Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2636-2642. doi:10.7150/jca.19873 This issue Cite
Short Research Communication
Metformin Sensitizes Leukemia Cells to Vincristine via Activation of AMP-activated Protein Kinase
Yong Yi1#, Linfeng Gao1#, Min Wu1,3#, Juan Ao1, Chunyan Zhang1, Xiaodong Wang2, Min Lin2, Johann Bergholz4, Yujun Zhang1, Zhi-Xiong Jim Xiao1 ![]()
1. Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment, Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, 610064. China
2. Hematology Department, Outpatient Department, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, Chengdu, 610072, China
3. Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing, 400038, China
4. Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
# These authors contributed equally to this work
Received 2017-3-1; Accepted 2017-6-30; Published 2017-8-21
Abstract
Vincristine is extensively used chemotherapeutic medicine to treat leukemia. However, it remains a critical clinical problem with regard to its toxicity and drug-resistance. AMP-activated protein kinase (AMPK) is an energy sensor that is pivotal in maintaining cell metabolic homeostasis. It is reported that AMPK is involved in vincristine-induced apoptosis. However, whether AMPK is involved in chemotherapy-resistance is largely unclear. It is well-documented that metformin, a widely used medicine to treat type II diabetes, possesses anti-cancer activities, yet whether metformin affects leukemia cell viability via vincristine is unknown. In this study, we showed that both AMPKα1 mRNA and phosphorylated AMPK protein levels were significantly decreased in clinical leukemia samples. We further demonstrated that metformin sensitized leukemia cells to vincristine-induced apoptosis in an AMPK-dependent manner. In addition, knockdown of AMPKα1 significantly reduced the effects of metformin on vincristine-induced apoptosis. Taken together, these results indicate that AMPK activation is critical in metformin effects on vincristine-induced apoptosis and suggest a putative strategy of a combination therapy using metformin and vincristine in treatment of leukemia.
Keywords: Leukemia, Vincristine, Metformin, AMP-activated Protein Kinase, Apoptosis, Combination Therapy
Introduction
Leukemia is a group of cancers that arise from the bone marrow and results in abnormal white blood cell proliferation and survival. In 2014, world health organization reported that leukemia developed in 352,000 people globally and caused 265,000 deaths. Chemotherapy is the primary treatment for leukemia. Vincristine (VCR) is a well-established cytotoxic drug extensively used in the treatment of leukemia [1]. Vincristine binds specifically to tubulin, which leads to microtubule depolymerization, cell cycle arrest and apoptosis in mitotic cells [2]. At the molecular level, vincristine has been shown to regulate p53, p21, Fas/Fas ligand and c-Myc expression [3, 4]. Moreover, vincristine inhibits NF-κB signaling [5] and JNK activity [3]. However, it remains a clinical problem for vincristine with regard to its toxicity and drug -resistance [6, 7].
Metformin is widely used to treat type II diabetes. It dramatically decreases blood glucose levels and enhances cell sensitivity to insulin [8]. It has been reported that metformin inhibits the mitochondrial respiratory chain (complex I), which leads to AMP-activated protein kinase activation [9]. In recent years, clinical data have indicated that metformin reduces the cancer incidence in patients with type II diabetes [10]. Metformin inhibits cancer cell proliferation, survival [11] and promotes cellular senescence [12]. Moreover, metformin has been shown to enhance chemotherapeutic drugs-induced apoptosis [13-15]. At the molecular level, metformin modulates various key proteins, including mTOR, p53 and SIRT 1 [16], as well as down-regulation of Her2 and cyclin D1 expression [17, 18].
AMP-activated protein kinase (AMPK) functions as an energy sensor, and plays an important role in maintaining cell metabolic homeostasis. AMPK is a heterotrimer that contains a α-catalytic subunit, a β-subunit, and a γ-subunit [19]. Metabolic stresses, such as glucose starvation and hypoxia, activate AMPK via threonine 172 phosphorylation by AMPK upstream kinases, including LKB1 and Calcium/calmodulin-dependent protein kinase kinase (CaMKK) [20, 21]. Upon activation, AMPK regulates a subset of downstream targets, including acetyl-CoA carboxylase (ACC) and mTOR [22, 23]. It is reported that AMPK plays a role in regulating growth and survival of multiple cancer cells including leukemia cells [24-26]. However, it remains unclear whether AMPK expression and/or activity is altered in leukemia cells and whether AMPK is involved in leukemia cell resistance to vincristine.
In this study, we show that metformin sensitizes leukemia cells to vincristine-induced apoptosis. Moreover, we found that AMPK plays a critical role in metformin-mediated enhancements of leukemia cell sensitivity to vincristine. Thus, this study suggests a new strategy of combination therapy using vincristine and metformin for treating leukemia.
Materials and Methods
Collection of patient samples
Leukemia patient blood samples (n=20) and healthy individuals (n=7) were collected at the Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, according to institutional regulation of Hospital Clinic Ethical Committee, and examined by pathologists. The blood samples were separated to obtain peripheral blood mononuclear cells (PBMCs) using a cell isolator (Ficoll-PaqueTM Plus, GE), which were subjected to Western blot analyses for phosphorylated AMPK.
Cell culture and drug treatment
Human leukemia chronic myelogenous leukemia K562 and T-Cell acute lymphoblastic leukemia A301 cells were grown in RPMI-1640 medium (GIBCO) supplemented with 10% fetal bovine serum (FBS; HyClone), 100 units/mL penicillin and 100 μg/mL streptomycin. Cells were maintained in a humidified 37°C incubator under a 5% CO2 atmosphere. Cells at 75-85% confluence were treated with vincristine (VCR, 1714018, Sigma) and metformin, as indicated (Met, PHR1084, Sigma).
Lentiviral infection and RNA interference
Lentiviruses were amplified by HEK 293T cells transfected with psPAX2 and pMD2.G packaging plasmids and lentiviral-based shRNAs specific for green fluorescent protein (GFP) (GAAGCAGCACGACTTCTTC) or AMPKα1 (GTATGATGTCAGATGGTGAATT) plasmids using Lipofectamine 2000. Viruses were collected at 60 hours after transfection. Cells at 50% confluence were infected with recombinant lentiviruses or empty vectors in the presence of 1 μg/mL polybrene before being subjected to 12 hours of incubation at 37°C with 5% CO2.
Western blot analysis
Cells were collected, washed with cold PBS, and resuspended in EBC250 lysis buffer (250 mM NaCl, 50 mM Tris [pH 8.0], 0.5% Nonidet P-40, 50 mM NaF, 0.5 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μg/mL aprotinin, and 2 μg/mL leupeptin). Equal amounts of protein were loaded onto gels, separated by SDS-PAGE, transferred to PVDF membranes (Millipore), and hybridized to the appropriate primary antibodies and HRP-conjugated secondary antibodies for subsequent detection via enhanced chemiluminescence. Antibody directed against actin (sc-8431) was purchased from Santa Cruz Biotech (CA, USA). Antibodies directed against phospho-AMPK (Thr172) (2535), AMPKα (2532) and PARP1 (9532) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell viability and FACS analyses
Cell viability was assessed by MTS assay using a CellTiter 96 Kit (Promega, USA) according to the manufacturer's instructions. Trypan blue exclusion assay (C0011, Beyotime, China) were performed according to the manufacturer's instructions. The percentages of viable cells were measured by Countess II (Life technologies, USA). For FACS analyses, cells were grown to approximately 80% confluence in 6-well plates, washed with cold PBS and then fixed in 70% ethanol at 4°C overnight. The cells were subsequently stained with 50 μg/mL propidium iodide (PI) supplemented with 80 μg/mL RNase A at 37°C in dark for 1 h. The cells were then subjected to FACS analyses using a FACSCalibur Low Cytometer (Becton Dickson).
Bioinformatic analysis of gene expression
Oncomine microarray database (Compendia Bioscience, Ann Arbor, MI, USA) was used to analyze AMPKα1 mRNA levels in human leukemia samples and visualized.
Statistical analysis
Quantitative data were analyzed using Student's t-test to determine significance. The data are presented as the mean ± SEM., as noted in the figure legends.
Results
AMPKα1 mRNA expression and phosphorylated AMPK protein are decreased in clinical leukemia samples
Deregulated cellular energetics is a characteristic of cancer cells. AMPK is a critical energy sensor and plays a pivotal role in energy homeostasis. Previous clinical studies have shown that AMPK activity is dramatically reduced in breast cancer [27]. To assess the role of AMPK in leukemia development, we first analyzed AMPK expression status in leukemia patients using Oncomine microarray database. As shown in Figure 1A, AMPKα1 mRNA levels were significantly decreased in T/B-cell acute lymphoblastic leukemia compared to normal bone marrow. We then examined levels of phosphorylated AMPK (pAMPK-T172) in normal (n=7) PBMC (peripheral blood mononuclear cells) and leukemia PBMC (n=20) by western blot analyses. The relative expression of phosphorylated AMPK over actin was plotted (Figure 1B), indicating significantly reduced AMPK phosphorylation in majority of leukemia PBMC.
Down-regulation of AMPK expression and activity in clinical leukemia samples and the effects of metformin or VCR on leukemia cell viability. (A) Oncomine analysis of Adersson leukemia data set is presented for AMPKα1 mRNA expression in clinical samples from patients with T-cell/B-cell acute lymphoblastic leukemia. (B) AMPK protein phosphorylation (pT172) in peripheral blood mononuclear cells (PBMC) from normal individuals or from leukemia patients were examined by Western blot analyses and quantified using Image Lab Analysis software. Actin protein levels in each sample were used as an internal control for normalization.(C-D) K562 or A301 cells were treated with 5 mM metformin for 0, 6, 12 or 24 hours. Western blot analyses were performed as indicated. (E-F) K562 cells were treated with metformin (0, 5, 10, 15, 20 or 25 mM) for 24 hours. Cell viability was assessed either by MTS assay (G) or by trypan blue exclusion assay (H). (G-H) A301 cells were treated with metformin (0, 5, 10, 15, 20 or 25 mM) for 24 hours. Cell viability was assessed either by MTS assay (G) or by trypan blue exclusion assay (H). (I-J) K562 cells were treated with VCR (0, 0.2, 0.4, 1.6 or 6.4 μM) for 24 hours. Cell viability was assessed either by MTS assay (G) or by trypan blue exclusion assay (H). (K-L) A301 cells were treated with VCR (0, 0.2, 0.4, 1.6 or 6.4 μM) for 24 hours. Cell viability was assessed either by MTS assay (G) or by trypan blue exclusion assay (H). The results are presented as the mean ± SEM from three independent experiments.
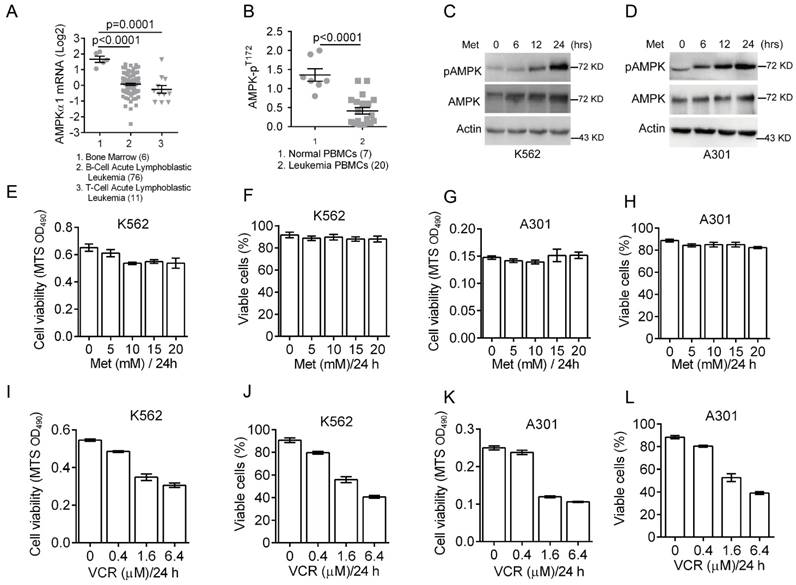
The effects of metformin or vincristine on viability of leukemia cells
The abovementioned data indicate that both AMPKα1 mRNA levels and AMPK phosphorylation are down-regulated in leukemia cells. To determine the role of AMPK activation in leukemia cell growth and survival, we examined the effects of metformin on AMPK activation in leukemia K562 and A301 cells. As shown in Figure 1C-1D, metformin significantly induced T172 phosphorylation of AMPK in both K562 and A301 cells in a time-dependent manner. Next, we examined cell viability after treatment with metformin (0, 5, 10, 15 and 20 mM) for 24 hours. As shown in Figure 1E-1H, metformin did not appear to significantly affect viability of K562 and A301 cells. Since vincristine, a first-line chemotherapy drug widely used to treat leukemia patients, we then examined effects of vincristine on cell viability. As shown in Figure 1I-1L, low concentration of vincristine (0.4 μM) had only a marginal effect on K562 and A301 cell viability, while increased dose of vincristine (beginning at 1.6 μM) significantly reduced K562 and A301 cell viability.
Metformin sensitizes leukemia cells to VCR-induced apoptosis
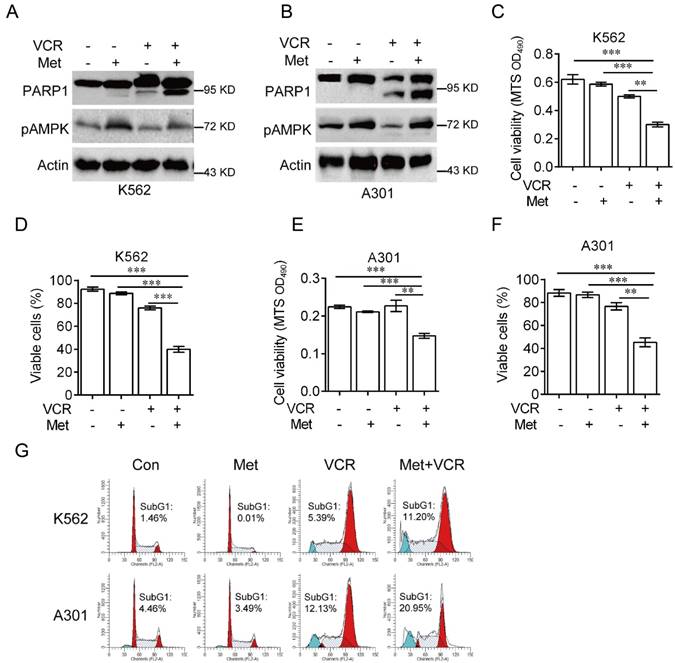
It has been reported that AMPK plays a role in vincristine-induced apoptosis in melanoma cells [28]. To investigate the role of AMPK in vincristine-induced leukemia cell apoptosis, K562 and A301 cells were treated with 5 mM metformin in the presence or absence of 0.4 μM vincristine for 24 hours. As shown in Figure 2, combination of metformin and vincristine, but not metformin or vincristine alone, significantly induced apoptosis (Figure 2A-2B), which was further supported by subsequent MTS or Trypan blue exclusion assays for cell viability (Figure 2C-2F) and FACS analyses for subG1 population (Figure 2G).
Metformin sensitizes leukemia cells to VCR-induced apoptosis. K562 or A301 cells were treated with or without 5 mM metformin in the presence or absence of 0.4 μM VCR for 24 h prior to Western blot analyses (A-B), MTS assay or trypan blue exclusion assay for cell viability (C-F) and FACS analyses for subG1 cell population (G). The data are presented as the mean ± SEM from three independent experiments (***P < 0.001; **P < 0.01).
Silencing AMPKα1 desensitizes leukemia cells to VCR-induced apoptosis enhanced by metformin
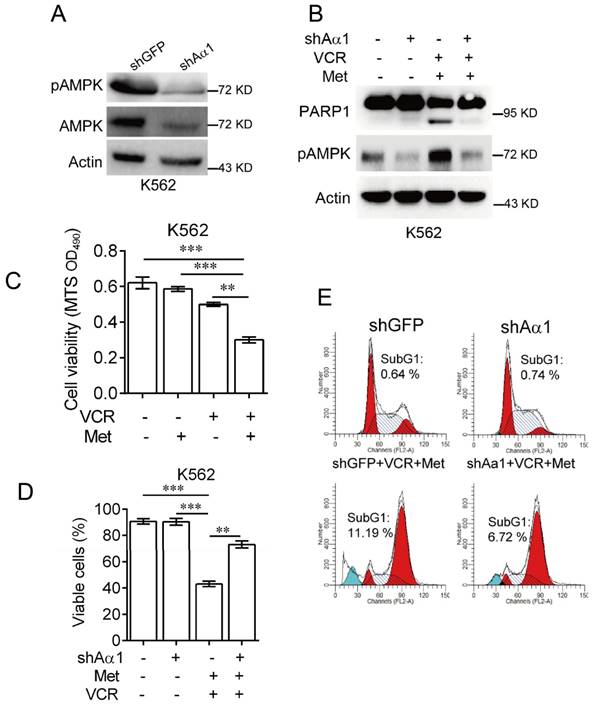
To confirm metformin sensitizes leukemia cells to vincristine-induced apoptosis, we used lentivirus expressing shRNA specifically against AMPKα. As shown in Figure 3A, knockdown of AMPKα1 was effective in K562 cells. Combination of metformin and vincristine, again, was able to induce apoptosis. By contrast, silencing AMPKα1 expression markedly inhibited apoptosis induced by combination of metformin and vincristine (Figure 3B). Data from subsequent examination of cell viability (Figure 3C-3D) and FACS analyses (Figure 3E) showed that AMPKα1 knock-down significantly, but not completely, reversed the combined effects of metformin and vincristine on K562 cell viability and subG1 cell population. Taken together, these findings indicate that metformin sensitizes leukemia cells to vincristine-induced apoptosis by activation of AMPK.
Knockdown of AMPKα1 desensitizes metformin-mediated enhancements of VCR-induced apoptosis.(A) K562 cells stably expressing shRNA against AMPKα1 were subjected to Western blot analyses, as indicated. K562 cells stably expressing shRNAs against AMPKα1 or GFP were treated with or without 5 mM metformin in the presence or absence of 0.4 μM VCR for 24 h prior to Western blot analyses (B), MTS assay or trypan blue exclusion assay for cell viability (C-D) and FACS analyses for subG1 cell population (E). The data are presented as the mean ± SEM from three independent experiments (***P < 0.001; **P < 0.01).
Discussion
Vincristine is extensively used to treat leukemia. However, it remains a clinical problem for its toxicity and drug-resistance. Therefore, it is important to explore strategies to better treat leukemia patients using vincristine regimen. In this study, we showed that both AMPKα1 mRNA and phosphorylated AMPK protein levels were significantly decreased in clinical leukemia samples. We further showed that metformin sensitizes leukemia cells to vincristine-induced apoptosis in an AMPK-dependent manner. Moreover, we demonstrated that knockdown of AMPK attenuates metformin-mediated enhancement for vincristine-induced apoptosis. Taken together, these findings indicate that AMPK plays an important role in vincristine-induced apoptosis in leukemia.
Energetic deregulation is an important hallmark of cancer. AMPK, a cellular energy sensor activated by metabolic stresses, has been shown as a potential target for cancer prevention and/or treatment since AMPK activation leads to inhibition of cell growth and survival. Indeed, AMPK is implicated in regulation of leukemia cell growth, apoptosis and autophagy [24, 29]. Several studies have shown that AMPK is regulated at post translational modification, For instance, AMPK activity is inhibited by AKT-mediated phosphorylation at AMPKα1 serine 485 [30, 31]. In this study, we found that not only phosphorylation of AMPK, but also AMPKα1 mRNA levels are significantly decreased in T-cell/B-cell acute lymphoblastic leukemia. However, the major causes that lead to the decrease of AMPKα1 mRNA levels are unknown, which deserves further investigation. Notably, knockout of AMPKα1 dramatically decreases mice survival in c-myc-driven lymphoma [32]. It would be interesting to investigate whether knockdown of AMPKα1 impacts leukemia development.
Vincristine binds specifically to tubulin, which leads to microtubule depolymerization, cell cycle arrest and apoptosis in mitotic cells [2]. However, long-term use of vincristine can lead to chemo-resistance. It is reported that metformin can enhance chemotherapeutic drugs, such as cisplatin and doxorubicin, to induce apoptosis in breast cancer, lung cancer and prostate cancer [13-15]. In this study, we demonstrate that metformin significantly sensitizes leukemia cell to vincristine-induced apoptosis, which may be the first to show that combination of metformin and vincristine synergistically inhibits leukemia cell viability.
How does metformin affect vincristine-induced cell apoptosis? Our study showed that silencing AMPKα1 significantly attenuates metformin-mediated enhancement for vincristine-induced apoptosis. Indicating that activation of AMPK plays a critical role in metformin effects, consistent with a recent study showing that AMPK activation plays a critical in vincristine-induced apoptosis in melanoma cells [28].
Our data indicates that phosphorylated AMPK and AMPKα1 mRNA levels are dramatically decreased in clinical leukemia cells, which might play a role in drug-resistance in leukemia. Notably, It is shown that inhibition of AMPK by compound C or expression of AMPKα siRNA dramatically inhibits vincristine-induced apoptosis in melanoma cells [28]. Thus, it is plausible that AMPK inactivation contributes to leukemia cell resistance to vincristine.
Together, in this study, we demonstrate that metformin sensitizes leukemia cells to vincristine-induced apoptosis and suggest that combination of metformin and vincristine maybe a new therapeutic strategy for treating leukemia.
Acknowledgements
We thank Drs. Qintong Li and Chenhua Li for helpful discussions. This work was supported in part by National Natural Science Foundation of China [81330054 and 81520108020] and by Science and Technology Department of Sichuan Province [2014SZ20116].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gidding CE, Kellie SJ, Kamps WA, de Graaf SS. Vincristine revisited. Critical reviews in oncology/hematology. 1999;29:267-87
2. Owellen RJ, Root MA, Hains FO. Pharmacokinetics of vindesine and vincristine in humans. Cancer research. 1977;37:2603-7
3. Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. The Journal of biological chemistry. 2000;275:29980-5
4. Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer chemotherapy and pharmacology. 1999;44:355-61
5. Huang Y, Fang Y, Wu J, Dziadyk JM, Zhu X, Sui M. et al. Regulation of Vinca alkaloid-induced apoptosis by NF-kappaB/IkappaB pathway in human tumor cells. Molecular cancer therapeutics. 2004;3:271-7
6. Pieters R, Huismans DR, Loonen AH, Hahlen K, van der Does-van den Berg A, van Wering ER. et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet. 1991;338:399-403
7. Wang W, Liu J, Qi J, Zhang J, Zhu Q, Ma J. et al. Downregulation of RLIP76 is associated with vincristine resistance in human colorectal cancer HCT-8/VCR cells. International journal of oncology. 2016;49:1505-12
8. Verma S, Yao L, Dumont AS, McNeill JH. Metformin treatment corrects vascular insulin resistance in hypertension. Journal of hypertension. 2000;18:1445-50
9. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. The Biochemical journal. 2000;348(Pt 3):607-14
10. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304-5
11. Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer research. 2009;69:6539-45
12. Menendez JA, Cufi S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L. et al. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging. 2011;3:1063-77
13. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM. et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:3297-302
14. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer research. 2011;71:3196-201
15. Qu C, Zhang W, Zheng G, Zhang Z, Yin J, He Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Molecular and cellular biochemistry. 2014;386:63-71
16. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer research. 2007;67:10804-12
17. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell cycle (Georgetown, Tex). 2009;8:88-96
18. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P. et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576-86
19. Dyck JR, Gao G, Widmer J, Stapleton D, Fernandez CS, Kemp BE. et al. Regulation of 5'-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. The Journal of biological chemistry. 1996;271:17798-803
20. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP. et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. Journal of biology. 2003;2:28
21. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM. et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell metabolism. 2005;2:9-19
22. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016-23
23. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology. 2012;13:251-62
24. Campas C, Lopez JM, Santidrian AF, Barragan M, Bellosillo B, Colomer D. et al. Acadesine activates AMPK and induces apoptosis in B-cell chronic lymphocytic leukemia cells but not in T lymphocytes. Blood. 2003;101:3674-80
25. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C. et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010;116:4262-73
26. Puissant A, Auberger P. AMPK- and p62/SQSTM1-dependent autophagy mediate resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy. 2010;6:655-7
27. Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G. et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC cancer. 2009;9:307
28. Chen MB, Shen WX, Yang Y, Wu XY, Gu JH, Lu PH. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. Journal of cellular physiology. 2011;226:1915-25
29. Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S. et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer research. 2010;70:1042-52
30. Valentine RJ, Coughlan KA, Ruderman NB, Saha AK. Insulin inhibits AMPK activity and phosphorylates AMPK Ser(4)(8)(5)/(4)(9)(1) through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Archives of biochemistry and biophysics. 2014;562:62-9
31. Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011;152:3143-54
32. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell metabolism. 2013;17:113-24
Author contact
![]() Corresponding author: Zhi-Xiong Jim Xiao; College of Life Sciences, Sichuan University, Chengdu, 610064, China; Email: jimzxedu.cn; Phone: 86-28-8541-0034; Fax: 86-28-8541-0034
Corresponding author: Zhi-Xiong Jim Xiao; College of Life Sciences, Sichuan University, Chengdu, 610064, China; Email: jimzxedu.cn; Phone: 86-28-8541-0034; Fax: 86-28-8541-0034