Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(14):2713-2719. doi:10.7150/jca.19458 This issue Cite
Research Paper
The Impact of Cetuximab Plus AKT- or mTOR- Inhibitor in a Patient-Derived Colon Cancer Cell Model with Wild-Type RAS and PIK3CA Mutation
Ju Sun Kim, Jung Eun Kim, Kyung Kim, Jeeyun Lee, Joon Oh Park, Ho Yeong Lim, Young Suk Park, Won Ki Kang, Seung Tae Kim ![]()
Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Received 2017-2-2; Accepted 2017-6-24; Published 2017-8-22
Abstract
Background: Anti-EGFR therapies have been recommended for advanced colorectal cancer (CRC) with wild-type RAS and PIK3CA mutation. However, PIK3CA mutations are a poor prognostic marker and a negative predictor of response to anti-EGFR therapies in RAS wild-type CRC. Therefore, new and advanced treatment strategies are needed for personalized medical treatment of patients with wild-type RAS and PIK3CA mutation.
Methods: Patient-derived tumor cells were collected from the ascites of a refractory colon cancer patient with wild-type RAS and PIK3CA mutation. We performed a cell viability assay for cetuximab, AZD5363 (AKT inhibitor), and everolimus (mTOR inhibitor) using PDCs. We also evaluated combinations of cetuximab plus AZD5363 or everolimus in a cell viability assay.
Results: Based on cellular proliferation by MTT assay, tumor cells were significantly inhibited by 1uM cetuximab (control vs. cetuximab, mean growth = 100.0% vs 58.07%, p = 0.0103), 1uM AZD5363 (control vs. AZD5363, mean growth = 100.0% vs 58.22%, p = 0.0123), and 1uM everolimus (control vs. everolimus, mean growth = 100.0% vs 52.17%, p = 0.0011). Tumor cell growth was more profoundly reduced by combinations of cetuximab plus AZD5363 (control vs. cetuximab plus AZD5363, mean growth = 100.0% vs 25.00%, p < 0.0001) or everolimus (control vs. cetuximab+everolimus, mean growth = 100.0% vs 28.24%, p < 0.0001).
Conclusions: Taken together, these results indicate that RAS wild-type and PIK3CA mutant PDCs originating from CRC are considerably inhibited by treatment with cetuximab plus AZD5363 or everolimus, with downregulation of the AKT and ERK pathways. These combinations may be considered as new options for advanced CRC patients with wild-type RAS and PIK3CA mutation in the context of clinical trials.
Keywords: Colon cancer, cetuximab, RAS wild-type, PIK3CA mutation.
Introduction
A combination of cytotoxic therapy with anti-epidermal growth factor (EGFR) monoclonal antibodies such as cetuximab or panitumumab is a standard approach for patients with a subset of metastatic colorectal cancers (mCRC) [1-3]. An activating mutation in exon 2 of the KRAS gene predicts mCRC resistance to anti-EGFR treatments, making it the first molecular biomarker in clinical use for mCRC. Other less frequent RAS gene mutations (KRAS exons 3 and 4 and NRAS exons 2, 3, and 4) are additional predictive markers of resistance to anti-EGFR treatments [4, 5]. Nevertheless, about 50% of mCRC patients with all wild-type RAS still do not respond to anti-EGFR treatments [6, 7]. Furthermore, most patients that obtain a clinical benefit from anti-EGFR treatments eventually develop acquired resistance.
PIK3CA mutation occurs in approximately 17% of CRC patients. Mutations in the p110a subunit of PI3K, called PIK3CA, are responsible for activation of the PI3K-AKT-mTOR pathway and have been implicated in various human cancers [8-11]. The PI3K-AKT-mTOR pathway is part of the downstream EGFR signaling pathways that are related to resistance mechanisms for anti-EGFR treatment [9, 12, 13]. Moreover, PIK3CA mutation might be a predictive marker of response to PI3K-AKT-mTOR inhibitors [14, 15]. Thus, PIK3CA mutation is an attractive molecular marker of overcoming resistance to anti-EGFR treatment and serves as a novel druggable target for PI3K-AKT-mTOR inhibitors.
Currently, anti-EGFR treatment with or without cytotoxic chemotherapy is considered in CRC patients with wild-type RAS and PIK3CA mutation. BRAF inhibitors have improved clinical outcomes in melanoma patients with a BRAF mutation [16, 17]. However, resistance develops in a majority of patients, and most reported resistance mechanisms involve reactivation of the MAPK pathway [18, 19]. The combination of a BRAF inhibitor and a MEK inhibitor could delay the emergence of resistance [20-22]. These results in melanoma suggest that that combined targeting with anti-EGFR therapy and a PI3K-AKT-mTOR inhibitor may apply to CRC with wild-type RAS and PIK3CA mutation.
In this study, we evaluated the antitumor activity of cetuximab, an AKT inhibitor (AZD5363), and an mTOR inhibitor (everolimus) in a patient-derived cell (PDC) line model with wild-type KRAS and PIK3CA mutation. We also evaluated the synergism of cetuximab plus an AKT inhibitor or an mTOR inhibitor and the effect of combined treatment on signaling pathways.
Materials and Methods
Patient consent form
The patients were enrolled as part of the SMC Oncology Biomarker study (NCT#01831609, clinicaltrials.gov), which is reported elsewhere. Effusions or tumor tissues were obtained for therapeutic purposes after obtaining written informed consent, and all procedures were carried out according to guidelines from the Declaration of Helsinki. The Institutional Review Board at Samsung Medical Center approved the protocol.
Patient-derived cell (PDC) culture
Tumor tissue was removed surgically or from biopsy from patients who provided informed consent. Collected tissue was minced and dissociated enzymatically. The cells were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum, 1% antibiotic-antimycotic solution (Gibco BRL, Paisley, UK), 0.5 g/mL hydrocortisone (Sigma Aldrich, St. Louis, MO, USA), 5 μg/mL insulin (PeproTech, Rocky Hill, NJ, USA), 5 ng epidermal growth factor (EGF), and 5 ng fibroblast growth factor (PeproTech). The media was changed every 2 to 3 days, and cells were maintained at 37°C in a humidified 5% CO2 incubator. PDCs were passaged using TrypLE Express (Gibco BRL) to detach cells when the culture was at 80 ~ 90% confluence.
DNA/RNA extractions from PDC
Cultured primary human cells (passage 1 to 2) were harvested with TrypLE Express. Genomic DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, GmBH, Hilden, Germany), and total RNA was isolated with an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. The concentrations of genomic DNA and RNA were measured using a NanoDrop ND-100 (Nano Drop Technologies, Wilmington, DE, USA). Genomic DNA and RNA were stored at -80°C.
Targeted sequencing
A SureSelect customized kit (Agilent Technologies, Santa Clara, CA, USA) was used to capture 381 cancer-related genes. An Illumina Hiseq 2500 instrument was used for sequencing with 100-bp paired-end reads. The sequencing reads were aligned to the human genome reference sequence (hg19) using BWA-mem (v0.7.5). SAMTOOLS (v0.1.18), Picard (v1.93), and GATK (v3.1.1) were used for sorting SAM/BAM files, duplicate marking, and local realignment, respectively. Local realignment and base recalibration were carried out based on dbSNP137, Mills indels, HapMap, and Omni. Single-nucleotide variations (SNVs) and insertion/deletions (InDels) were identified using Mutect (v1.1.4) and Pindel (v0.2.4), respectively. ANNOVAR was used to annotate the detected variants. Only variants with higher than 1% allele frequency were included in the results, as previously described
Cell viability assays in the patient tumor-derived cell (PDC) line
We conducted the experiment with PDCs from advanced CRC patients with wild-type RAS and PIK3CA mutation. After pathologic re-confirmation, 1 X 10^5 cells were seeded on 96-well plates, incubated for 5 days at 37℃, and treated with 0.1 and 1 uM/ml of cetuximab, AZD5363, or everolimus at 37℃. We also tested combinations of cetuximab with AZD5363 and everolimus. Cells were harvested after drug treatment and pelleted by centrifugation. Cell growth inhibition was determined with a CellTiter 96 Aqueous One Solution assay (Promega, Madison, WI, USA) according to the manufacturer's protocol.
Immunoblot analysis
Total protein from PDCs was isolated using RIPA buffer (Sigma-Aldrich) containing a protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Roche), and protein concentrations were determined according to the Bradford procedure using a Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA, USA). Thirty micrograms of proteins were subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis on 10% gels and then electrotransferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% v/v Tween 20 and were probed overnight at 4°C with specific antibodies targeting PI3K, AKT, mTOR, phospho-PI3K p85(Tyr458)/p55(Tyr199), phospho-mTOR (Ser2448), phospho-AKT (Ser473), or phospho-S6 ribosomal protein (Ser235/236) from Cell Signaling Technology (Beverly, MA, USA). Membranes were blotted with anti-beta actin antibodies (Sigma Aldrich) as a loading control. Horseradish peroxidase-conjugated anti-rabbit or mouse IgG (Vector, Burlingame, CA, USA) was used as a secondary antibody, and signals were detected by chemiluminescence using ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA), followed by visualization with an LAS-4000 (Fujifilm, Tokyo, Japan).
Statistical methods
In the PDC viability curves, results are expressed as mean. Paired Student's t-tests were used to calculate the P values. Statistical significance was assessed using two-tailed Student's t-tests and is described in each figure.
Results
Patient history
A 70-year-old man presented with constipation and lower abdominal pain in May 2012, with a computed tomography (CT) scan showing rectosigmoid wall thickening, hepatic masses, and multiple pulmonary nodules. He was diagnosed with colon cancer with hepatic and pulmonary metastasis after colonoscopic biopsies for a colonic mass. He initially achieved partial remission with CapeOx (capecitabine, oxaliplatin), but an increased hepatic mass appeared on the follow-up CT scan in November 2012, after the fifth treatment cycle. He underwent second-line chemotherapy with XELIRI (capecitabine, irinotecan) in November 2012. After the second treatment-cycle, he achieved stable disease and then continued to receive XELIRI until disease progression. In August 2013, a CT scan demonstrated progressive disease with newly developed ascites and increased hepatic masses. At that time, patient-derived tumor cells from ascites were collected and cultured. The patient was on supportive care until death due to disease progression.
Patient-derived colon cancer cells - Monotherapy with cetuximab, AZD5363, and everolimus
Malignant tumor cells were harvested from malignant ascites developed during resistance to second-line chemotherapy (XELIRI) in this patient, as described in the Methods section. Simultaneously, the patient's tumor tissue was acquired from colonoscopic biopsies. Patient tumor tissues demonstrated a PIK3CA E545A mutation and all wild-type RAS (KRAS, NRAS, and HRAS). This genotyping was identified by targeted deep sequencing of patient tumor DNA.
To evaluate the anti-tumor effects of cetuximab (anti-EGFR agent), AZD5363 (AKT inhibitor), and everolimus (mTOR inhibitor) in wild-type RAS and PIK3CA-mutant CRC, we performed a cell viability assay using PDCs established from the matched tumor tissue. The PDC model was also confirmed with targeted deep sequencing for wild-type RAS and PIK3CA mutation. Based on cellular proliferation by MTT assay with cetuximab, an AKT inhibitor (AZD5363) and an mTOR inhibitor (everolimus) (Fig. 1A), tumor cells were significantly inhibited by 1uM cetuximab (control vs. cetuximab, mean growth = 100.0% vs 58.07%, difference = 41.81%, 95% CI = 13.52% to 70.1%, p = 0.0103), 1uM AZD5363 (control vs. AZD5363, mean growth = 100.0% vs 58.22%, difference = 41.67%, 95% CI = 15.00% to 68.34%, p = 0.0123), and 1uM everolimus (control vs. everolimus, mean growth = 100.0% vs 52.17%, difference = 23.57%, 95% CI = 15.84% to 31.30%, p = 0.0011). Treatment with 0.1 uM AZD5363 and everolimus also had a significant inhibitory effect on PDC. However, 0.1uM cetuximab did not significantly inhibit the growth of PDC.
(A) Cellular proliferation by MTT assay with cetuximab, an AKT inhibitor (AZD5363,) and an mTOR inhibitor (everolimus). (B) Combination of cetuximab with an AKT inhibitor or an mTOR inhibitor
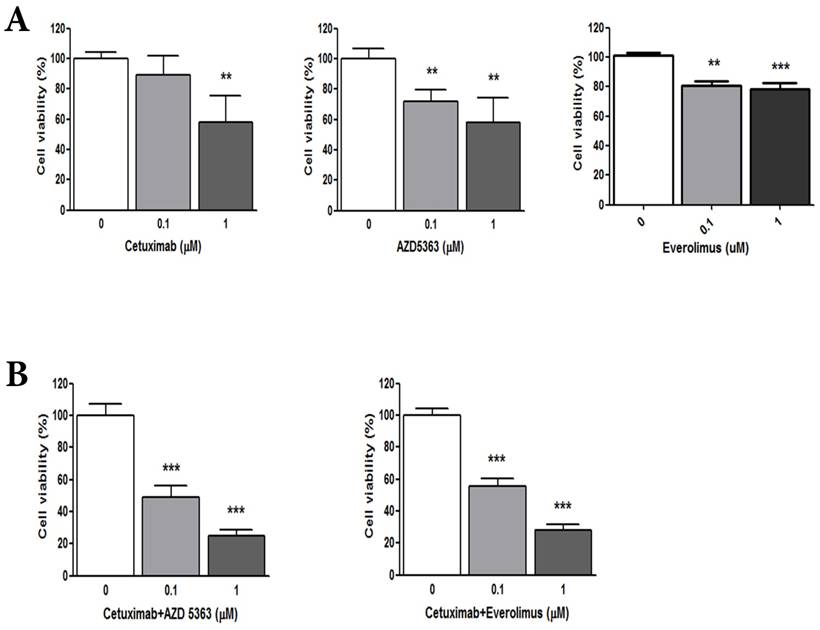
Patient-derived colon cancer cells - Combinations of cetuximab with AZD5363 or everolimus
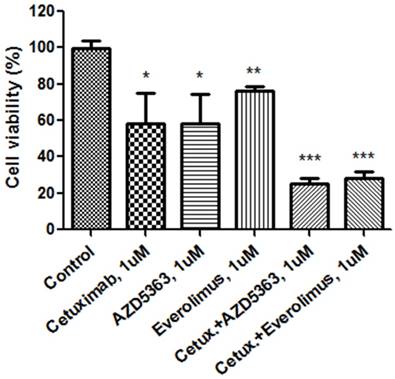
We evaluated the synergism of cetuximab with an AKT inhibitor or an mTOR inhibitor (Fig. 1B). Tumor cell growth was significantly reduced by exposure to 1.0 μM cetuximab plus 1.0 uM AZD5363 compared with the control (control vs. cetuximab plus AZD5363, mean growth = 100.0% vs. 25.00%, difference = 74.88%, 95% CI = 66.26% to 83.51%, p < 0.0001). Similarly, 1.0 μM cetuximab plus 1.0 μM everolimus profoundly inhibited tumor cell growth in vitro compared to the control (control vs. cetuximab plus everolimus, mean growth = 100.0% vs. 28.24%, difference = 71.65%, 95% CI = 63.07% to 80.22%, p < 0.0001). A significant inhibitory effect on PDC growth was also observed at 0.1 uM dose combinations. At 1.0 uM, combinations of cetuximab plus AZD5363 and cetuximab plus everolimus had more potent inhibitory effects on PDC than did monotherapy, such as cetuximab, AZD5363, or everolimus (Fig. 2).
Cellular proliferation by MTT assay with cetuximab, an AKT inhibitor (AZD5363), an mTOR inhibitor (everolimus) and combinations at 1.0 uM.
Patient-derived colon cancer cells - The effects of combined treatment on signaling pathways
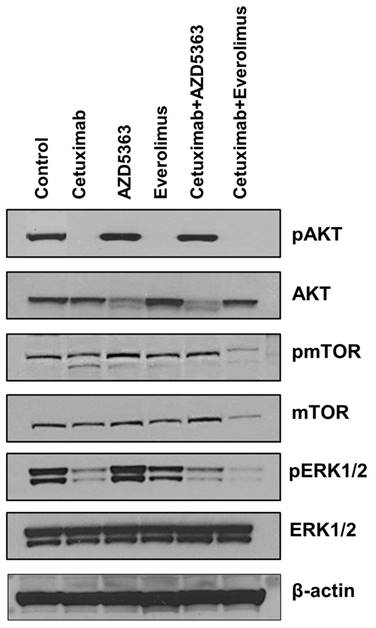
Next, we tested whether downstream targets were downregulated upon exposure to each drug and drug combination. As shown in Fig 3, cetuximab, everolimus, and cetuximab plus everolimus completely downregulated AKT phosphorylation of tumor cells. However, AZD5363 and cetuximab plus AZD5363 up-regulated AKT phosphorylation of tumor cells, indicating engagement of AZD5363 with its target. Phosphorylated mTOR was downregulated by the combination of cetuximab plus everolimus, revealing a robust block of feedback loops and cross-talk within mTOR signaling. Extracellular signal-regulated kinase (ERK) phosphorylation was inhibited by cetuximab alone or in combinations, but it was not inhibited by AZD5363 alone or everolimus alone.
Western blot analysis for evaluating the down-stream targets by drugs
Discussion
The introduction of molecular targeted agents, namely the development of antibodies against the epidermal growth factor receptor (EGFR), has raised new hope for successful treatment of advanced CRC [3]. RAS gene mutations (in KRAS and NRAS) are a major predictive marker of response to anti-EGFR therapies [4, 5]. However, only a subgroup of patients, especially those with a RAS wild-type tumor, benefit from anti-EGFR therapies [6, 7]. Therefore, the presence of wild-type RAS is not sufficient to predict clinical response to anti-EGFR therapies. PIK3CA mutations are present in 10-20% of CRC [10, 23-25]. In RAS wild-type CRC, the frequency of PIK3CA mutation was reported at about 10% [26]. Currently, anti-EGFR therapies have been recommended for advanced CRC with wild-type RAS and PIK3CA mutation. However, PIK3CA mutation is a poor prognostic factor and a negative predictive marker of response to anti-EGFR therapies in RAS wild-type CRC [9, 12, 27]. Thus, advances in treatment strategies for patients with wild-type RAS and PIK3CA mutation are required to take steps toward personalized medicine for CRC patients. The concept of dual EGFR and PI3K/mTOR pathway blockade has been explored. In metastatic CRC, phase I and phase II trials using cetuximab plus everolimus have been conducted (NCT00522665). The outcome of phase II trial will be released
We have established patient-derived tumor cells (PDCs) from an advanced CRC patient who was refractory to oxaliplatin, irinotecan, and 5-fluorouracil (FU). Genomic consistency between PDCs and patient tumor sample was identified by targeted deep sequencing. We previously demonstrated that patient derived tumor cells (PDCs) are feasible preclinical model which closely resemble the patient tumor genome and clinical response [28]. Using a conventional MTT assay with PDCs, we demonstrated that cetuximab, AZD5363, and everolimus were effective in inhibiting cell proliferation of the established PDCs with wild-type RAS and PIK3CA mutation. Moreover, cetuximab plus AZD5363 or everolimus resulted in more potent inhibition of cell proliferation compared to any single treatment. The ERK pathway was significantly downregulated upon drug treatment with cetuximab, alone or in combinations. The AKT pathway was significantly downregulated by drug treatments without AZD5363. AZD5363 induces phosphorylation of AKT, perhaps reflecting a homeostatic feedback mechanism by which the cell attempts to maintain AKT activity. This finding was consistent with previous studies using AKT inhibitors [14, 15]. Our results suggest that cetuximab plus an AKT inhibitor or an mTOR inhibitor may offer a new therapeutic intervention in CRC with wild-type RAS and PIK3CA mutation. However, this finding must be interpreted with the caution. This finding was drawn in PDC from only one patient without the additional validation. No previous study has also reported the use of cetuximab plus AZD5363 or everolimus in CRC with wild-type RAS and PIK3CA mutation. This needs further validation with a larger number of samples to be considered a potential therapy.
Oncogenic activation of intracellular signaling pathways downstream of EGFR, including the RAS-RAF-MAPK and PI3K-PTEN-AKT signaling pathways, are important mechanisms for predicting sensitivity to anti-EGFR therapies. Therefore, in CRC with wild-type RAS and PIK3CA mutation, BRAF mutation and/or PTEN loss might affect the antitumor activity of anti-EGFR therapies, AKT inhibitors, and mTOR inhibitors [13, 15, 29, 30]. Approximately 15% of CRCs have the BRAF mutation, which is a negative prognostic indicator in CRC and a negative predictor of response to EGFR inhibitors [3, 4, 31]. Furthermore, PTEN is a key tumor suppressor gene involved in AKT signaling. PTEN loss is present in 15-42% of CRC and often coincides with KRAS, BRAF, and PIK3CA mutations [32]. The loss of PTEN protein expression is a biomarker of nonresponsiveness to EGFR inhibitors [33]. We confirmed the status of the BRAF mutation and PTEN loss in both PDCs and the tumor sample in order to clarify our findings. PDCs and tumor tissue in this study did not have any BRAF mutations or PTEN loss.
Our study demonstrated that the model we developed can be a useful preclinical screening tool to interrogate chemosensitivity to targeted agents based on genomic aberrations. Although our results are limited, as this is an observation based on a single case, our study also demonstrates the potential insights that can be drawn from a single patient case when combined with preclinical models and genomic sequencing.
Conclusions
Taken together, these results indicate that RAS wild-type and PIK3CA mutant PDCs originating from CRC are considerably inhibited by treatment with cetuximab plus AZD5363 or everolimus, with downregulation of the AKT and ERK pathways. These combinations may be considered as new options for advanced CRC patients with wild-type RAS and PIK3CA mutation in the context of clinical trials.
Abbreviations
CRC: Colorectal cancer; EGFR: Epidermal growth factor; PDC: Patient-derived cell; CT: Computed tomography; CapeOx: capecitabine, oxaliplatin; XELIRI: capecitabine, irinotecan
Acknowledgements
This work was supported by funding from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1A6A3A11934805 and 2016R1A6A3A11932444). Support was also provided by a grant from the 20 by 20 project of Samsung Medical Center (GF01160111) and the Samsung Medical Center (SMX1161251).
Authors' contributions
All authors made substantial contributions to the conception and design of the study, and acquisition, analysis, and interpretation of the data. All authors were involved in drafting the manuscript (or revising it), and all read and approved the final manuscript.
Ethics approval and consent to participate
The Ethics Committee at Samsung Medical Center approved the study in accordance with the Declaration of Helsinki. All individuals gave written informed consent for participation in the study. The Institutional Review Board at Samsung Medical Center approved the protocol.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bokemeyer C, Bondarenko I, Makhson A. et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663-671
2. Tol J, Punt CJ. Monoclonal antibodies in the treatment of metastatic colorectal cancer: a review. Clin Ther. 2010;32:437-453
3. Van Cutsem E, Kohne CH, Hitre E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408-1417
4. Seymour MT, Brown SR, Middleton G. et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749-759
5. De Roock W, Claes B, Bernasconi D. et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753-762
6. Allegra CJ, Jessup JM, Somerfield MR. et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091-2096
7. Douillard JY, Oliner KS, Siena S. et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023-1034
8. Herreros-Villanueva M, Gomez-Manero N, Muniz P. et al. PIK3CA mutations in KRAS and BRAF wild type colorectal cancer patients. A study of Spanish population. Mol Biol Rep. 2011;38:1347-1351
9. Perrone F, Lampis A, Orsenigo M. et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20:84-90
10. Nosho K, Kawasaki T, Ohnishi M. et al. PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia. 2008;10:534-541
11. Prenen H, De Schutter J, Jacobs B. et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15:3184-3188
12. Sartore-Bianchi A, Martini M, Molinari F. et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851-1857
13. Jhawer M, Goel S, Wilson AJ. et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953-1961
14. Li J, Davies BR, Han S. et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med. 2013;11:241
15. Davies BR, Greenwood H, Dudley P. et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873-887
16. Chapman PB, Hauschild A, Robert C. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516
17. Hauschild A, Grob JJ, Demidov LV. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365
18. Van Allen EM, Wagle N, Sucker A. et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94-109
19. Shi H, Hugo W, Kong X. et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80-93
20. Larkin J, Ascierto PA, Dreno B. et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867-1876
21. Robert C, Karaszewska B, Schachter J. et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39
22. Long GV, Stroyakovskiy D, Gogas H. et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888
23. Liao X, Lochhead P, Nishihara R. et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596-1606
24. Ogino S, Nosho K, Kirkner GJ. et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477-1484
25. Iida S, Kato S, Ishiguro M. et al. PIK3CA mutation and methylation influences the outcome of colorectal cancer. Oncol Lett. 2012;3:565-570
26. Mao C, Yang ZY, Hu XF. et al. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23:1518-1525
27. Sood A, McClain D, Maitra R. et al. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11:143-150
28. Lee JY. Patient-derived cell models as preclinical tools for genome-directed targeted therapy. Oncotarget. 2015;6:25619-25630
29. Georgescu MM. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer. 2010;1:1170-1177
30. Cathomas G. PIK3CA in Colorectal Cancer. Front Oncol. 2014;4:35
31. Bokemeyer C, Bondarenko I, Hartmann JT. et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535-1546
32. Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M. et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One. 2009;4:e7287
33. Frattini M, Saletti P, Romagnani E. et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97:1139-1145
Author contact
![]() Corresponding author: Seung Tae Kim, M.D., Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul, 135-710 (Korea) Tel. +82 2 3410 0297, Fax +82 2 3410 1754 Email: seungtae1.kimcom
Corresponding author: Seung Tae Kim, M.D., Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul, 135-710 (Korea) Tel. +82 2 3410 0297, Fax +82 2 3410 1754 Email: seungtae1.kimcom