanoma have shown the effectiveness of IFN-γ in cellular immunity, especially CTL-mediated immunity, as an adjuvant treatment (3, 15-25). In addition, we observed that the growth of B16 melanoma cells was inhibited with recombinant IFN-γ in vitro as the previous study demonstrated (data not shown) (3). As a result, GluOC administration at 7.5 μg/mouse/day significantly increased serum IFN-γ levels both in serum and tumor mass compared with saline administration (Fig. 1F-G), and CD3-popsitive T cell population was greater in GluOC-treated mouse tumors than in control ones (Fig. 1H). IFN-γ, a type II interferon and pleiotropic cytokine that has diverse biological functions (26), is secreted by CTLs and activated natural killer (NK) cells to increase antitumor activity by enhancing antigen presentation and promoting the proliferation, expansion and survival of CD8+T cells (27, 28). It also binds to cognate receptors at the cell surface and activates the JAK (Janus kinase)-STAT (signal transducer and activator of transcription) pathway (29). We observed that Stat1 was activated in response to recombinant IFN-γ in vitro (data not shown), which satisfied with the previous report (30). Take these results into consideration, JAK-STAT pathway was potentially activated in B16 melanoma cells treated with GluOC.
We also found that similar GluOC administration using athymic Balb/c nu/nu mice did not show an antitumor activity against human prostate cancer cells in vivo and serum IFN-γ concentrations in those mice were not increased (data not shown), even though GluOC has significantly suppressed the growth of the same cancer cells by reducing phosphorylation of RTKs in vitro (10).
Considering these data, we speculated that cell-mediated immunity, especially T cell-mediated production of IFN-γ, is essential for GluOC to show an antitumor activity in vivo.
Effects of GluOC on ConA-specific lymphocyte proliferation and cytokine production
We next tested the effects of GluOC on ConA-stimulated lymphocyte proliferative (blastogenesis) activation and cytokine production. Splenocytes from mice, which were administrated with GluOC (7.5 μg/mouse/day) via a subcutaneous micro-osmotic pump for 3 weeks, showed an increased proliferative response at 15, 45, and 65 h after ConA stimulation compared with those from saline-treated mice (Fig. 2A). In addition, splenocytes of GluOC-treated mice produced significantly higher amounts of IFN-γ following ConA stimulation compared with the control (Fig. 2B), while the amount of the interleukin (IL)-6, another proinflammatory cytokine (31), was unaffected by GluOC treatment (Fig. 2C).
Effects of GluOC on ConA-stimulated splenocytes of C57BL/6 mice ex vivo (A-C) and in vitro (D-F). For ex vivo experiments, splenocytes were collected from C57Bl/6N female mice administrated saline or GluOC (7.5 µg/mouse/day) through an osmotic pump for 3 weeks. A lymphocyte blast transformation assay (A) and ELISAs of IFN-γ (B) and IL-6 (C) in culture supernatants were performed at 15 h (white columns), 45 h (gray columns), and 65 h (black columns) after ConA stimulation. For in vitro experiments, splenocytes collected from C57Bl/6N female mice were stimulated by ConA in the presence or absence of GluOC (12.5, 25, or 50 ng/ml) for 15 h, 45 h, and 65 h. A lymphocyte blast transformation assay (D) and ELISAs of IFN-γ (E) and IL-6 (F) in culture supernatants were performed at 15, 45, and 65 h after stimulation. Each experiment was repeated three times. Data represent the mean ± SEM. *P < 0.05 and ***P < 0.001 versus the control (n=8-9).
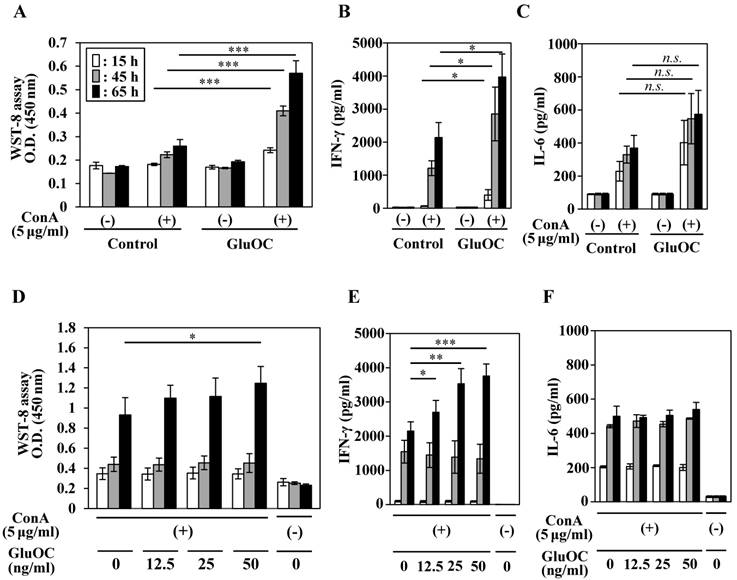
Next, we examined whether these results were reflected by the direct effects of GluOC on splenocytes. Splenocytes from C57Bl/6N mice were treated with ConA in the presence or absence of GluOC at 0, 12.5, 25, 50 ng/ml in vitro. ConA-stimulated proliferative response of T cells (Fig. 2D) and IFN-γ production (Fig. 2F) were increased by in vitro administration of GluOC at 65 h after stimulation, as well as in vivo GluOC administration. In addition, the level of IL-6 in the supernatant was unaltered (Fig. 2E). Both IFN-γ and IL-6 are regarded as proinflammatory cytokines that regulate immune responses, cell proliferation, and tumor development and progression (3, 31, 32). However, these cytokines frequently have functionally different roles. IFN-γ is a major Th1 cytokine associated with the cellular response (3, 32), while IL-6 is a Th2 cytokines associated with the humoral response to extracellular pathogens (31). Although it is currently unknown whether GluOC contributes to the predominance of the Th1 response in cancer, there is the possibility that GluOC might regulate the Th1/Th2 paradigm in several pathophysiological processes including cancer.
It has been reported that GPRC6A, a receptor for GluOC, is highly expressed not only in organs where it is involved in glycolipid metabolism (33-35), but also in the spleen (34, 35). Although its function in the spleen is unknown, recent reports suggested that the receptor is associated with immune responses (36, 37). Our immunological experiments also showed the expression of GPRC6A in mouse spleen and splenocytes (data not shown), that include a variety of immune cell populations. It could not be concluded without further detailed analysis, but there is a possibility that GluOC increased IFN-γ secretion via GPRC6A expression in certain splenic immune cells. Because IFN-γ is produced by CTLs and NK cells (26-29), we examined whether NK cells were needed for GluOC to perform immunopotentiative functions under ConA stimulation. GluOC did not increase lymphocyte proliferation or IFN-γ production of NK cell-depleted splenocytes treated with ConA (Fig. S1A, B). This result indicates that not only T cell but also NK cells are essential for the strong GluOC-mediated immunostimulation. In addition, such immunostimulating effects were specific to GluOC, because no changes occurred in ConA-stimulated splenocytes treated with GlaOC (Fig. S2A, B). These results indicate that GluOC suppresses cancer cell growth in vivo by upregulating the anticancer effect of cellular immunity, especially increased secretion of IFN-γ.
Direct effects of GluOC and GlaOC on B16 cell viability in vitro
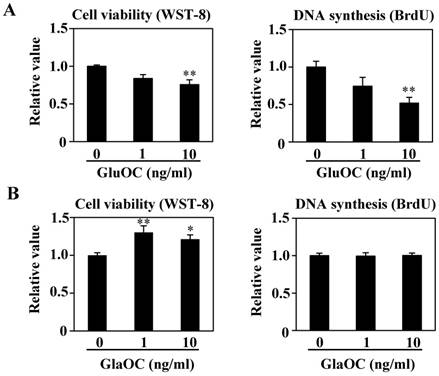
We have reported that GluOC directly suppresses, while GlaOC promotes, human prostate cancer cell growth in vitro (10). We therefore investigated the effect of each OC type on B16 cell viability in vitro. WST-8 and BrdU uptake assays showed that GluOC significantly suppressed B16 cell viability (Fig. 3A). In contrast, GlaOC increased B16 cell viability in the WST-8 assay, although there were no significant differences in the BrdU uptake assay (Fig. 3B). These results indicate a functional difference between GlaOC and GluOC in B16 mouse melanoma cell growth, and that GluOC or GlaOC has suppressing or promoting effects on these cells, respectively, which is consistent with our previous report (10).
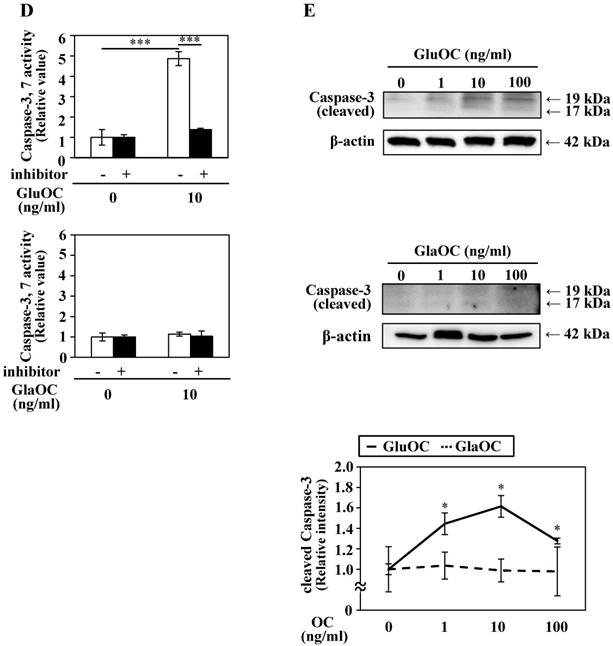
GluOC, but not GlaOC, suppresses B16 cell growth in vitro. B16 cells were incubated with GluOC (A) or GlaOC (B) for 24 h, followed by WST-8 (left panels) and BrdU uptake (right panels) assays. Mean data are expressed as the ratio to the control. Each experiment was repeated three times. Data represent the mean ± SEM. *P < 0.05 and **P < 0.01 versus the control. (C) Phospho-RTK array. After B16 cells were treated with the vehicle, GluOC, or GlaOC for 6 h, the cell lysate was applied to the array. Each pair of kinase dots that increased (red) or decreased (blue) compared with the controls is enclosed by a square. Quantitation of the dot densities of phospho-RTKs was performed using scanned images and ImageQuant LAS 4000 software (GE Healthcare). (D) Fluorimetric assay of caspase-3 and -7 activities detected in B16 cells treated with the vehicle, GluOC, or GlaOC for 6 h. Fluorescence was monitored in the presence or absence of DEVD-CHO. Each experiment was repeated three times. (E) Immunoblot analysis of cleaved caspase-3 after stimulation with GluOC or GlaOC for 6 h in B16 cells. β-actin was used as an internal control. Quantitation of cleaved caspase-3 expression (normalized by the amount of β-actin) in an immunoblot is shown in the bottom panel. Each experiment was repeated three times. Data represent the mean ± SEM. *P < 0.05 and ***P < 0.001 versus the control.
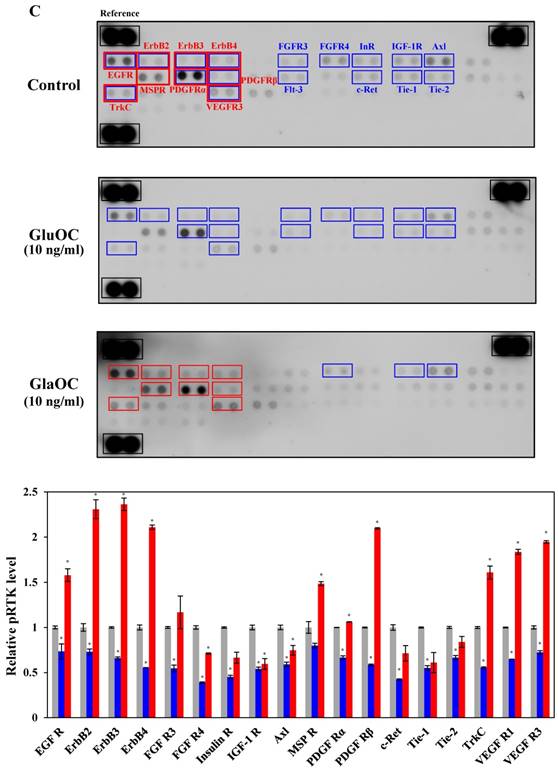
We next subjected B16 mouse melanoma cells to phospho-RTK arrays. RTKs are a family of cell surface receptors that mediate key signaling pathways involved in cell proliferation (38). As shown in Fig. 3C, GluOC reduced phosphorylation levels of many kinds of RTKs including epidermal growth factor receptor (EGFR), ErbB2, ErbB3, ErbB4, fibroblast growth factor receptor (FGFR) 3, FGFR4, InR (insulin-like receptor), insulin-like growth factor 1 receptor, Axl, platelet-derived growth factor receptor (PDGFR) α, PDGFRβ, Flt-3 (fms-like tyrosine kinase), c-Ret, tyrosine kinase with immunoglobulin-like and EGF-like domains (Tie)-1, Tie-2, tropomyosin receptor kinase C (TrkC), and vascular endothelial growth factor receptor 3 (VEGFR3). These results indicate that GluOC directly suppresses melanoma cell growth by reducing RTK activation as shown in human prostate cancer cells (10). In contrast, GlaOC enhanced the phosphorylation levels of many kinds of RTKs, particularly EGFR, ErbB2, ErbB3, ErbB4, macrophage-stimulating protein receptor, PDGFRα, PDGFRβ, TrkC, and VEGFR3. These nine RTKs are growth factors belonging to the families of EGFR (39), MET proto-oncogene (40), PDGFR (41, 42), Trk (43), and VEGFR (44), which are closely related to tumor progression of melanoma (38, 43, 45-51). These data were consistent with the results of proliferation assays, although whether the phosphorylation is a direct effect or a secondary effect has not been established. A multi-kinase inhibitor, which inhibits many growth factors, has been greatly focused on in clinical cancer therapy (45), and some RTK multi-inhibitors have already been used in clinical pathology (46-49, 52-54). However, they also have the risk of side effects by downregulating kinase activity required in normal physiological functions. Considered that administration of GluOC did not result in any observable toxic effects on normal tissues or cells of the treated mice in vivo or in vitro (10), GluOC might be a promising multi-targeted inhibitor of RTKs in cancer therapy.
We also analyzed another molecular mechanism of the inhibitory effect on cell growth by GluOC. In a caspase-3/7 fluorescence assay, GluOC, but not GlaOC, elevated the proteolytic activity of caspase-3 and -7, which was inhibited by Ac-DEVD-CHO, an inhibitor of caspase-3 and -7 (Fig. 3D). This result was also supported by immunoblot analysis in which cleaved caspase-3 was visualized in GluOC-treated B16 cell lysates in a dose-dependent manner, but not in GlaOC-treated B16 cell lysates (Fig. 3E). These results indicate that GluOC suppresses B16 cell growth in vitro, which directly involves downregulating phosphorylation of multiple RTKs as well as promotion of apoptosis.
Conclusion
In summary, we propose a new therapeutic strategy using GluOC, which is not only a multi-kinase inhibitor and apoptosis-inducer, but also an adjuvant in tumor immunotherapy. Although further studies are needed to elucidate the potential molecular mechanism of GluOC in cancer progression, studies are now in progress to evaluate the applied dosage and an effective method for clinical application.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (KAKENHI grants 24229009 to M. Hirata, 26861554 and 16K11496 to T. Kawakubo-Yasukochi, and 26861553 and 16K20421 to A. Mizokami), the Central Research Institute of Fukuoka University (157103 to T. Kawakubo-Yasukochi. and M. Hazekawa), and Fukuoka Foundation for Sound Health Cancer Research Fund. The authors acknowledge useful discussion with Prof. M. Nakashima (Fukuoka University). Y. Hayashi was a recipient of The Iwadare Scholarship Foundation and Morita Scholarship Foundation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Owens B. Melanoma. Nature. 2014;515:7527
2. Kleffel S, Posch C, Barthel SR. et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242-1256
3. Kakuta S, Tagawa Y, Shibata S. et al. Inhibiton of B16 melanoma experimental metastasis by interferon-γ through direct inhibition of cell proliferation and activation of antitumor host mechanisms. Immunology. 2002;105:92-100
4. Gardner TA, Lee SJ, Lee SD. et al. Differential expression of osteocalcin during the metastatic progression of prostate cancer. Oncol Rep. 2009;21:903-908
5. Mizokami A, Yasutake Y, Gao J. et al. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS One. 2013;8:e57375
6. Mizokami A, Yasutake Y, Higashi S. et al. Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone. 2014;69:68-79
7. Kawakubo-Yasukochi T, Kondo A, Mizokami A. et al. Maternal oral administration of osteocalcin protects offspring from metabolic impairment in adulthood. Obesity. 2016;24:895-907
8. Otani T, Mizokami A, Hayashi Y. et al. Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell Signal. 2015;27:532-544
9. Lee NK, Sowa H, Hinoi E. et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456-469
10. Hayashi Y, Kawakubo-Yasukochi T, Mizokami A. et al. Differential roles of carboxylated and uncarboxylated osteocalcin in prostate cancer growth. J Cancer. 2016;12:1605-1609
11. Kansara M, Teng MW, Smyth MJ. et al. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14:722-735
12. Li YP, Stashenko P. Proinflammatory cytokines tumor necrosis factor-alpha and IL-6, but not IL-1, down-regulate the osteocalcin gene promoter. J Imuunol. 1992;148:788-794
13. Mera P, Laue K, Wei J. et al. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol Metab. 2016;5:1042-1047
14. Brinkmann V, Sharma SD, Remington JS. Different regulation of the L3T4-T cells subset by B cells in different mouse strains bearing the H-2K haplotype. J Immunol. 1986;137:2291-2997
15. Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends in Immunology. 2015;36:257-264
16. Gioarelli M, Cofano F, Vecchi A. et al. Interferon-activated tumor inhibition in vivo. Small amounts of interferon-γ inhibit tumor growth by eliciting host systemic immunoreactivity. Int J Cancer. 1986;37:141-148
17. Maekawa R, Kitagawa T, Hojo K. et al. Distinct antitumor mechanisms of recombinant murine interferon-γ against two murine tumor models. J Interferon Res. 1988;8:227-239
18. Gansbacher B, Bannerji R, Daniels B. et al. Retroviral vector-mediated γ-interferon gene transfer intro tumor cells generates potent and long lasting antitumor immunity. Cancer Res. 1990;50:7820-7825
19. Kaplan DH, Shankaran V, Dighe AS. et al. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556-7561
20. Yim JH, Wu SJ, Lowney JL. et al. Enhancing in vivo tumorigenicity of B 16 melanoma by overexpressing interferon regulatory factor-2: resistance to endogenous IFN-γ. J Interferon Cytokine Res. 1999;19:723-729
21. Mule JJ, Custer MC, Travis WD. et al. Cellular mechanisms of the antitumor activity of recombinant IL-6 in mice. J Immunol. 1992;148:2622-2629
22. Xu YR, Shen Y, Ouahab A. et al. Antitumor activity of TNF-alpha after intratumoral injection using an in situ thermosensitive hydrogel. Drug Dev Ind Pharm. 2015;41:369-374
23. Ramirez-Montagut T, Turk MJ, Wolchok JD. et al. Immunity to melanoma: unraveling the relation of tumor immunity and autoimmunity. Oncogene. 2003;22:3180-3187
24. Davar D, Kirkwood JM. Adjuvant Therapy of Melanoma. Cancer Treat Res. 2016;167:181-208
25. Vesely MD, Kershaw MH, Schreiber RD. et al. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol. 2011;29:235-271
26. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat Rev Immunol. 2005;5:375-386
27. Smyth MJ. Type I interferon and cancer immunoediting. Nat Immunol. 2005;6:646-648
28. Dighe AS, Richards E, Old LJ. et al. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447-456
29. Liu SY, Sanchez DJ, Aliyari R. et al. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA. 2012;109:4239-4244
30. Heikkilä K, Ebrahim S, Lawlor DA. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur J Cancer. 2008;44:937-945
31. Lugade AA, Sorensen EW, Gerber SA. et al. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180(5):3132-3139
32. Dias DS, Fontes LBA, Crotti AEM. et al. Copaiba oil suppresses inflammatory cytokines in splenocytes of C57Bl/6 mice induced with experimental autoimmune encephalomyelitis (EAE). Molecules. 2014;19:12814-12826
33. Pi M, Wu Y, Quales LD. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J Bone Miner Res. 2011;26:1680-1683
34. Clemmensen C, Smajilovic S, Wellendorph P. et al. The GPCR, class C, group 6, subtype A (GPRC6A) receptor: from cloning to physiological function. Br J Pharmacol. 2014;171:1129-1141
35. Kuang D, Yao Y, Lam J. et al. Cloning and characterization of a Family C orphan G-protein coupled receptor. J Neurochem. 2005;93:383-391
36. Dehghan A, Dupuis J, Barbalic M. et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123:731-738
37. Rossol M, Pierer M, Raulien N. et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature commun. 2012;3:1329
38. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-1134
39. Boone B, Jacobs K, Ferdinande L. et al. EGFR in melanoma: clinical significance and potential therapeutic target. J Cutan Pathol. 2011;38:492-502
40. Molhoek KR, Shada AL, Smolkin M. et al. Comprehensive analysis of RTK activation in human melanomas reveals autocrine signaling through IGF-1R. Melanoma Res. 2011;21(4):274-284
41. Nazarian R, Shi H, Wang Q. et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973-977
42. Sanna PS, Piia T, Michael E. et al. Melanoma cell-derived factors stimulate hyaluronan synthesis in dermal fibroblasts by upregulating HAS2 through PDGFR-PI3K-AKT and p38 signaling. Histochem Cell Biol. 2012;138:895-911
43. Marchetti D, Murry B, Galjour J. et al. Human melanoma TrkC: its association with a purine-analog-sensitive kinase activity. J Cell Biochem. 2003;88:865-872
44. Rajabi P, Neshat A, Mokhtari M. et al. The role of VEGF in melanoma progression. J Res Med Sci. 2012;17:534-539
45. Easty DJ, Gray SG, O'Byrne KJ. et al. Receptor tyrosine kinases and their activation in melanoma. Pigment Cell Melanoma Res. 2011;24:446-461
46. Clark JW, Eder JP, Ryan D. et al. Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin Cancer Res. 2005;11:5472-5480
47. Rini Brian I. Sunitinib. Expert Opin Pharmacother. 2007;8:2359-2369
48. Orestis L, Annett M, Florian H. et al. Analysis of anti-proliferative and chemosensitizing effects of sunitinib on human esophagogastric cancer cells: synergistic interaction with vandetanib via inhibition of multi-receptor tyrosine kinase pathways. Int J Cancer. 2010;127:1197-1208
49. Shi H, Kong X, Ribas A. et al. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71:5067-5074
50. Tarik R. Targeting RTK Signaling Pathways in Cancer. Cancers. 2015;7:1758-1784
51. Garay T, Molnár E, Juhász É et al. Sensitivity of melanoma cells to EGFR and FGFR activation but not inhibition is influenced by oncogenic BRAF and NRAS mutations. Pathol Oncol Res. 2015;21:957-968
52. Yang LPH, McKeage K. Axitinib. Drugs. 2012;72:2375-2384
53. Hong DS, Kurzrock R, Falchook GS. et al. Phase 1b study of lenvatinib (E7080) in combination with temozolomide for treatment of advanced melanoma. Oncotarget. 2015;6:43127-43134
54. Dayer LE, Hutchins LF, Johnson JT. Treatment of metastatic melanoma with pazopanib: A report of five patient cases. J Oncol Pharm Practice. 2015;21:224-231
Author contact
![]() Corresponding authors: Tomoyo Kawakubo-Yasukochi DDS, PhD, Department of Immunological and Molecular Pharmacology, Faculty of Pharmaceutical Science, Fukuoka University, Fukuoka 814-0180, Japan. Email: tomoyoyasuac.jp; Masato Hirata DDS, PhD, Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan, and Fukuoka Dental College, Fukuoka 814-0193, Japan. Email: hirata1kyushu-u.ac.jp
Corresponding authors: Tomoyo Kawakubo-Yasukochi DDS, PhD, Department of Immunological and Molecular Pharmacology, Faculty of Pharmaceutical Science, Fukuoka University, Fukuoka 814-0180, Japan. Email: tomoyoyasuac.jp; Masato Hirata DDS, PhD, Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan, and Fukuoka Dental College, Fukuoka 814-0193, Japan. Email: hirata1kyushu-u.ac.jp