Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2542-2553. doi:10.7150/jca.19918 This issue Cite
Research Paper
Identification of Novel Epitopes with Agonistic Activity for the Development of Tumor Immunotherapy Targeting TRAIL-R1
Lu Guo1, 2*, Xin Sun1*, Zhichao Hao1, Jingjing Huang1, Xiaojian Han1, Yajie You1, Yaying Li1, Meiying Shen3, Tatsuhiko Ozawa4, Hiroyuki Kishi4, Atsushi Muraguchi4, Aishun Jin1 ![]()
1. Department of Immunology, Harbin Medical University, Harbin, Heilongjiang 150081, China;
2. Department of Basic Medical Sciences, Heilongjiang Nursing College, Harbin, Heilongjiang 150086, China;
3. Department of Breast Surgery, Harbin Medical University Cancer Hospital, Harbin, Heilongjiang 150000, China;
4. Department of Immunology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama 930-0194, Japan.
* These authors contributed equally to this work.
Received 2017-3-4; Accepted 2017-5-30; Published 2017-8-2
Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor-1/2 (TRAIL-R1/R2), also known as death receptors, are expressed in a wide variety of tumor cells. Although TRAIL can induce cell apoptosis by engaging its cognate TRAIL-R1/R2, some tumor cells are or become resistant to TRAIL treatment. Monoclonal antibodies (mAbs) against TRAIL-R1/R2 have been developed to use as potential antitumor therapeutic agents instead of TRAIL. However, TRAIL-R1/R2-based tumor therapy has not yet been realized. We previously generated a series of fully human monoclonal antibodies against TRAIL-R1 (TR1-mAbs) that induced tumor cell apoptosis. In this study, we identified the antigenic binding sites of these TR1-mAbs and proposed two major epitopes on the extracellular domain of TRAIL-R1. The analysis revealed that the epitopes of some TR1-mAbs partially overlaps with the beginning of TRAIL-binding sites, and other epitopes are located within the TRAIL-binding region. Among these mAbs, TR1-422 and TR1-419 mAbs have two antigenic binding sites that bound to the same binding region, but they have different essential amino acid residues and binding site sizes. Furthermore, we investigated the apoptosis activity of TR1-419 and TR1-422 mAbs in the form of IgG and IgM. In contrast to the IgG-type TR1-419 and TR1-422 mAbs, which enhanced and inhibited TRAIL-induced apoptosis, respectively, both IgM-type TR1-419 and TR1-422 mAb strongly induced cell apoptosis with or without soluble TRAIL (sTRAIL). Moreover, the results showed that IgM-type TR1-419 and TR1-422 mAbs alone can sufficiently activate the extrinsic and intrinsic apoptosis signaling pathways and suppress tumor growth in vivo. Consequently, we identified two antigenic binding sites with agonistic activity, and their specific IgM-type mAbs exhibited strong cytotoxic activity in tumor cells in vitro and in vivo. Thus, these agonistic antigenic binding sites may be useful for the development of effective Ab-based drugs or Ab-based cell immunotherapy for various human solid tumors.
Keywords: epitope, TRAIL-R1, monoclonal antibody, apoptosis, tumor immunotherapy.
Introduction
Cancer is one of the leading causes of death worldwide, accounting for most of the mortality associated with solid tumors. Current approaches in tumor therapy predominantly target tumor-associated antigens of tumor cells to avoid the toxicity of traditional chemotherapeutic drugs. Recent tumor immunotherapy, such Ab-based drugs or genetically engineered antibody-based immune cell therapy, has successfully been applied in hematological malignancies [1-8]. However, applications in the treatment of solid tumors are lacking. In approaches for the treatment of solid tumors, screening targeting epitopes or antigens uniquely expressed on tumor cells has been assessed.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of the TNF superfamily [9,10]. TRAIL has four transmembrane receptors expressed on the cell surface. Among them, only TRAIL-R1 and TRAIL-R2 can trigger apoptosis [9,11,12]. Over the past decade, binding of the TRAIL trimer to TRAIL-R1 and/or TRAIL-R2 has been shown to selectively induce apoptosis in tumor cells by activating extrinsic and intrinsic apoptosis pathways [11-23]. These studies laid the foundation for the development of TRAIL-R-targeting therapies for clinical use. However, tumor cells were later shown to become resistant following TRAIL treatment [19,24-28]. TRAIL is an unstable cytokine, which may limit its pharmacodynamic activity in the body [19]. And TRAIL's two decoy receptors, TRAIL-R3 and TRAIL-R4, have been shown to be involved in inhibiting TRAIL-induced apoptosis [29-31]. Thus, several mAbs targeting TRAIL-R1 or TRAIL-R2, which have antitumor activity, have been established as another therapeutic strategy instead of TRAIL [32-34] and have shown promising activity in preclinical observations [35].
Several studies have reported that TRAIL-R1 and TRAIL-R2 exist as preassembled receptor oligomers on the cell surface [36,37]. The binding of TRAIL and/or agonist antibodies to TRAIL-R1 or R2 causes a conformational change in the TRAIL-R1 or R2 ectodomain and the formation of a death-inducing signaling complex (DISC), which triggers activation of apoptosis signaling pathways [17,36,38-40]. A recent report showed that the human monoclonal agonistic antibody KMTR2 enhanced TRAIL-R2 superoligomerization, leading to strong agonistic activity [41]. Another study demonstrated that the TRAIL-R2-specific mAb AMG 655 in cooperation with TRAIL promoted the formation of a trimeric complex, which strongly enhanced antitumor activity [42]. In general, crosslinking of TRAIL-Rs on the tumor cell surface is essential for effective induction of cell apoptosis. These studies further supported the development of Ab-based drugs and Ab-based immune cell therapy targeting TRAIL-R1 or TRAIL-R2.
Recently, we developed a series of TRAIL-R1-specific human mAbs (TR1-mAbs) [43,44]. We found that some mAbs enhanced TRAIL-induced apoptosis, while others blocked TRAIL-induced apoptosis [43,45,46]. In this study, we described the antigenic binding sites of the TR1-mAbs and characterized these sites in the TR1-419 and TR1-422 mAbs. Furthermore, we investigated the epitope-mediated antitumor activity in vitro and in vivo. As a result, we identified two novel epitopes with agonistic activity on the extracellular domain of TRAIL-R1, suggesting that these epitopes may be useful in the development of effective immunotherapies for a range of human cancers.
Materials and Methods
Cells and cell culture
HeLa and SW480 cells were maintained in DMEM (Dulbecco's modified Eagle's medium, Applichem, Germany) supplemented with 10% fetal calf serum, 1% L-glutamate and 1% penicillin/streptomycin. All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Analysis of antigenic binding sites of mAbs to TRAIL-R1
For identification of the antigenic epitopes recognized by the TR1-mAbs, cDNA coding for the extracellular region of TRAIL-R1 was cloned from K562 cells. Eight fragments (P1-P8), which contained 45-mer peptides with fifteen overlapping amino acid residues, were in turn amplified from the cDNA of TRAIL-R1. Then, they were cloned into bacterial display vectors, and the vectors were transformed into E. coli to obtain protoplasts. The protoplasts were incubated with 5 μg/ml of TR1-mAbs for 30 min on ice and then washed and stained with a FITC-conjugated human IgG-specific goat antibody for 30 min on ice. After being washed, 10,000 cells were collected and analyzed by flow cytometry (BD Biosciences, MI, USA) to determine the antigenic binding site of TR1-mAbs. For further antigen binding site analysis of TR1-419 and TR1-422, we constructed two fragments containing 25-mer peptides with 5 overlapping amino acid residues (named P5-up and P5-down) from the P5 fragment and analyzed the exact binding sites of these TR1-mAbs as described above. To specify the active amino acid residues in the TR1-419 and TR1-422 binding sites, we constructed variants with alanine substituted for various amino acid residues in the TR1-422 binding site using a site-directed mutagenesis strategy and analyzed as described above.
Flow cytometry analysis
To assess the binding of TR1-IgGs or TR1-IgMs to TRAIL-R1 on the cell surface, we incubated HeLa and SW480 cells with 1 µg/ml TR1-IgGs (TR1-IgG-419 or IgG-422) or TR1-IgMs (TR1-IgM-419 or IgM-422) for 30 min on ice, followed by incubation with FITC-conjugated antibodies to human IgG or Allophycocyanin (APC)-conjugated antibodies to human IgM (Biolegend, San Diego, USA) for 30 min on ice. After the cells were washed, they were analyzed by flow cytometry (BD Biosciences, MI, USA).
To investigate the effects of TR1-IgGs or TR1-IgMs on the binding of TRAIL to TRAIL-R1, we treated HeLa and SW480 cells with various doses of soluble TRAIL (sTRAIL) and with TR1-IgGs or TR1-IgMs, respectively, and then analyzed them as described above.
Annexin V/PI staining
After the HeLa cells were treated with TR1-IgMs (1 μg/ml) and TR1-IgGs (1 μg/ml) plus crosslinking Ab (10 μg/ml) for 6 h, the fraction of apoptotic cells was determined by staining cells suspending in Annexin-V binding buffer (BD Pharmingen, CA, USA) with FITC-conjugated Annexin-V and propidium iodide (PI) according to the manufacturer's instructions. The samples were analyzed by flow cytometry. Apoptotic cells were defined as Annexin V and PI-positive cells.
Cell viability assays
To examine cell viability, we seeded cells in triplicate in 96-well plates at 5×103 cells/well in cell culture medium containing 10% FCS. For analysis of the apoptosis induced by TR1-IgMs, HeLa and SW480 cells were cultured with TR1-IgMs at the indicated concentrations for 24 h. Cell viability was determined by MTT assays. With respect to TR1-IgGs, HeLa and SW480 cells were incubated with 1 μg/ml TR1-IgG-419 and IgG-422 mAbs in the absence or presence of 10 μg/ml anti-human IgG Fc secondary crosslinking Abs for 48 h, and cell viability was measured.
To investigate the effects of TR1-IgMs on TRAIL-induced apoptosis, we cultured HeLa and SW480 cells with 1 μg/ml TR1-IgMs in the absence and presence of 1μg/ml TRAIL for 24 h, and cell viability was measured. Then we further cultured HeLa and SW480 cells with 1 μg/ml TR1-IgMs in the presence of 0-5 μg/ml TRAIL or with TRAIL alone at the indicated concentrations for 24h, and cell viability was measured.
Western blot analysis
HeLa cells were stimulated with TR1-IgMs alone or with TR1-IgGs in the presence of crosslinking Ab for 3 h. Western blot analyses were carried out with antibodies against caspase-8 (9746S, Ref:03/2015), caspase-3 (9662S, Ref:12/2015), caspase-7 (9494S, Ref:11/2015), PARP (9532S, Ref:12/2015), Smac (15108S, Ref:09/2015), Bid (2002S, Ref:01/2016), Bax (2774S, Ref:12/2015) (Cell Signaling Technology, Beverly MA, USA) and β-actin (Abcam, ab119716, Cambridge, MA,USA) and with a horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (Biosharp, Hefei, Anhui, China) or goat anti-mouse secondary antibody (Bioss, Shanghai, China). Densitometry analysis was performed using the Talon ECL system.
Analysis of tumoricidal activity in vivo
Six-week-old female BALB/cA-nude mice (SLAC Laboratory, Shanghai, China) were used for the experiments, and animal protocols were approved by the Committee on Animal Experiments at Harbin Medical University (Harbin, Heilongjiang, China). BALB/cA-nude mice were inoculated subcutaneously with HeLa cells (5×105) in the lower right flank. The treatments were initiated when the tumor volume reached 100 mm3, and ten mice were used for each treatment group. TR1-IgG-419, IgG-422, TR1-IgM-419, IgM-422 and control-PBS were administered to the animals intravenously (i.v.) via the tail vein at a dose of 12.5 mg/kg every two days (Supplemental Figure 1). Body weight and tumor weight were measured. The tumor mass size was measured with the two largest dimensions, and the tumor volumes in mm3 were calculated using the following formula: tumor volume= length×width2×1/2.
Histological analysis of the xenograft tumors
The mice were sacrificed after the experimental treatment, and immunohistochemical staining was employed to determine the expression of TRAIL-R1 (Abcam, Cambridge, MA, USA) in tumor tissues. Microscopy analysis was performed using 4-20× objectives.
Statistical analysis
All the data are presented as the mean ± SEM. When there were two treatment groups, Student's t-test was used. For three or more treatment groups, one-way ANOVA was used with a Bonferroni post-test for the comparison of two selected treatment groups as well as a Dunnett post-test for the comparison of all treatment groups to the corresponding control. A value of *p < 0.05 or even **p<0.01 was considered significant. Data were analyzed using GraphPad Prism 5.0 and SPSS 20.0
Results
Antigenic binding site analysis of the mAbs to TRAIL-R1
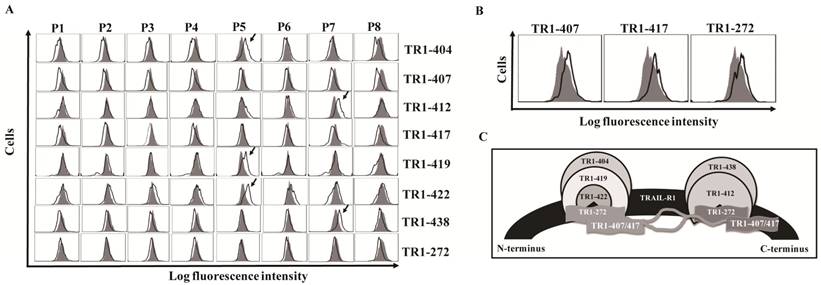
We previously generated ten fully human monoclonal Abs (TR1-mAbs) to human TRAIL-receptor 1 (hTRAIL-R1) [43]. We found that some TR1-mAbs enhanced TRAIL-induced apoptosis, while others inhibited TRAIL-induced apoptosis [43,45,46]. To clarify the mechanisms underlying the distinct effects of these TR1-mAbs on TRAIL-induced apoptosis, we carried out antigenic binding site mapping analysis of the TR1-mAbs using bacterial display technology. We amplified cDNA of the ectodomain of TRAIL-R1 from K562 cells and expressed TRAIL-R1 with no mutations. We then divided the ectodomain of TRAIL-R1 into eight peptide fragments (named Parts 1-8, P1-8), which were designed to overlap by 15 amino acids (Supplemental Figure 2). We transformed bacterial display vectors containing the eight peptides into E. coli, and analyzed them with the TR1-mAbs using flow cytometry. The results showed that the TR1-404, TR1-419 and TR1-422 mAbs bound to the P5 fragment (129-173 aa) and TR1-412 and TR1-438 mAbs to P7 (189-233 aa) (Figure 1A). TR1-272, TR1-407 and TR1-417 mAbs only bound to the full-length ectodomain of TRAIL-R1 but not to any of the truncated peptides (Figure 1A, B), suggesting that TR1-272, TR1-407 and TR1-417 mAbs might recognize conformational epitopes on the ectodomain of TRAIL-R1. Based on previous data indicating that the binding of the TR1-272, TR1-407 and TR1-417 mAbs to TRAIL-R1 was in part blocked by either TR1-419 or TR1-412 mAb using competitive antibody-binding assays [43], we hypothesized that the conformational epitopes of TR1-272, TR1-407 and TR1-417 mAbs were involved in two major binding domains of the external portion of TRAIL-R1. A schematic diagram shows the relationships among the binding regions of these TR1-mAbs on the TRAIL-R1 ectodomain (Figure 1C). These results indicated that there are two predominant binding domains on the extracellular domain of TRAIL-R1, and some TR1-mAbs bound to the same epitopes.
Identification and relationships of the epitopes of TR1-mAbs. (A) Screening antigenic binding sites of TR1-mAbs. The extracellular domain of TRAIL-R1 was divided into eight overlapping fragments named P1-P8, which were cloned into bacterial display vectors. After transformation of each fragment into E. coli and treatment of the protoplasts, we incubated the protoplasts with 5 μg/ml of TR1-mAbs, followed by incubation with a FITC-conjugated human IgG-specific goat antibody. The samples were analyzed with flow cytometry. (B) TR1-407, -417 and -272 bound to the full-length extracellular domain of TRAIL-R1. (C) Epitopes of TR1-mAbs on the ectodomain of TRAIL-R1. The diagram shows the relationship of TR1 mAb-binding sites on TRAIL-R1 expressed on the tumor cell surface.
Identification of two novel epitopic peptides on the extracellular region of TRAIL-R1
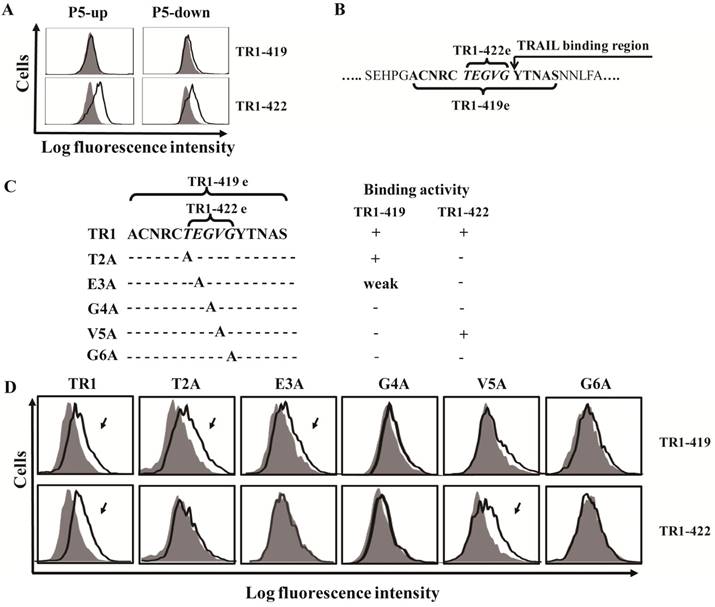
In previous studies [43,45,46], we found that TR1-419 and TR1-422 had different effects on TRAIL-induced apoptosis. However, the above results showed that both the TR1-419 and TR1-422 mAbs bound to the P5 fragment of the TRAIL-R1 ectodomain. To further determine their exact epitope sites, we divided the P5 fragment into two parts with 5 overlapping amino acid residues (named P5-up and P5-down). The results of epitope analysis showed that TR1-419 did not bind to either truncated peptide from P5 fragment, whereas TR1-422 bound to both the P5-up and P5-down fragments (Figure 2A). We confirmed that binding site of TR1-419 (named TR1-419e) contains 15 amino acid residues (144ACNRCTE GVGYTNAS158), and the binding site of TR1-422 (named TR1-422e) consists of only 5 amino acid residues (149TEGVG153), which were located in a central site of the TR1-419e (Figure 2B). Based on the binding regions of TRAIL to TRAIL-R2 [37,47], we speculated that the binding region of TRAIL to TRAIL-R1 starts from 154YTNAS158. Therefore, the binding site peptides of TR1-419 partially overlap the beginning of the TRAIL-binding region, and that of TR1-422 are just adjacent to the beginning of the TRAIL-binding region. Figure 2B shows the amino acid sequences of TR1-419e and TR1-422e as well as the relationships among TR1-419e and TR1-422e and the beginning of the TRAIL-binding site. In addition, we found that the G153 amino acid residue determines the specific binding of TR1-419 and TR1-422 only to TRAIL-R1 but not to other TRAIL-Rs.
Determination of the binding sites and active amino acid residues of TR1-IgG-419 and TR1-IgG-422. (A) Exact epitope analysis of TR1-419 and TR1-422. The P5 fragment was divided into two parts with 5 overlapping amino acid residues (named P5-up and P5-down). Data are presented as the representative results of three assays. (B) Amino acid sequences of the two epitopes and the relationships among the binding sites of TR1-419 and TR1-422 and the epitopes of TRAIL on the extracellular region of TRAIL-R1. (C) Schematic of variants of the TR1-422 binding site. Variants with substitution of amino acid residues with alanine were designed and named T2A, E3A, G4A, V5A and G6A. Binding activity of TR1-419 and TR1-422 to the variants is indicated. ''+'' indicates strong binding activity; “weak” indicates weak binding activity; ''-'' indicates no binding to the variant. (D) Flow cytometric analysis of the binding activity of TR1-419 and TR1-422 to various epitope variants. Representative histograms in two independent experiments are shown.
To further verify that the TR1-422e site was located within that of TR1-419e, we investigated whether TR1-422 inhibited the binding of TR1-419 to tumor cells. The results revealed that TR1-422 blocked the binding of TR1- 419 to TRAIL-R1 on tumor cells (Supplemental Figure 3). In recent studies, we found that TR1-419 enhanced TRAIL-induced apoptosis in some tumor cells [45]. Next, we investigated whether TR1-422 inhibited the effect of TR1-419 on TRAIL-induced cell apoptosis in tumor cells. As expected, TR1-422 could block TR1-419-enhanced TRAIL-induced apoptosis in a dose-dependent manner (Supplemental Figure 4). These results further confirmed that TR1-422e is embedded in TR1-419e, suggesting that the amino acid resides of TR1-422e are necessary for the binding of the TR1-419 and TR1-422 mAbs and for the cell death induced by the TR1-419 and TR1-422 mAbs. Taken together, we identified two novel epitopes of the ectodomain of TRAIL-R1.
To specify key amino acids that determine the binding activity of the TR1-419 and TR1-422 mAbs, we produced a series of variants with substitution of alanine at various residues in TR1-422e using a site-directed mutagenesis strategy (Figure 2C). We then detected the binding activity of TR1-419 and TR1-422 mAbs to the variants of TR1-422e. The results demonstrated that TR1-419 and TR1-422 mAbs bound to the T2A and V5A variants, respectively, but neither bound to the G4A nor to G6A variants. TR1-419 reacted to E3A but TR1-422 did not. Consequently, in the consensus TR1-422e sequence (TEGVG), TEGxG is a prerequisite for the binding of TR1-422 to TRAIL-R1, and at least xEGVG is required for that of TR1-419 (Figure 2C, D). Overall, although both TR1-419 and TR1-422 recognize the TEGVG residues, their binding activity to TRAIL-R1 is determined by different amino acid residues as well as the peptide length.
TR1-419e- and TR1-422e-mediated cell apoptosis
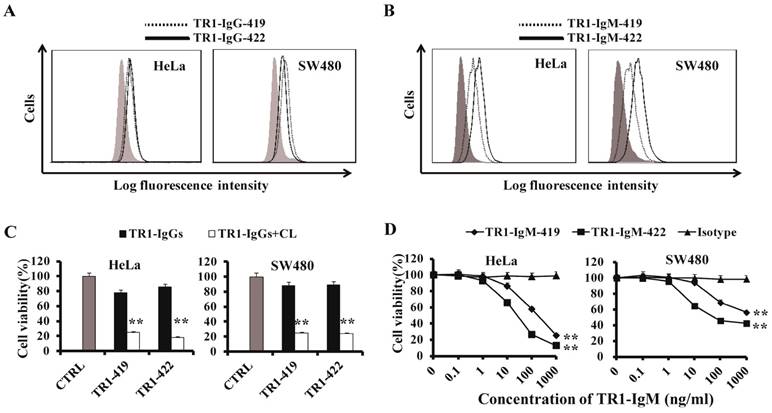
To compare the TR1-419e- and TR1-422e-mediated cell apoptosis, we employed their corresponding IgG and IgM-type monoclonal antibodies for analysis of cell apoptosis. IgM-type TR1-419 and TR1-422 mAbs were constructed by combining the variable region of TR1-IgGs with the constant region of IgM [48]. First, we detected whether TR1-IgMs could bind to TRAIL-R1 expressed on the surface of tumor cells. Similar to TR1-IgGs (Figure 3A), TR1-IgMs bound to TRAIL-R1 expressed on HeLa cells and SW480 cells, and TR1-IgM-422 appeared to have stronger binding activity to tumor cells than that of TR1-IgM-419 (Figure 3B). The Log fluorescence intensity (%) of binding activity of IgM and IgG was indicated in Supplementary Table 1. Next, we investigated the cytotoxic activity of TR1-mAbs. TR1-IgG -419 and IgG-422 strongly induced cell death in the presence of crosslinking Ab and maximally induced cell death in approximately 80% of the cells but had little effect on HeLa and SW480 cells in the absence of crosslinking Ab (Figure 3C). To assess the cytotoxic activity of TR1-IgMs, we incubated various tumor cells with different concentrations of TR1-IgM-419 or IgM-422 alone for 24 h. The results demonstrated that induction of cell death by TR1-IgM-419 and IgM-422 was significantly increased at a high concentration of 1000 ng/ml in HeLa and SW480 cells (Figure 3D). We observed that cell apoptosis induced by TR1-IgMs alone was similar to that by the TR1-IgGs in the presence of crosslinking Ab. Additionally, we observed that TR1-IgM-422 induced stronger cell apoptosis than TR1-IgM-419, even at a low concentration of 10 ng/ml, in HeLa and SW480 cells. Taken together, these results demonstrated that in contrast to the IgG-type mAbs of TR1-419e and TR1-422e, the IgM-type TR1-mAbs alone strongly induced cell death; in particular, the IgM-type Ab TR1-422e with a short peptide binding site appeared to trigger complete apoptosis.
Characteristics of TR1-mAbs with two antigenic binding sites. (A, B) Binding activity of TR1-IgG-419 and IgG-422 (A) or TR1-IgM-419 and IgM-422 (B) to HeLa and SW480 cells was detected by flow cytometry. (C) Cell death induced by TR1-IgGs in the absence or presence of secondary crosslinking antibody in HeLa and SW480. Cell viability was detected by MTT analysis. (D) Cell death induced by TR1-IgM-419 and TR1-IgM-422 alone in HeLa and SW480 cells. Cell viability was detected. The data presented are representative of three independent experiments. “CTRL” represents a negative control. Isotype represents an irrelevant IgM-type Ab as a negative control. Statistical significance is defined as **P<0.01 vs. CTRL or Isotype.
TRAIL inhibits TR1-IgG- but not TR1-IgM -induced cell apoptosis
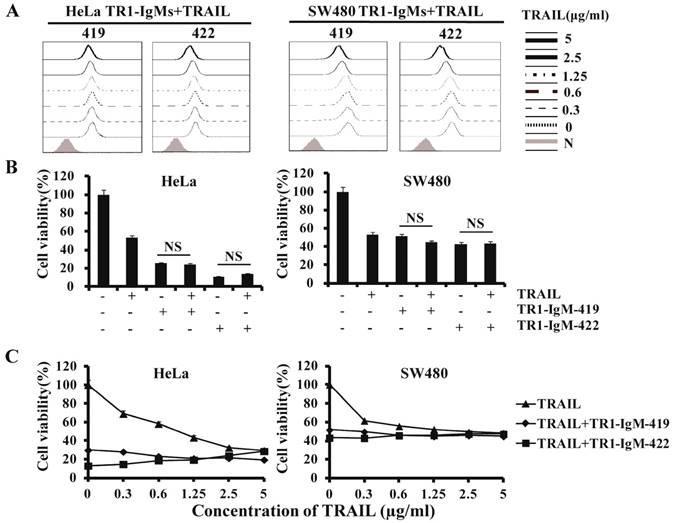
In previous studies, we found that the binding of TR1-IgG-422 to TRAIL-R1 was blocked by sTRAIL in a dose-dependent manner, but the binding of TR1-IgG-419 had no significant effects on HeLa and SW480 cells in the absence or presence of sTRAIL, although a high concentration of sTRAIL (5 μg/ml) slightly blocked binding of TR1-419 to TRAIL-R1 [45] (Supplemental Figure 5). TR1-IgG-419 in combination with TRAIL increased induction of cell apoptosis, while a combination of TR1-IgG-422 and TRAIL inhibited cell apoptosis as previously reported (Supplemental Figure 6). To investigate the effects of TR1-IgMs on TRAIL-mediated binding activity and cell apoptosis, we examined the binding activity and apoptosis induction of the IgM-type TR1-419 and TR1-422 mAbs in the presence of sTRAIL. We observed that the binding of TR1-IgM-419 or IgM-422 to TRAIL-R1 was slightly blocked only at high concentrations (5 μg/ml and 2.5 μg/ml) of sTRAIL but not at intermediate (1 μg/ml) or low doses (<1 μg/ml) of sTRAIL (Figure 4A). With regard to cytotoxic activity, in contrast to TR1-IgG-419 or IgG-422 Ab, which enhanced or blocked TRAIL-induced apoptosis, respectively (Supplemental Figure 6), cell apoptosis induction by TR1-IgM-mAbs was not significantly changed in the presence of 1 μg/ml compared to the absence of TRAIL in HeLa and SW480 cells (Figure 4B). Furthermore, we confirmed that both the TR1-IgM-419 and TR1-IgM-422 mAbs strongly induced cell apoptosis even in the presence of TRAIL, although a slight reduction in cell apoptosis induced by TR1-IgM-422 in high doses (2.5 μg/ml -5 μg/ml) of sTRAIL was observed in HeLa cells (Figure 4C). Taken together, these results demonstrated that bivalent IgG-type TR1-mAbs but not multivalent IgM-type TR1-mAbs inhibited sTRAIL binding avidity or induction of cell apoptosis. IgM-type TR1-mAbs alone strongly induced cell apoptosis.
TR1-IgGs but not TR1-IgMs inhibit TRAIL-induced cell apoptosis. (A) Analysis of the competitive inhibition by TRAIL of TR1-IgM binding to TRAIL-R1 on HeLa and SW480 cells. Cells were pre-cultured with different concentrations of soluble TRAIL, and then, 1 μg/ml of TR1- IgM-419 or IgM-422 was added. The results were assessed by flow cytometry. (B, C) TRAIL interferes with TR1-IgM-induced tumor cell apoptosis. HeLa and SW480 cells were incubated alone or in combination with TRAIL (1 μg/ml) and TR1-IgMs (1 μg/ml) (B) or with different concentrations of TRAIL (0-5 μg/ml) and TR1-IgMs (1 μg/ml) (C) for 24 h. Cell viability was detected by MTT assays. The data presented are representative of three independent experiments. “NS” indicates no statistical significance.
The activation of intrinsic and extrinsic apoptosis signaling pathways mediated by TR1-419e and TR1-422e
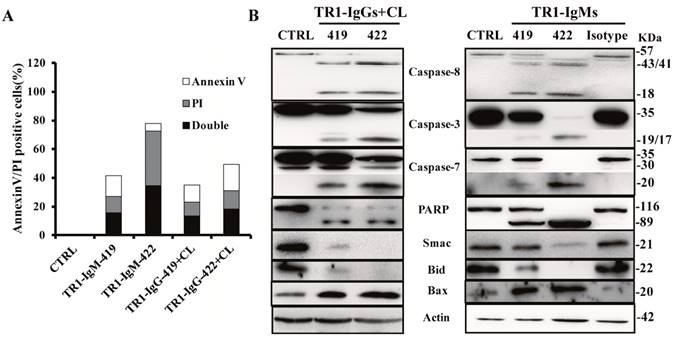
Next, we investigated the molecular mechanisms of cell apoptosis mediated by two epitopes. First, we detected Annexin V/propidium iodide (PI)-positive cells by FITC-Annexin V/PI staining at 6 h after the treatments with TR1-IgM alone and TR1-IgG plus crosslinking in HeLa cells. As shown in Figure 5A, Annexin V/PI-positive cells significantly increased in HeLa cells treated with the TR1-mAbs compared to those of the negative control (CTRL), particularly TR1-IgM-422 compared to other TR1-mAbs (Figure 5A). Many reports have confirmed that TRAIL-R1 or TRAIL-R2 mediated by TRAIL or agonistic antibodies triggered cell apoptosis via activation of extrinsic and intrinsic apoptosis pathways [11-23]. Our results indicated that TR1-IgGs plus crosslinking Ab or TR1-IgMs alone resulted in significant cleavage of caspase-8, caspase-3, caspase-7 and PARP but not negative controls of the irrelevant isotype Abs, and in particular, pro-caspase-8, caspase-3, caspase-7 and PARP were barely observed in the cells treated with TR1-IgM-422 (Figure 5B). Furthermore, we investigated the involvement of mitochondria and its associated proapoptotic and antiapoptotic proteins. Both the addition of TR1-IgGs as well as the TR1-IgMs promoted the cleavage of Bid and upregulated the expression of the proapoptotic protein Bax, while the release of the antiapoptotic protein Smac was reduced (Figure 5B). The results suggested that similar to IgG-type TR1-mAbs, IgM-type TR1-mAbs induced cell apoptosis via the activation of the extrinsic and intrinsic apoptosis pathways; in particular, TR1-IgM-422, with a short peptide binding site, showed sufficient activation of the two apoptotic pathways compared with other TR1-mAbs.
Mechanisms of the induction of cell apoptosis by TR1-mAbs. (A) Annexin V and PI double staining apoptosis detection in HeLa cells. The treated cells were stained with Annexin V and PI, and then, Annexin V, PI, and Annexin V and PI double positive cells were analyzed by flow cytometry. “CTRL” represents a negative control. (B) Analysis of apoptotic signaling pathways. HeLa cells were treated with TR1-IgGs in the presence of crosslinking or TR1-IgMs alone for 3 h. Cell lysates were analyzed by Western blot. The data presented are representative of three independent experiments. “CTRL” represents a negative control; Isotype represents an irrelevant IgM-type Ab as a negative control.
The tumoricidal activity of TR1-419 and TR1-422 in vivo
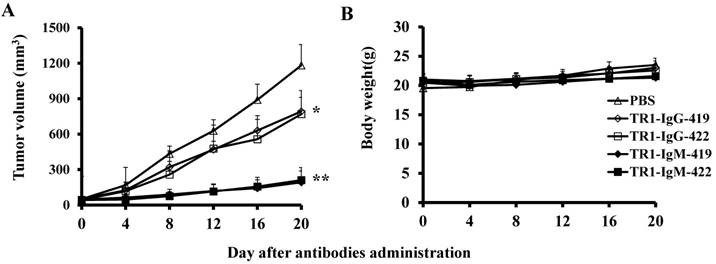
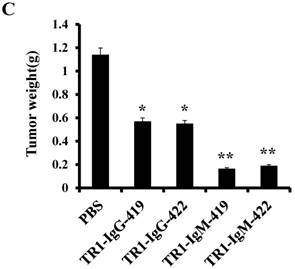
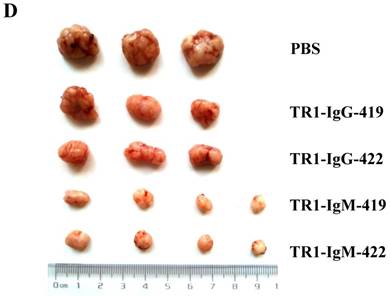
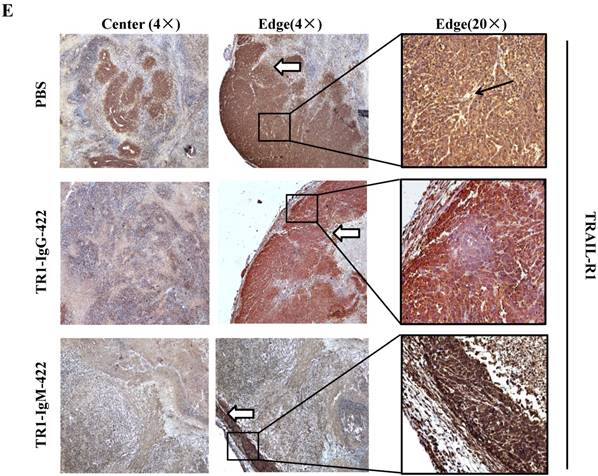
To assess the tumoricidal activity of TR1-mAbs in vivo, we assessed TR1-IgGs (-419 or -422) and TR1-IgMs (-419 or -422) as therapeutic mAb drugs in a xenograft tumor-bearing mouse model. BALB/cA-nude mice were inoculated subcutaneously with HeLa cells. The mice were treated with a single intravenous dose of TR1-IgGs (-419 or -422) and TR1-IgMs (-419 or -422) alone when solid tumor formation reached a volume of approximately 100 mm3. We observed that the tumors grew much more slowly in mice treated with TR1-IgMs compared with those of the control-PBS and TR1-IgGs mice (Figure 6A). In experimental groups, there were no obvious abnormalities in body weight and appearance (Figure 6B). After administration, tumors were harvested and measured. The results showed that tumors were substantially smaller and lighter in mice treated with TR1-IgMs (either IgM-419 or IgM-422) alone or TR1-IgGs (either IgG-419 or IgG-422) alone than those in mice treated with PBS. In particular, TR1-IgMs significantly suppressed tumor growth compared to that of the control-PBS group (Figure 6C, D). Furthermore, we isolated and analyzed the tumor tissues after administration of the drugs. H&E staining showed that tumor cells and tumor mass clusters were widely distributed in the tumor tissues of the PBS-treated mice. In contrast, the tumor cell layer in the surrounding tumor tissue was thinner in the mice treated with TR1-IgMs than that in the mice treated with PBS, and widespread tumor debris was observed in the center of the tumor tissues in the mice treated with TR1-IgMs (Supplemental Figure 7). Moreover, we stained the tumor tissues with antibody against TRAIL-R1 to confirm its expression in the tumors. We observed more abundant expression of TRAIL-R1 in the PBS-treated mice, in either in the central area or in the edge of the tumor tissues (Figure 6E, first row). In the TR1-IgG-treated mice, reduced TRAIL-R1 expression was observed in the center or in the edge of tumor tissues compared with that in the mice treated with PBS (Figure 6E, second row). In contrast, in the TR1-IgM-treated mice, we observed a thin layer of tumor cells expressing TRAIL-R1 in the surrounding tumor tissues but rarely detected tumor cells in the center of the tissues (Figure 6E, third row). These data indicated that the TR1-IgMs penetrate tumor tissue to suppress tumor growth in vivo. Taken together, these results showed that IgM-type TR1-419 and TR1-422 targeting two epitopes had strong tumoricidal activity, suggesting that the two epitopes may be ideal targets for tumor therapy in tumors expressing TRAIL-R1.
Tumoricidal activity of TR1-mAbs with two binding sites in a xenograft tumor-bearing mouse model. (A) Antitumor efficacy of TR1-IgGs (IgG-419 and IgG-422) and TR1-IgMs (IgM-419 and IgM-422). Ten mice were included in each group. Each time point represents the mean value (±S.E.M.) of the tumor sizes within the treated group on the day of measurement. (B) The body weight of the mice was measured every four days. (C) Tumor tissues were harvested, and (D) tumor weight was measured. (E) Histological analysis of tumor sections after treatment with TR1-IgG-422 and IgM-422. Tumor tissue paraffin sections were stained with antibodies specific for TRAIL-R1. The left column indicates the tumor center position, the middle column indicates the tumor edge, and the right column shows the magnified black boxes. The white arrows indicate TRAIL-R1 expression in the tumor edge, and the black arrow indicates the blood vessels in the tumor tissue. Statistical significance is defined as *P<0.05 vs. control-PBS; **P<0.01 vs. control-PBS.
Discussion
Recently, TRAIL-R1/R2-targeting therapeutic efficiency has being extensively studied in clinical trials. We developed a series of TRAIL-R1-specific human mAbs (TR1-mAbs) [43]. These TR1-mAbs (IgG type) exhibited different characteristics in binding activity and cell apoptosis in the presence of TRAIL [43,45]. In this study, we accurately identified the different antigenic binding sites of TR1-419 and TR1-422 mAbs. Using IgG-type and IgM-type mAbs targeting two epitopes, we clarified that TR1-IgG-mAbs but not IgM-mAbs enhanced or inhibited TRAIL-mediated apoptosis, while both IgM-type TR1-mAbs targeting two epitopes strongly induced cell apoptosis in vitro and in vivo, particularly IgM-TR1-422 with a short peptide binding site.
Through antigenic epitope mapping analysis, we hypothesized that there are two major binding domains for linear epitopes (129-173 aa for binding of TR1-404 and -419 and -422, as well as 189-233 aa for TR1-412 and -438, respectively) on the extracellular domain of TRAIL-R1, and conformational epitopes were involved in two binding domains (TR1-417 and -407 and -272 mAbs). The results of epitopic analysis showed that TR1-419 binds to TRAIL-R1 at the junction between cysteine-rich domains CRD 1 and CRD2, and TR1-422 only binds at CRD2. Based on the binding activity and affinity of the TR1-mAbs as well as the cytotoxic activity from our previous data [43,45], we hypothesized that the binding regions of TR1-419 and TR1-422 may be major antigenic domains. Furthermore, because IgG-type TR1-419 and TR1-422 had different effects on TRAIL-induced apoptosis (enhancing and blocking apoptosis), we accurately analyzed their antigenic binding sites. The results showed that the TR1-419 site contains 15 amino acids, whereas that of TR1-422 consists of only 5 amino acids that overlaps with the central 5 amino acids of the TR1-419 binding site. Based on the binding sites of TRAIL on TRAIL-R2, we hypothesized that the TR1-419 peptide partially overlaps the beginning of the TRAIL binding region and that of TR1-422 is just adjacent to the beginning of the TRAIL binding region. Therefore, TR1-419 (IgG type), with a long binding peptide, and TRAIL simultaneously bind to TRAIL-R1 at overlapping residues, promoting the aggregation of TRAIL-R1 and increasing cell apoptosis by activation of apoptotic pathways. Several studies have demonstrated that antibodies against TRAIL-R2 competed with TRAIL for binding to TRAIL-R2 at overlapping epitopes, thereby preventing them from acting in synergy [49], but other antibodies against TRAIL-R2 did not compete with TRAIL for binding to TRAIL-R2, and they bound to TRAIL-R2 simultaneously, resulting in enhancing TRAIL-R2 crosslinking and apoptosis-inducing synergy [42,49]. Ultimately, the aggregation and clustering of TRAIL-Rs are required for sufficient activation of apoptosis pathways. TR1-422 (IgG type) binds to TRAIL-R1 at an epitope that interferes with TRAIL-R1's concomitant interaction with TRAIL so that both proteins cannot bind to TRAIL-R1 simultaneously, resulting in an inhibitory effect on tumor cell apoptosis. However, the inhibitory effect on TRAIL-induced apoptosis could be reversed by IgM-type TRA-422 Ab, suggesting that the binding of TR1-422 Ab had agonistic activity rather than antagonistic activity.
To further confirm this hypothesis, we investigated the effects of IgM-type TR1-419 and TR1-422 on TRAIL-mediated binding activity and TRAIL-induced apoptosis. In contrast to IgG-type TR1-419 and TR1-422, both IgM-type TR1-419 and TR1-422 mAbs strongly induced cell apoptosis even in the presence of TRAIL. In particular, IgM-type TR1-422 strongly promoted cell apoptosis and sufficiently triggered the activation of the extrinsic and intrinsic apoptosis pathways in vitro, indicating that a short peptide may have a spatial advantage in the binding of multivalent IgM type Abs compared to that of a long peptide. TR1-IgM-422 was not more effective than TR1-IgM-419 in suppressing tumor growth in vivo, which may be due to the doses and duration of treatment strategies. In general, the proteins, such as multivalent IgM, interact with the major antigenic domain to facilitate the aggregation of TRAIL-R1 on the surface of tumor cells and efficiently induce tumor cell apoptosis. Although the affinity of TR1-422 (1.4x10-8 M) is higher than that of TR1-419 (2.1x10-7 M), our previous study suggested that the antitumor effect was not correlated with the affinity of TR1-IgMs [48]. With respect to the antigenic binding sequences, the TR1-422 peptide is encompassed and located in the central site of the TR1-419 peptide, and there are differences in the active amino acid residues of their binding sites; TEGxG is required for the binding of TR1-422, and at least xEGVG is required for TR1-419. Whether these active amino acid residues contribute to the activation of apoptosis signaling pathways will require further confirmation.
Our results also indicated that both the extrinsic and intrinsic apoptotic signaling pathways are activated by TR1-IgGs plus crosslinking or TR1-IgMs alone. In particular, TR1-IgM-422 sufficiently induced cleavage of caspase-8, caspase-3, caspase-7, and PARP, as well as Bid and Smac release. These results demonstrated that the two binding sites of the TR1-419 and TR1-422 mAbs had agonistic activity. However, the low tissue penetrance of IgM-type mAbs limits their clinical applications [50-55]. Our results indicated that TR1-IgMs strongly suppressed tumor growth in vivo compared with that of TR1-IgGs alone. IHC analysis indicated that tissues expressing TRAIL-R1 were completely eradicated in the center of the tumor, and there was a thin cell layer of the surrounding tumor in TR1-IgM-treated mice, although the tumor size was not notably smaller after administration. These observations suggested that tumor size alone may be insufficient for evaluating pharmacotherapeutic efficacy, and long-term observation of tumor growth is required.
In conclusion, for the first time, we identified two epitopes with agonistic activity on the extracellular region of TRAIL-R1, one of which is xEGVG for the recognition of TR1-419 and the other is TEGxG for TR1-422. We demonstrated that the effects of IgG-type but not IgM-type TR1-mAbs on binding activity and cell apoptosis were blocked by the presence of TRAIL. TR1-IgM-419 and IgM-422 strongly induced antitumor activity in vitro through the extrinsic and intrinsic pathways and suppressed tumor growth in vivo. We therefore conclude that two epitopes exhibit agonistic activity, suggesting that Ab-based reagents or genetically engineered antibody-based immune cell therapy targeting the epitopes on tumor cells may be applicable for the development of effective immunotherapy for a range of human cancers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank the Translational Medicine Research and Cooperation Center of Northern China, Heilongjiang Academy of Medical Sciences, Harbin China and the Key Laboratory of Infectious and Immunology of Heilongjiang province for providing the experimental platform. We also thank members of Atsushi Muraguchi's laboratory, Department of Immunology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Japan for their help and support. This research was supported by grants from the Natural Science Foundation of China (81171973, 81572807) and the Natural Science Foundation of Heilongjiang Province, China (ZD201118).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361-70
2. Fuenmayor J, Montano RF. Novel antibody-based proteins for cancer immunotherapy. Cancers. 2011;3:3370-93
3. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024-8
4. Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192-8
5. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525-41
6. Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550-7
7. van de Donk NW, Janmaat ML, Mutis T. et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016;270:95-112
8. Vasekar M, Rizvi S, Liu X. et al. Novel immunotherapies for hematological malignancies. Curr Mol Pharmacol. 2016;9:264-71
9. Wiley SR, Schooley K, Smolak PJ. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673-82
10. Pitti RM, Marsters SA, Ruppert S. et al. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687-90
11. Pan G, Ni J, Wei YF. et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815-8
12. Pan G, O'Rourke K, Chinnaiyan AM. et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111-3
13. Koschny R, Walczak H, Ganten TM. The promise of TRAIL-potential and risks of a novel anticancer therapy. J Mol Med. 2007;85:923-35
14. Walczak H, Degli-Esposti MA, Johnson RS. et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386-97
15. Walczak H, Miller RE, Ariail K. et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157-63
16. Dickens LS, Boyd RS, Jukes-Jones R. et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291-305
17. Kischkel FC, Lawrence DA, Chuntharapai A. et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611-20
18. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897-907
19. Mahalingam D, Oldenhuis CN, Szegezdi E. et al. Targeting TRAIL towards the clinic. Curr Drug Targets. 2011;12:2079-90
20. Mahalingam D, Szegezdi E, Keane M. et al. TRAIL receptor signalling and modulation: are we on the right TRAIL? Cancer Treat Rev. 2009;35:280-8
21. Srivastava RK. TRAIL/Apo-2L: mechanisms and clinical applications in cancer. Neoplasia. 2001;3:535-46
22. Suliman A, Lam A, Datta R. et al. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122-33
23. Zhang H, Xu Q, Krajewski S. et al. BAR: an apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc Natl Acad Sci U S A. 2000;97:2597-602
24. Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist Updat. 2004;7:139-56
25. Harsha Raj M, Yashaswini B, Rossler J. et al. Combinatorial treatment with anacardic acid followed by TRAIL augments induction of apoptosis in TRAIL resistant cancer cells by the regulation of p53, MAPK and NFkappabeta pathways. Apoptosis. 2016;21:578-93
26. Dyer MJ, MacFarlane M, Cohen GM. Barriers to effective TRAIL-targeted therapy of malignancy. J Clin Oncol. 2007;25:4505-6
27. MacFarlane M, Harper N, Snowden RT. et al. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene. 2002;21:6809-18
28. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417-21
29. Marsters SA, Sheridan JP, Pitti RM. et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003-6
30. Degli-Esposti MA, Smolak PJ, Walczak H. et al. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165-70
31. Degli-Esposti MA, Dougall WC, Smolak PJ. et al. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813-20
32. Ichikawa K, Liu W, Zhao L. et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954-60
33. Ishida I, Tomizuka K, Yoshida H. et al. Production of human monoclonal and polyclonal antibodies in TransChromo animals. Cloning Stem Cells. 2002;4:91-102
34. Pukac L, Kanakaraj P, Humphreys R. et al. HGS-ETR1, a fully human TRAIL-receptor 1 monoclonal antibody, induces cell death in multiple tumour types in vitro and in vivo. Br J Cancer. 2005;92:1430-41
35. Luster TA, Carrell JA, McCormick K. et al. Mapatumumab and lexatumumab induce apoptosis in TRAIL-R1 and TRAIL-R2 antibody-resistant NSCLC cell lines when treated in combination with bortezomib. Mol Cancer Ther. 2009;8:292-302
36. Hymowitz SG, O'Connell MP, Ultsch MH. et al. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry. 2000;39:633-40
37. Hymowitz SG, Christinger HW, Fuh G. et al. Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell. 1999;4:563-71
38. Pennarun B, Meijer A, de Vries EG. et al. Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys Acta. 2010;1805:123-40
39. Walczak H, Haas TL. Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol Biol. 2008;414:221-39
40. Falschlehner C, Emmerich CH, Gerlach B. et al. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462-75
41. Motoki K, Mori E, Matsumoto A. et al. Enhanced apoptosis and tumor regression induced by a direct agonist antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 2. Clin Cancer Res. 2005;11:3126-35
42. Graves JD, Kordich JJ, Huang TH. et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell. 2014;26:177-89
43. Jin A, Ozawa T, Tajiri K. et al. Generation of TRAIL-receptor 1-specific human monoclonal Ab by a combination of immunospot array assay on a chip and human Ab-producing mice. Eur J Immunol. 2010;40:3591-3
44. Jin A, Ozawa T, Tajiri K. et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat Med. 2009;15:1088-92
45. Hao Z, Han X, Sun X. et al. Fully human monoclonal antibodies to TRAIL-R1 enhance TRAIL-induced apoptosis via activation of caspase-8 pathway. Biochem Biophys Res Commun. 2016;475:238-44
46. Lin Z, Jin A, Ozawa T. et al. Post-translational modification of TRAIL receptor type 1 on various tumor cells and the susceptibility of tumors to TRAIL-induced apoptosis. Biochem Biophys Res Commun. 2010;395:251-7
47. Adams C, Totpal K, Lawrence D. et al. Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ. 2008;15:751-61
48. Piao X, Ozawa T, Hamana H. et al. TRAIL-receptor 1 IgM antibodies strongly induce apoptosis in human cancer cells in vitro and in vivo. Oncoimmunology. 2016;5:e1131380
49. Tuthill MH, Montinaro A, Zinngrebe J. et al. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene. 2015;34:2138-44
50. Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147-57
51. Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48:7022-32
52. Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645-68
53. Petrusic V, Zivkovic I, Stojanovic M. et al. Antigenic specificity and expression of a natural idiotope on human pentameric and hexameric IgM polymers. Immunol Res. 2011;51:97-107
54. Tchoudakova A, Hensel F, Murillo A. et al. High level expression of functional human IgMs in human PER.C6 cells. MAbs. 2009;1:163-71
55. Cao Y, Lan Y, Qian J. et al. Targeting cell surface beta2 -microglobulin by pentameric IgM antibodies. Br J Haematol. 2011;154:111-21
Author contact
![]() Corresponding author: Aishun Jin, Department of Immunology, Harbin Medical University, 157 Baojian Road, Nangang District, Harbin, Heilongjiang 150081, China. E-mail: aishunjinhrbmu.edu.cn or aishunjin53com; Tel.: 86-451-86674566; Fax: 86-451-86674566.
Corresponding author: Aishun Jin, Department of Immunology, Harbin Medical University, 157 Baojian Road, Nangang District, Harbin, Heilongjiang 150081, China. E-mail: aishunjinhrbmu.edu.cn or aishunjin53com; Tel.: 86-451-86674566; Fax: 86-451-86674566.