Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2575-2586. doi:10.7150/jca.19980 This issue Cite
Research Paper
Differentially Expressed lncRNAs in Gastric Cancer Patients: A Potential Biomarker for Gastric Cancer Prognosis
Xianglong Tian*, Xiaoqiang Zhu*, Tingting Yan, Chenyang Yu, Chaoqin Shen, Jie Hong ![]() , Haoyan Chen
, Haoyan Chen ![]() , Jing-Yuan Fang
, Jing-Yuan Fang ![]()
Division of Gastroenterology and Hepatology; Key Laboratory of Gastroenterology and Hepatology, Ministry of Health; State Key Laboratory for Oncogenes and Related Genes; Renji Hospital, School of Medicine, Shanghai JiaoTong University; Shanghai Institute of Digestive Disease; 145 Middle Shandong Road, Shanghai 200001, China.
*contributed equally
Received 2017-3-8; Accepted 2017-5-10; Published 2017-8-3
Abstract
Current studies indicate that long non-coding RNAs (lncRNAs) are frequently aberrantly expressed in cancers and implicated with prognosis in gastric cancer (GC). We intended to generate a multi-lncRNA signature to improve prognostic prediction of GC. By analyzing ten paired GC and adjacent normal mucosa tissues, 339 differentially expressed lncRNAs were identified as the candidate prognostic biomarkers in GC. Then we used LASSO Cox regression method to build a 12-lncRNA signature and validated it in another independent GEO dataset. An innovative 12-lncRNA signature was established, and it was significantly associated with the disease free survival (DFS) in the training dataset. By applying the 12-lncRNA signature, the training cohort patients could be categorized into high-risk or low-risk subgroup with significantly different DFS (HR = 4.52, 95%CI= 2.49-8.20, P < 0.0001). Similar results were obtained in another independent GEO dataset (HR=1.58, 95%CI=1.05 - 2.38, P=0.0270). Further analysis showed that the prognostic value of this 12-lncRNA signature was independent of AJCC stage and postoperative chemotherapy. Receiver operating characteristic (ROC) analysis showed that the area under receiver operating characteristic curve (AUC) of combined model reached 0.869. Additionally, a well-performed nomogram was constructed for clinicians. Moreover, single-sample gene-set enrichment analysis (ssGSEA) showed that a group of pathways related to drug resistance and cancer metastasis significantly enriched in the high risk patients. A useful innovative 12-lncRNA signature was established for prognostic evaluation of GC. It might complement clinicopathological features and facilitate personalized management of GC.
Keywords: gastric cancer, lncRNA, prognosis, survival, ssGSEA, nomogram.
Introduction
GC is a common and highly lethal malignancy, being the fourth most common cancer and the second leading cause of cancer death in the world [1]. Although the tendency of incidence rates declines, it is still concerned worldwide with the highest estimated mortality rates in Eastern Asian [2]. Surgery is the only curative treatment strategy and conventional chemotherapy has shown limited efficacy. Despite the recent therapeutic advances, the overall outcome of GC remains undesirable [3, 4]. For the risk stratification of GC, the TNM Staging System has been widely used, which is developed and maintained by American Joint Committee on Cancer (AJCC) and adopted by the Union International Committee on Cancer (UICC). Although TNM staging system is of great value clinically, it has not adequate prognostic and predictive capabilities to guide patient management [5, 6]. Thus, new biomarkers are needed to discriminate the high-risk patients with GC and consequently improve personalized cancer care.
Currently, substantial studies have focused on the roles of dysregulated functional long non-coding RNAs (lncRNAs) in human cancers [7, 8]. LncRNAs range from 200 nucleotides to multiple kilobases in length, but have no protein-coding capability [9]. The aberrant expression of lncRNAs is implicated in diverse cancers and some of them act as biomarkers for diagnosis and prognostication [10, 11]. Compared with single biomarker, integrating multiple biomarkers into a single model would be much better [12, 13], so it is of concrete predictive and prognostic value to identify a multi-lncRNA signature in GC.
Presently, a large group of lncRNA-specific probes were represented on the commonly used microarray platform (Affymetrix HG-U133 plus 2.0), we mined previously published gene expression microarray data from the Gene Expression Omnibus (GEO), and conducted lncRNA profiling on large cohorts of GC patients. The Cox proportional hazards regression analysis is one common approach to assess the prognostic factors in survival analysis, however, it is not suitable for high-dimensional microarray data when the ratio between sample size and variables is too low [14]. The least absolute shrinkage and selection operator method (LASSO) can conquer this limitation and has been widely adopted for optimal selection of prognostic genes [15-17]. By this way, we identified a 12-lncRNA signature in training set GSE62254 to predict survival probability for patients with GC. We validated it in another independent set GSE15459, and assessed the prognostic value and accuracy of this classifier in training set.
Material and methods
GC datasets preparation
GC gene expression data and corresponding clinical information data used in this study were obtained from the publicly available GEO (http://www.ncbi.nlm.nih.gov/geo/). The gene expression data were from the same chip platform (Affymetrix Human Genome U133 Plus 2.0 chips). Dataset GSE79973 including ten paired GC and normal mucosa tissues was used to identify the differentially expressed lncRNAs. First, the training set (GSE62254) was used to screen out the prognostic multi-lncRNA signature from the differentially expressed lncRNAs by LASSO Cox regression model. Then the GC samples in GSE15459 were analyzed as an independent validation set. After filtering out samples without clinical survival information, there were a total of 491 samples, including 300 from GSE62254, 191 from GSE15459 (Table S1). Supplementary Fig. 1 depicts the schematic diagram of work flow.
Microarray data processing and lncRNA profile mining
The raw CEL files were downloaded from GEO database and background adjusted using Robust Multichip Average (RMA) [18], which was a potent measure tool for lncRNA profiling data [19]. The lncRNA profile mining approach was mainly described by Zhang et al. [20]. First, the Affymetrix HG-U133 Plus 2.0 probe set IDs was mapped to the NetAffx Annotation Files. Second, based on the Refseq transcript ID and/or Ensembl gene ID, non-coding protein genes were extracted and were further filtered through excluding pseudogenes. Finally, we produced the 2448 lncRNA transcripts annotated with corresponding Affymetrix probe IDs.
Construction and assessment of the nomogram
The nomogram and calibration plots were generate using “rms” package of R software (version 3.3.1). Calibration was used to assess the performance of the nomogram. Nomogram-predicted survival and observed outcome were plotted on the x-axis and y-axis respectively, and the 45-degree line represented the best prediction. ROC analysis was also performed to estimate the predictive accuracy of the DFS nomogram using the “pROC” package of R software. Additionally, decision curve analysis (DCA) was also performed to assess the clinical utility of the nomogram. The DCA could be used to assess and compare prediction models which incorporated clinical consequences [21, 22]. The x-axis indicated the percentage of threshold probability, and the y-axis represented the net benefit.
Gene enrichment analysis
Single-sample gene-set enrichment analysis (ssGSEA) was performed to identify the differentially expressed gene sets between the low and high- risk cohorts. The enrichment score stands for the degree of absolute enrichment of a gene set in each sample within a certain dataset [23, 24]. Using GSVA package and its ssGSEA method (http://www.bioconductor.org), enrichment scores in each sample were calculated as the normalized difference in empirical cumulative distribution functions of gene expression ranks inside and outside the gene set [25]. The most significantly differentially expressed gene sets (p-value <0.001) were generated for further analysis.
Statistical analysis
We used the R software version 3.0.2 and the “glmnet” package (R Foundation for Statistical Computing, Vienna, Austria) to perform the LASSO Cox regression model analysis. The risk scores were calculated according to the formula generated through LASSO Cox regression model. Using the median risk score as the cutoff point, patients in each dataset were divided into low-risk or high-risk group correspondingly. For the outcome analysis, five-year recurrence was the primary endpoint in GSE62254 [26], and DFS was defined as the time of surgery to the first confirmed relapse; Overall survival (OS) was measured in GSE15459 [27]. Survival differences between the low-risk and high-risk groups in each dataset were assessed by the Kaplan-Meier estimator and the log-rank test. To test whether the risk score was independent of AJCC stage, or postoperative chemotherapy, we conducted multivariable Cox regression and stratification analysis. ROC analysis was introduced to assess the sensitivity and specificity of the survival prediction based on the risk score, AJCC stage, and the combined model of risk score and AJCC stage. To generate ROC curves, the patients whose durations were less than the median DFS needed to be excluded, if they still did not recur at last follow-up. The rest patients were classified as surviving either longer or shorter than the median DFS [28]. During all the statistical analysis, including the log-rank test, Cox regression analysis and ROC analysis, P value being less than 0.05 was defined as the significant difference.
Results
Identification of prognostic lncRNAs from the training dataset
By using “limma” package, we identified a set of 339 differentially expressed lncRNAs whose parameter p-value was less than 0.01 from dataset GSE79973 (Supplementary Fig. 2). We further analyzed those 339 genes by LASSO Cox regression model in the training dataset GSE62254 (Supplementary Fig. 3). Consequently, we identified the 12-lncRNA signature that was significantly correlated with DFS in GC patients. Table 1 showed a list of probes with their obtained coefficients which were derived from the LASSO analysis. The higher risk score indicated unfavorable prognosis in GC. Thus, the higher expression levels of seven genes with positive coefficients indicated (CHST9-AS1, TPT1-AS1, MIR100HG, LOC400043, LINC00340, LOC283174, LOC401093) meant higher risk score and accordingly worse outcome. The negative coefficients for the remaining five genes (ENSG00000251538, LOC100133985, Hs.93194, ENSG00000233236, ENSG00000229565) indicated that their higher levels of expression were associated with better prognosis.
LncRNAs significantly associated with the disease free survival in the test series patients (N=300)
| Probe | Gene symbol | Coefficient | Description | Expression profiles |
|---|---|---|---|---|
| 1560751_at | CHST9-AS1 | 0.1243 | CHST9 antisense RNA 1 (non-protein coding) | high expression in normal tissue |
| 1562801_at | ENSG00000251538 | -0.4656 | NA | high expression in normal tissue |
| 1563983_at | TPT1-AS1 | 0.2788 | TPT1 antisense RNA 1 (non-protein coding) | high expression in normal tissue |
| 225381_at | MIR100HG | 0.034 | mir-100-let-7a-2 cluster host gene (non-protein coding) | high expression in tumor tissue |
| 226582_at | LOC400043 | 0.1696 | competing endogenous RNAs | high expression in normal tissue |
| 229280_s_at | LINC00340 | 0.0243 | long intergenic non-protein coding RNA 340 | high expression in tumor tissue |
| 229734_at | LOC283174 | 0.0051 | NA | high expression in tumor tissue |
| 230325_at | LOC100133985 | -0.5749 | NA | high expression in normal tissue |
| 231694_at | Hs.93194 | -0.0659 | Apolipoprotein A-I | high expression in normal tissue |
| 232298_at | LOC401093 | 0.0008 | NA | high expression in tumor tissue |
| 235824_at | ENSG00000233236 | -1.3684 | NA | high expression in normal tissue |
| 238251_at | ENSG00000229565 | -0.0054 | NA | high expression in normal tissue |
The 12-lncRNA signature and patients' survival in the training dataset
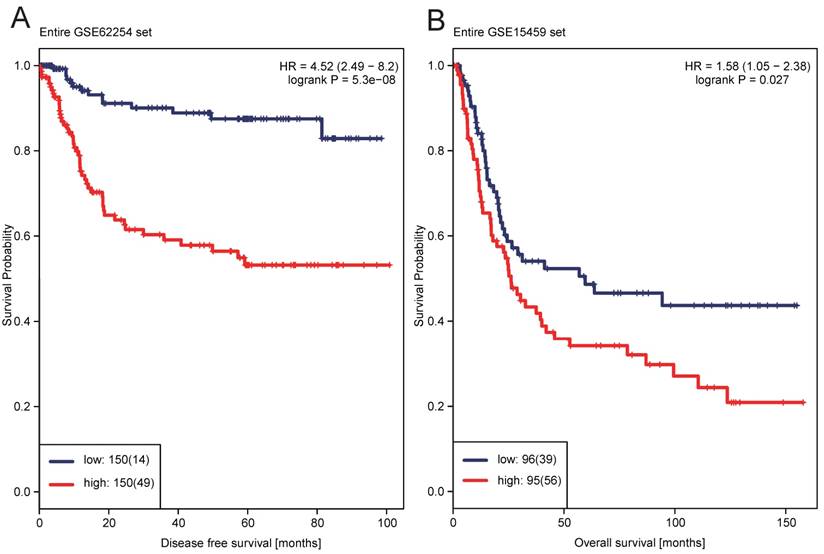
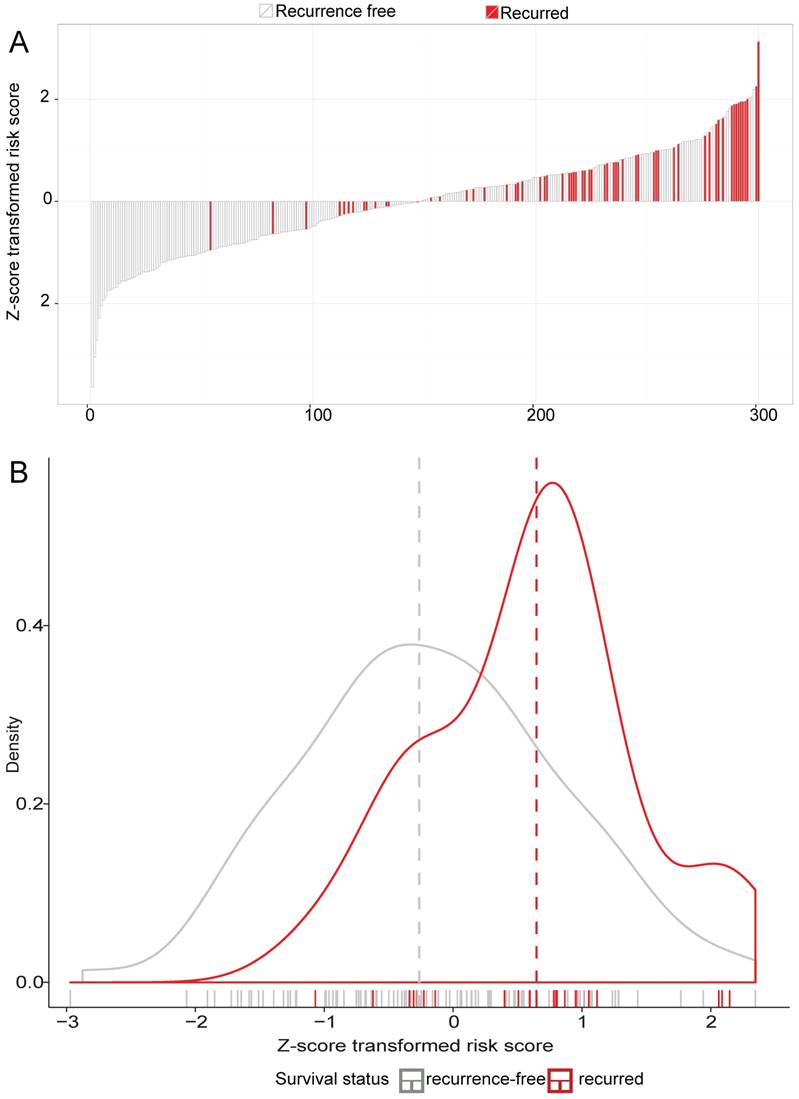
A risk-score formula was created according to the expression of these 12 lncRNAs for DFS prediction, as follows: Risk score = (0.1243*expression level of CHST9-AS1) + (-0.4656*expression level of ENSG00000251538) + (0.2788*expression level of TPT1-AS1) + (0.0340*expression level of MIR100HG) + (0.1696*expression level of LOC400043) + (0.0243*expression level of LINC00340) + (0.0051*expression level of LOC283174) + (-0.5749*expression level of LOC100133985) + (-0.0659*expression level of Hs.93194) + (0.0008*expression level of LOC401093) + (-1.3684*expression level of ENSG00000233236) + (-0.0054*expression level of ENSG00000229565). We then calculated the 12-lncRNA signature risk score for each patient in the training dataset GSE62254. The patients were classified into low-risk group (n=150) and high-risk group (n=150) using the median risk score as the cutoff point. Patients in the high-risk group had significantly shorter median DFS than those in the low-risk group (HR = 4.52, 95%CI= 2.49-8.20, P < 0.0001) (Figure 1A). The association of the 12-lncRNA risk score with DFS was also significant when it was assessed by the multivariable Cox regression model as a continuous variable (HR = 6.9340, 95%CI= 3.655-13.156, P < 0.0001) (Table 2). As shown in Figure 2A, there were apparently more recurred patients in high risk group, and the distribution of Z-score transformed risk score observably shifted to right in recurred patients compared with recurrence-free ones (Figure 2B).
Kaplan-Meier estimates of the patients' survival using the 12-lncRNA signature. The Kaplan-Meier plots were used to visualize the patients' survival probabilities for the low-risk versus high-risk group of patients based on the median risk score from corresponding GEO datasets patients. (A) Kaplan-Meier curves for GSE62254 training series patients (N=300); (B) Kaplan-Meier curves for GSE15459 patients (N=191). The tick marks on the Kaplan-Meier curves represent the censored subjects. The differences between the two curves were determined by the two-side log-rank test
Multivariable Cox regression analysis in each data set.
| GSE62254 training set(N=300) | GSE15459 set (N=191) | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI of HR | P value | HR | 95% CI of HR | P value | |
| 12-lncRNA risk score | 6.934 | 3.655 - 13.156 | < 0.0001 | 1.476 | 1.071 - 2.037 | 0.0175 |
| Age | 1.017 | 0.993 - 1.041 | 0.1719 | 1.014 | 0.998 - 1.031 | 0.0909 |
| Gender (male vs. female) | 1.027 | 0.600 - 1.757 | 0.9226 | 1.244 | 0.509 - 1.271 | 0.3506 |
| AJCC stages (II / III / IV vs. I) | ||||||
| Stage (I) | 1.00(referent) | 1.00(referent) | ||||
| Stage(II) | 1.059 | 0.228 - 4.922 | 0.9417 | 2.394 | 0.73 7 - 7.781 | 0.1465 |
| Stage(III) | 2.684 | 0.624 - 11.556 | 0.1849 | 9.294 | 3.267 - 26.435 | < 0.0001 |
| Stage(IV) | 5.515 | 1.274 - 23.873 | 0.0224 | 24.568 | 8.417 - 74.714 | < 0.0001 |
| Postoperative chemotherapy (yes vs. no) | 0.468 | 0.271 - 0.809 | 0.0065 | \ | \ | \ |
Abbreviations: HR, hazard ratio; CI, confidence interval.
Risk score and age were evaluated as continuous variables.
In GSE15459 set, there was no information available related to postoperative chemotherapy
LncRNA risk score analysis of GSE62254. The distribution of 12-lncRNA Z-score transformed risk score and patients' DFS status were analyzed in the GSE62254 series patients (N=300). (A) the distribution of patients' DFS status and time; (B) the density distribution of LncRNA Z-score transformed risk score
Validation of the 12-lncRNA signature for survival prediction in the validation dataset
To verify the ability of the 12-lncRNA signature in predicting the survival of GC patients, we further validated our findings in another independent dataset GSE15459, which yielded the similar results as above. Through the same risk score-based classification, patients were categorized into high-risk group (N=95) and low-risk group (N=96). Patients with GC in high-risk group had significantly shorter median OS than those in low-risk group (HR=1.58, 95%CI=1.05 - 2.38, P=0.0270) (Figure 1B). The multivariable Cox regression analysis showed that the 12-lncRNA risk score also had statistical significance as a continuous variable in the GSE15459 cohorts (HR=1.476, 95%CI=1.071 - 2.037, P=0.0175) (Table 2).
Prognostic value of the 12-lncRNA signature
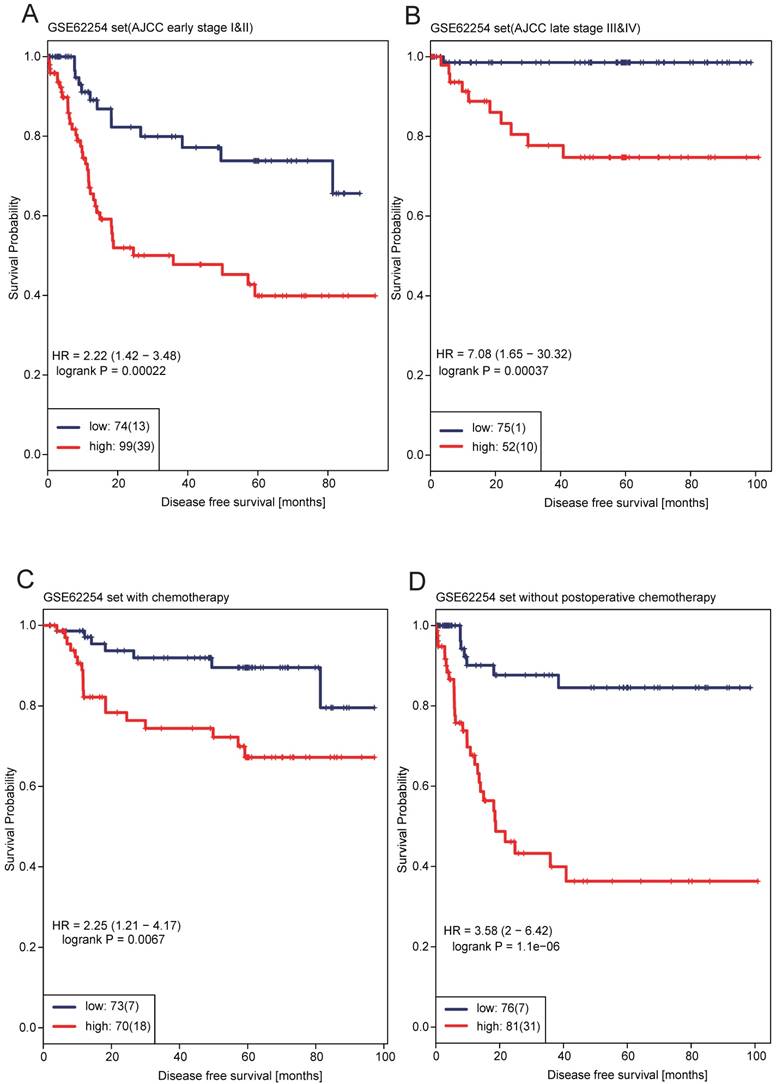
We performed multivariable Cox regression analysis in the two datasets. The 12-lncRNA risk score and other clinicopathological factors, including age, gender, AJCC stage and postoperative chemotherapy were used as covariates. It showed that even adjusted by AJCC stage and other covariates in each dataset, the 12-lncRNA risk score remained to be significantly associated with patients' survival (P < 0.0001 in GSE62254, P = 0.0175 in GSE15459) (Table 2). Consistent with risk score, AJCC stage was also significantly associated with patients' survival (Table 2). In order to test whether the prognostic value of the 12-lncRNA signature was independent of AJCC stage, stratification analysis was performed. Patients in dataset GSE62254 (N=300) were factitiously stratified into early stage stratum (stage I&II) and late stage stratum (stage III&IV). The results showed that 12-gene risk score remained the ability of predicting the prognosis within each stage stratum. Figure 3A showed that high-risk patients in early stage stratum had significantly shorter median DFS than low-risk patients (HR = 2.22, 95%CI= 1.42-3.48, P = 0.0002), patients in late stage stratum yielded similar results (HR = 7.08, 95%CI= 1.65-30.32, P = 0.0004) (Figure 3B), indicating that the prognostic value of 12-lncRNA signature was independent of AJCC stage. We also investigated whether the 12-lncRNA signature was independent of postoperative chemotherapy. The same approaches were adopted as above. Multivariable Cox regression analysis showed that postoperative chemotherapy was also significantly associated with DFS in dataset GSE62254 (HR = 0.468, 95%CI= 0.271-0.809, P = 0.0065) (Table 2), indicating that postoperative chemotherapy was a protective factor. Figure 3C showed that patients with postoperative chemotherapy in the low-risk group had significantly longer median DFS than high-risk patients (HR = 2.25, 95%CI= 1.21-4.17, P = 0.0067), similar results was generated in patients without postoperative chemotherapy (HR = 3.58, 95%CI= 2.00-6.42, P < 0.0001) (Figure 3D). The results indicated that regardless of the postoperative chemotherapy status, the 12-lncRNA signature could discriminate high risk GC patients from low risk ones.
Kaplan-Meier survival analysis to evaluate the independence of the 12-lncRNA signature from AJCC stage and postoperative chemotherapy. The patients from GSE62254 were stratified into four subgroups based on AJCC stage or postoperative chemotherapy. The 12-lncRNA signature was applied to early-stage patients (A), late-stage patients (B), the patients with postoperative chemotherapy (C) or the patients without postoperative chemotherapy (D) separately
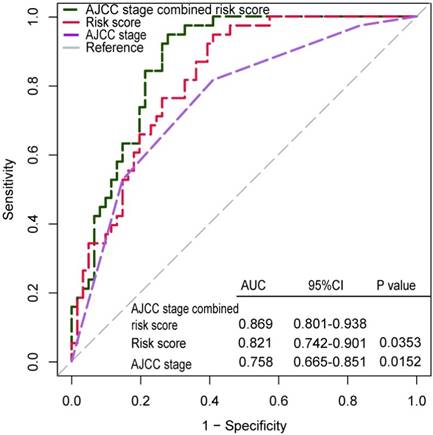
Additionally, ROC analysis was performed to demonstrate the sensitivity and specificity of survival prediction. AUC was evaluated and compared between the 12-lncRNA risk score model and AJCC stage. As shown in Figure 4, both AJCC stage and 12-lncRNA risk score model owned valuable predicted power to estimate the prognosis of GC patients, and there was no significant difference between them. If combined the 12-lncRNA risk score model with AJCC stage together, the AUC of combined model was significantly greater than that of AJCC stage alone (0.869 versus 0.758, 95%CI: 0.665-0.851, P =0.0152).
Receiver operating characteristic (ROC) analysis of the sensitivity and specificity of the survival prediction by the 12-lncRNA risk score, AJCC stage in GSE62254 dataset patients (N=300). P values were from the comparisons of the area under the ROC (AUC) of 12-lncRNA risk score combined with AJCC stage versus AUC of 12-lncRNA risk score or AJCC stage separately
Clinical utility of the 12-lncRNA signature
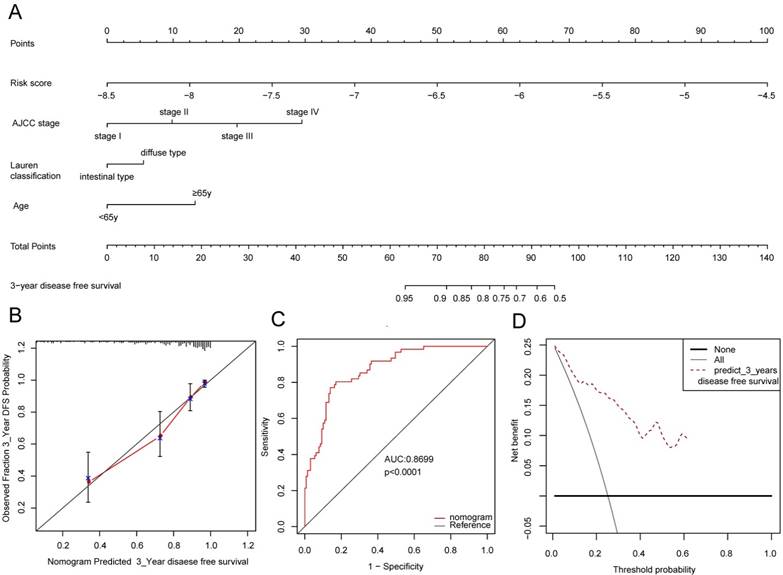
In order to provide a quantitative method for the clinicians to predict the probability of 3-year DFS in GC, a nomogram was constructed in GSE62254 dataset which integrated the 12-lncRNA signature, age, tumor stage and the Lauren classification (Figure 5A). Figure 5B showed that the nomogram did well in the calibration plots compared with the ideal model. ROC analysis was performed to calculate the predictive accuracy of the nomogram, and the AUC of nomogram is 0.8699(Figure 5C). DCA was introduced to estimate the clinical utility of the nomogram, and it performed well as shown in Figure 5D.
The nomogram to predict 3-year DFS in GSE62254. (A) The nomogram for predicting proportion of patients with 3-year DFS. (B) The calibration plots for predicting patient 3-year DFS. Nomogram-predicted probability of survival is plotted on the x-axis; actual survival is plotted on the y-axis. (C) ROC curve based on the nomogram for 3-year DFS probability. (D) Decision curve analysis (DCA) for assessment of the clinical utility of the nomogram. The x-axis represents the percentage of threshold probability, and the y-axis represents the net benefit
Identification of 12-lncRNA signature associated biological pathways
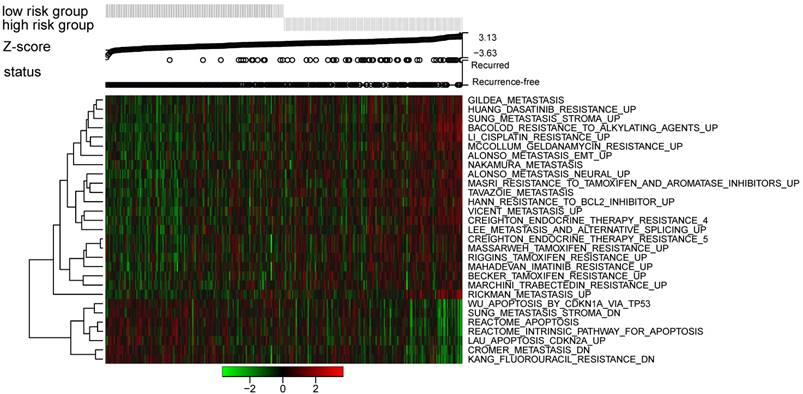
To identify the 12-lncRNA associated pathways, we performed ssGSEA to analyze the GSE62254 dataset using risk score for classification. As shown in Figure 6, a group of pathways related to drug resistance and cancer metastasis significantly enriched in the high risk patients; however, some apoptosis-related pathways were up-regulated in the low risk cohorts. These pathways were found to be significantly associated with the risk score, which was validated through Pearson's correlation analysis (Figure 7A). As cancer metastasis is an important factor that exerts influence on the disease occurrence and patients' prognosis [29], the risk scores were further analyzed in patients with and without distant metastases in GSE62254 dataset (TNM stage IV patients were considered to be with distant metastases). In accordance with the results above, patients were inclined to get higher risk scores in the cohorts with distant metastases compared to the ones without distant metastases (Figure 7B).
Pathway profiles across dataset GSE62254. Rows represent pathways, and columns represent patients. Each grid represents a score of pathway activity calculated by single-sample GSEA. No further adjustment of the ssGSEA score was performed. The upper horizontal bar marked the information related to every patient, including its risk group, risk score (ranked from low to high), and the DFS status)
Correlation analysis of risk score, pathways and distant metastases across dataset GSE62254. Pearson's correlation analysis of risk score and pathways (A), distribution of risk score based on the status of distant metastases (B)

Discussion
Numerous reports indicate that dysregulated lncRNA expression may be implicated in various aspects of tumor, including carcinogenesis, progression, and metastasis [30-32]. Some lncRNAs are considered to be useful biomarkers to predict prognosis in GC patients, such as HULC and LINC00668 [11, 33]. However, several limits are still concerned including small number of lncRNAs screened, inadequate samples, and lack of independent validation, the reliability and utility of prognostic predication in GC need further investigation. To establish a prognostic multi-lncRNA signature, we mined the existing microarray gene expression data to profile lncRNAs. In our study, LASSO analysis was introduced, which was a popular tool for regression with high-dimensional predictors [17]. By exploring the relevance between lncRNA expression profiles and clinical outcome of GC patients in dataset GSE62254, we constructed a 12-lncRNA signature that was significantly associated with patients' DFS.
In this study, a novel prognostic 12-lncRNA signature was developed and validated to improve the ability of predicting prognosis for GC patients. Our results revealed that this classifier could successfully classify GC patients into high-risk and low-risk groups with significant differences in DFS in training set. The prognostic value of this 12-lncRNA signature could be verified in another independent dataset GSE15459, indicating the reproducibility and utility of this multi-lncRNA signature for the prognostic prediction in GC.
Stratification analysis revealed that prognostic power of this 12-lncRNA signature was independent of AJCC stage, which was currently the most important prognostic factor for GC. AJCC staging system could provide effective prognostic information and contribute to the selection of proper therapeutic regimen. Our study revealed that AJCC stage was a strong prognostic factor through the multivariable Cox regression analysis, which was consistent with previous studies [34, 35]. Therefore, it was necessary to further evaluate whether the prognostic value of 12-lncRNA signature was independent of AJCC stage. The patients were divided into early stage and late stage stratums factitiously to facilitate analysis. The 12-lncRNA signature could successfully divide the stratified patients into low risk and high risk subgroups in GSE62254, and there was a clear separation in the survival curves between them. Based on these results, we could conclude that the prognostic value of the 12-lncRNA signature was independent of AJCC stage in our study.
Moreover, 12-lncRNAs risk model remained strong prognostic ability when stratified by postoperative chemotherapy. In Asian countries, postoperative chemotherapy have been extensively used as the standard treatment, and two import clinic trials showed that patients could benefit from postoperative chemotherapy with prolonged survival [36, 37]. It was consistent with our results of multivariable Cox regression analysis. Further stratification analysis demonstrated that the 12-lncRNA signature could also allow a discrimination of GC patients' prognosis, having nothing with its postoperative chemotherapy stratum. This further demonstrated that the 12-lncRNA signature might be an independent prognostic factor for GC.
In order to assess the predictive ability of the 12-lncRNA signature, ROC analysis was performed. An AUC was used as a measure of the accuracy in diagnostic test [38]. ROC analysis revealed that the 12-lncRNA signature had a similar survival predictive power as AJCC stage. Interestingly, the prognostic power was superior to AJCC stage alone when we combined 12-lncRNAs risk model with AJCC stage together. Moreover, the AUC of combined model reached 0.869, indicating it might complement clinicopathological features and improve the accuracy of prognostic prediction in GC.
As the 12-lncRNA signature could discriminate the patients with high risk of recurrence from GC patients, we hypothesized that this gene signature might be associated with some signaling pathways that could impact the prognosis of GC. Currently, cancer metastasis and drug resistance were the main challenges in clinical practice and badly affected patients' prognosis [29, 39]. Interestingly, according to the results of ssGSEA, these pathways were highly enriched in the high risk group. Furthermore, our finding showed that the risk score was closely related to these pathways, providing some insight into the molecular mechanisms that underlie the pathological process and cancer progression in GC.
Our study has showed that the 12-lncRNA signature was strongly associated with the prognosis of GC. However, the biological functions of 12 lncRNAs have not been clarified completely in GC. Some of the lncRNAs used in our signature have been reported in previous studies. MIR100HG acted as regulators of hematopoiesis and oncogenes in myeloid leukemia [40]. LOC400043 was one of “miRNA sponges”, it controlled several biological functions via sequestering miR-28-3p and miR-96-5p [41]. Interestingly, LINC00340 was found to act both as a tumor suppressor and pro-metastasis factor in cancer [42, 43]. The reports with respect to the other lncRNAs have been extremely rare, further researches about the biological functions of the lncRNAs are needed. In our study, MIR100HG, LINC00340, LOC283174, LOC401093 are up-regulated in GC samples compared with their paired normal tissues and correlated with shorter survival, indicating a detrimental role in GC biogenesis. Contrariwise, ENSG00000251538, LOC100133985, Hs.93194, ENSG00000233236, ENSG00000229565 are down-regulated in GC and might be protective factors. There may be some biases in the course of selecting prognostic multi-lncRNA signature from view of biogenesis; however, because of its strong relevance with prognosis, the roles of these genes deserve further investigations, especially in GC.
Some limitations in our study need to be acknowledged. First, the primary endpoints in the two datasets are not exactly the same. DFS was used in the training set, but OS in the validation set, so the robustness of the 12-lncRNA signature on prognostic prediction requires further validation in large prospective clinic trials. Second, we have no experimental data about the lncRNAs in the signature, and most of which have been rarely reported, so it is in need of more evidence to elucidate the inherent association between the 12-lncRNA signature and prognosis in GC. Despite these drawbacks, our findings still showed the significant and consistent correlation of the 12-lncRNA signature with OS in independent dataset, implying it is a useful prognostic biomarker for GC.
In conclusion, we have generated an innovative prognostic 12-lncRNA signature in GC. It might complement clinicopathological features and facilitate personalized management of GC. In future, large-scale prospective researches are needed to further assess the robustness of this signature before clinical application, and the underlying biological mechanisms associated with this signature warrant further study.
Abbreviations
AJCC: American Joint Committee on Cancer; AUC: area under receiver operating characteristic. CI: confidence interval; DCA: decision curve analysis; DFS: disease free survival; GC: gastric cancer; GEO: Gene Expression Ominus; HR: hazard ratio; lincRNA: large intergenic non-coding RNAs; lncRNAs: long non-coding RNAs; ncRNAs: non-coding RNAs; OS: overall survival; ROC: receiver operating characteristic; ssGSEA: single-sample gene-set enrichment analysis; UICC: Union International Committee on Cancer.
Supplementary Material
Supplementary figures.
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (Grant No: 81421001, 81320108024, 31371420, 81530072, 81402347 and 31371273); The Program for Professor of Special Appointment (2015 Youth Eastern Scholar NO. QD2015003) at Shanghai Institutions of Higher Learning; Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (NO.20161309); and Chenxing Project of Shanghai Jiao-Tong University to H. Chen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends-An Update. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2016;25:16-27
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015;136:E359-86
3. Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. Journal of the National Comprehensive Cancer Network: JNCCN. 2010;8:437-47
4. Jung KW, Won YJ, Kong HJ, Oh CM, Shin A, Lee JS. Survival of korean adult cancer patients by stage at diagnosis, 2006-2010: national cancer registry study. Cancer research and treatment: official journal of Korean Cancer Association. 2013;45:162-71
5. Marano L, Boccardi V, Braccio B, Esposito G, Grassia M, Petrillo M. et al. Comparison of the 6th and 7th editions of the AJCC/UICC TNM staging system for gastric cancer focusing on the "N" parameter-related survival: the monoinstitutional NodUs Italian study. World journal of surgical oncology. 2015;13:215
6. Kim BS, Park YS, Yook JH, Kim BS. Comparison of the prognostic values of the 2010 WHO classification, AJCC 7th edition, and ENETS classification of gastric neuroendocrine tumors. Medicine. 2016;95:e3977
7. Mitra SA, Mitra AP, Triche TJ. A central role for long non-coding RNA in cancer. Frontiers in genetics. 2012;3:17
8. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 2009;10:155-9
9. Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nature reviews Drug discovery. 2013;12:433-46
10. Mehra R, Udager AM, Ahearn TU, Cao X, Feng FY, Loda M. et al. Overexpression of the Long Non-coding RNA SChLAP1 Independently Predicts Lethal Prostate Cancer. European urology. 2015
11. Jin C, Shi W, Wang F, Shen X, Qi J, Cong H. et al. Long non-coding RNA HULC as a novel serum biomarker for diagnosis and prognosis prediction of gastric cancer. Oncotarget. 2016
12. Venook AP, Niedzwiecki D, Lopatin M, Ye X, Lee M, Friedman PN. et al. Biologic determinants of tumor recurrence in stage II colon cancer: validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1775-81
13. Agesen TH, Sveen A, Merok MA, Lind GE, Nesbakken A, Skotheim RI. et al. ColoGuideEx: a robust gene classifier specific for stage II colorectal cancer prognosis. Gut. 2012;61:1560-7
14. Simon R, Altman DG. Statistical aspects of prognostic factor studies in oncology. British journal of cancer. 1994;69:979-85
15. Zhang JX, Song W, Chen ZH, Wei JH, Liao YJ, Lei J. et al. Prognostic and predictive value of a microRNA signature in stage II colon cancer: a microRNA expression analysis. The Lancet Oncology. 2013;14:1295-306
16. Gui J, Li H. Penalized Cox regression analysis in the high-dimensional and low-sample size settings, with applications to microarray gene expression data. Bioinformatics. 2005;21:3001-8
17. Tibshirani R. The lasso method for variable selection in the Cox model. Statistics in medicine. 1997;16:385-95
18. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic acids research. 2003;31:e15
19. Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX. et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679-89
20. Zhang X, Sun S, Pu JK, Tsang AC, Lee D, Man VO. et al. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiology of disease. 2012;48:1-8
21. Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Medical decision making: an international journal of the Society for Medical Decision Making. 2006;26:565-74
22. Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC medical informatics and decision making. 2008;8:53
23. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545-50
24. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12
25. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics. 2013;14:7
26. Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nature medicine. 2015;21:449-56
27. Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J. et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS genetics. 2009;5:e1000676
28. Kang J, D'Andrea AD, Kozono D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. Journal of the National Cancer Institute. 2012;104:670-81
29. Chau I, Norman AR, Cunningham D, Waters JS, Oates J, Ross PJ. Multivariate prognostic factor analysis in locally advanced and metastatic esophago-gastric cancer-pooled analysis from three multicenter, randomized, controlled trials using individual patient data. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:2395-403
30. Sas-Chen A, Aure MR, Leibovich L, Carvalho S, Enuka Y, Korner C. et al. LIMT is a novel metastasis inhibiting lncRNA suppressed by EGF and downregulated in aggressive breast cancer. EMBO molecular medicine. 2016
31. Ma HW, Xie M, Sun M, Chen TY, Jin RR, Ma TS. et al. The pseudogene derived long noncoding RNA DUXAP8 promotes gastric cancer cell proliferation and migration via epigenetically silencing PLEKHO1 expression. Oncotarget. 2016
32. Zhou T, Pan F, Cao Y, Han Y, Zhao J, Sun H. et al. R152C DNA Pol beta mutation impairs base excision repair and induces cellular transformation. Oncotarget. 2016;7:6902-15
33. Zhang E, Yin D, Han L, He X, Si X, Chen W. et al. E2F1-induced upregulation of long noncoding RNA LINC00668 predicts a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically silencing of CKIs. Oncotarget. 2016;7:23212-26
34. Marrelli D, Morgagni P, de Manzoni G, Coniglio A, Marchet A, Saragoni L. et al. Prognostic value of the 7th AJCC/UICC TNM classification of noncardia gastric cancer: analysis of a large series from specialized Western centers. Annals of surgery. 2012;255:486-91
35. Kawaguchi T, Komatsu S, Ichikawa D, Kubota T, Okamoto K, Shiozaki A. et al. Comparison of prognostic compatibility between seventh AJCC/TNM of the esophagus and 14th JCGC staging systems in Siewert type II adenocarcinoma. Anticancer research. 2013;33:3461-5
36. Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A. et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. The New England journal of medicine. 2007;357:1810-20
37. Bang YJ, Kim YW, Yang HK, Chung HC, Park YK, Lee KH. et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet. 2012;379:315-21
38. Bunger R, Mallet RT. Metabolomics and Receiver Operating Characteristic Analysis: A Promising Approach for Sepsis Diagnosis. Critical care medicine. 2016;44:1784-5
39. Wicki A, Mandala M, Massi D, Taverna D, Tang H, Hemmings BA. et al. Acquired Resistance to Clinical Cancer Therapy: A Twist in Physiological Signaling. Physiological reviews. 2016;96:805-29
40. Emmrich S, Streltsov A, Schmidt F, Thangapandi VR, Reinhardt D, Klusmann JH. LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Molecular cancer. 2014;13:171
41. Militello G, Weirick T, John D, Doring C, Dimmeler S, Uchida S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Briefings in bioinformatics. 2016
42. Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M. et al. CASC15-S Is a Tumor Suppressor lncRNA at the 6p22 Neuroblastoma Susceptibility Locus. Cancer research. 2015;75:3155-66
43. Lessard L, Liu M, Marzese DM, Wang H, Chong K, Kawas N. et al. The CASC15 Long Intergenic Noncoding RNA Locus Is Involved in Melanoma Progression and Phenotype Switching. The Journal of investigative dermatology. 2015;135:2464-74
Author contact
![]() Corresponding authors: Jie Hong: jiehong97edu.cn; Haoyan Chen:haoyanchenedu.cn; Jing-Yuan Fang: jingyuanfangedu.cn
Corresponding authors: Jie Hong: jiehong97edu.cn; Haoyan Chen:haoyanchenedu.cn; Jing-Yuan Fang: jingyuanfangedu.cn