Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(14):2802-2808. doi:10.7150/jca.19142 This issue Cite
Research Paper
HES1 Promotes Colorectal Cancer Cell Resistance To 5-Fu by Inducing Of EMT and ABC Transporter Proteins
Lei Sun1*, Jia Ke2* ![]() , Zhen He2*, Zexian Chen2, Qinghua Huang3, Wenjia Ai1, Guoqiang Wang1, Yisheng Wei1, Xiangcai Zou1, Shi Zhang1, Ping Lan2, Chuyuan Hong1
, Zhen He2*, Zexian Chen2, Qinghua Huang3, Wenjia Ai1, Guoqiang Wang1, Yisheng Wei1, Xiangcai Zou1, Shi Zhang1, Ping Lan2, Chuyuan Hong1 ![]()
1. Department of Gastrointestinal Surgery, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou City, Guangdong Province, PR China;
2. Department of Colorectal Surgery, The Sixth Affiliated Hospital of Sun Yat-Sen University, Guangzhou City, Guangdong Province, PR China;
3. Department of Breast Surgery, Affiliated Tumor Hospital of Guangxi Medical University, Nanning, Guangxi Province, PR China.
* These authors contributed equally to this work.
Received 2017-1-11; Accepted 2017-6-24; Published 2017-8-23
Abstract
Background and Aim: Hairy enhancer of split-1 (HES1) is a downstream transcriptional factor of Notch signaling pathway, which was found to be related to chemoresistance. This study was aimed to investigate the role of HES1 in chemoresistance of colorectal cancer (CRC).
Methods: Tissue microarray was used to analyze the clinical significance of HES1 in radical resected (R0) stage II/III CRC patients that received adjuvant chemotherapy. 5-fluorouracil (5-Fu) chemoresistance was examined in CRC cell lines (RKO and HCT8, LOVO) with stable over-expression and inhibition of HES1 gene by cytotoxicity test. Gene expression microarray was used to investigate the enriched pathways and different expressed of genes in cells with over-expressed HES1. Expression changes of the chemoresistance related genes were confirmed by qPCR and western blot analysis.
Results: Stage II CRC patients with higher HES1 expression showed higher recurrence rate after chemotherapy. Colon cancer cell lines which over-expressed HES1 were more resistant to 5-Fu treatment in vitro. Gene expression microarray revealed that HES1 was related to the signaling pathways of epithelial-mesenchymal transition (EMT) and drug metabolism. Immunofluorescence assay showed HES1 over-expression lead to depressed E-cadherin and elevated N-cadherin. QPCR and western blot analysis confirmed that ABCC1, ABCC2 and P-gp1 were induced after HES1 over-expression.
Conclusions: HES1 promotes chemoresistance to 5-Fu by prompting EMT and inducing of several ABC transporter genes. HES1 might be a novel therapeutic target in CRC treatment.
Keywords: HES1, colorectal cancer, chemoresistance, EMT, ABC transporter proteins.
Introduction
Colorectal cancer (CRC) remains a major cause of cancer-related morbidity and mortality in the world. 5-Fu based adjuvant chemotherapy after curative surgery is considered as standard therapy for stage II/III CRC. Unfortunately, approximately 40% of stage these patients develop local recurrence or metastatic disease which mainly due to chemoresistance [1]. To date, the mechanisms of chemoresistance in CRC have not been fully elucidated.
Notch signaling pathway plays an essential role in promoting cell survival [2, 3]. Activation of Notch pathway leads to the release of the Notch intracellular domain (NICD), which translocates to the nucleus and activates transcription of numerous downstream target genes, including HES1 [4, 5]. Aberrant activation of Notch signaling pathway is involved in chemoresistance in multiple cancers including CRC, and blocking Notch signaling pathway by a γ-secretase inhibitor could enhance chemosensitivity [6-9].
As a downstream target of canonical Notch signaling pathway, HES1 plays a vital role in chemoresistance. In ovarian cancer, inhibiting the Notch pathway by γ-secretase inhibitor could decrease expression of HES1 mRNA and sensitize cells to paclitaxel [6]. HES1 might modulate the therapeutic resistance by mediating Gli1 expression in medulloblastoma and glioblastoma [10]. However, except for Notch signaling pathway, HES1 signaling could be activated by other pathways, including Hedgehog, c-Jun N terminal kinase and TGF-a/Ras/MAPK pathways, which are also involved in chemoresistance [11-13]. In addition, HES1 acts as a marker of colon cancer stem cells (CSCs), which might also contribute to tumor recurrence after 5-Fu based adjuvant chemotherapy [14-16]. Thus, the role of HES1 in CRC chemoresistance is unpredictable upon Notch signaling pathway pathway status.
In this study, we investigated the clinical significance of chemo-response of HES1 in stage II/III CRC patients (n=121) using tissue microarray. Chemosensitivity was examined in colorectal cancer cells with over-expression and inhibition of HES1. Furthermore, the enriched pathways and different expression of genes in cells which over-expressed HES1 were investigated by gene expression microarray. Expression changes of the main genes related to chemoresistance were confirmed by qPCR and western blot analysis.
Methods
Patients and tissues
This retrospective study included 121 staged II/III CRC patients who underwent radical resection (R0) and received 5-Fu based adjuvant chemotherapy. Overall survival (OS) period was defined as the period between diagnosis and death or the last follow-up. Disease free survival (DFS) was defined as the period between diagnosis and the first clinical or pathologic evidence of local or distant recurrent disease. Written informed consent was obtained from each patient before surgery. The study was approved by the Institutional Review Board of Sun Yat-Sen University.
Immunohistochemistry analysis
Immunohistochemistry analysis was carried out according to the Envision System (Dako Cytomation, Glostrup, Denmark) guidance. In brief, each TMA slides was deparaffinzed and rehydrated through graded ethanol. Sodium citrate was used for antigen retrieval. Slides underwent 0.3% hydrogen peroxide solution to block endogenous peroxidase activity. Then samples was incubated with the primary antibody anti-HES1 (1:400; Abcam, ab71559), at 4°C overnight. After incubation with secondary (goat) antibody, slides were developed in diaminobenzine (EnVision, DAKO) and counterstained with haematoxylin.
Cell culture and lentivirus infection
RKO, HCT8 and LOVO were obtained from Chinese Academy of Science Cell Bank (Shanghai, China). All cell lines were cultured in RPMI-1640 or DMEM-F12 medium (Gibco, China) with 10% FBS (Gibco, China), 100U/mL penicillin and 100ug/mL streptomycin (Gibco, China) at 37°C with 5% CO2. Lentiviral plasmid for over-expressing HES1 and plasmid for negative control (disrupted DNA-binding domain) were purchased from Addgene (No.17624 and No.24982). The plasmids were verified via sequencing. ShRNA lentivirus was constructed by GENECHEM CO. (Shanghai, China). GV248 backbone was used, with the shRNA sequences: SCR (scramble): 5'-TTCTCCGAACGTGTCACGT-3'; shRNA-1: 5'-AGATCAAT GCCATGACCTA-3'; shRNA-2: 5'- GGACATTCTGGAAATGACA -3'. In brief, the lentiviruses were produced by transfecting 293FT cells with pGC-LV, pPAX2 and pMD2.G plasmids mix and lipofectamine-2000 (Invitrogen, NY) in Opti-MEM medium (Gibco, USA). Virus was collected and stored at -80°C before using, and titrated with Lenti-X qRT-PCR Titration Kit (Clontech, CA). Cell lines were infected at a MOI (Multiplicity of Infection) of 10 with 8ug/mL polybrene (Millipore, MA). Knockdown efficiency higher than 80% was considered acceptable. For the following experiments, the positive cells population was enriched using puromycin selection or FACS sorting for GFP.
Cell viability assay
Cells were seeded 2000 per well in 96-well plates for 24 hours, then treated with 5-Fu at various concentrations (0, 0.5, 1, 5, 10, 50μmol/L). Sixty hours later, 10μl of CCK-8 solution (Cell counting kit-8, Sigma, St. Louis, MO, USA) was added to each well. Absorbance was determined at 450 nm after 3 hours of incubation. Cell viability was calculated as following: Viability = (OD test group-OD blank group) / (OD control group-OD blank group) × 100 %, and IC50 (half maximal inhibitory concentration) was calculated from the dose-response curves. All experiments were repeated in triplicate.
Gene expressional profiles and analysis
RNAs were extracted from RKO-HES1 and RKO-Mutant cells. RNA integrity was assessed by standard denaturing agarose gel electrophoresis. The Human 12x135K Gene Expression Array was manufactured by Roche NimbleGen. 45,033 genes are collected from the authoritative data source including National Center of Biotechnology Information (NCBI). Double-strand cDNA (ds-cDNA) was synthesized from total RNA, which was then cleaned and labeled before hybridization. Differentially expressed genes were identified through Fold change filtering. Genes with fold-change≥2.0 expression were enrolled. Realtime RT-PCR was used to confirm the results.
Pathways enrichment analysis was based on KEGG (Kyoto Encyclopedia of Genes and Genomes) database, which identified the biological pathways that had a significant enrichment of differently expressed genes. The P-values denote the significance of the pathways, with cut-off at 0.05.
Immunofluorescence and western blot analysis
For immunofluorescence assay, cells were grown on glass slices and fixed in 4 % formaldehyde for 10 min, permeabilized through 0.3 % Triton X-100. Then the slices were blocked in goat serum for 15 min, 37°C and incubated overnight at 4°C with anti-E-cadherin (1:80, Bioworld, MN, USA), anti-N-cadherin (1:80, Bioworld, MN, USA). Samples were washed three times before incubated with goat TRITC labeled secondary antibody (1:70, Bioworld, MN, USA) at 37°C for 1 h. DAPI (Genview Inc, Shanghai, China) was used for counterstaining. Fluorescence was visualized with a microscope under ×400 magnification.
Total protein was extracted using RIPA Lysis Buffer (Beyotime, China) and PMSF (Sigma-Aldrich). The proteins were transferred to NC membranes (Millipore Corp, MA USA) using the TransBlot System (Bio-Rad, CA, USA). The membranes were blocked in 5% w/v non-fat milk in TBS and incubations were performed overnight at 4°C. The membranes were then washed using TBST and incubated with secondary antibodies (1:10000, IRDye Goat IgG, LI-COR Bioscience, NE USA) for 1h at room temperature. Protein staining was detected using the Odyssey Imaging System (LI-COR Biosciences, NE USA). The following primary antibodies were used: GAPDH (1:10000, Proteintech Group, Chicago USA), ABCC1 (1:100, Abcam, ab24102), ABCC2 (1:100, Abcam, ab3373), P-gp1 (1:2000, Abcam, ab129450).
Statistics
An open source software TMAJ (Johns Hopkins, Baltimore, USA) was applied to measure the HES1 expression index as described elsewhere [17]. The median of HES1 expression was employed for the cut-point.
Statistical analysis was carried out using SPSS 17.0 (SPSS, Chicago, IL, USA). Correlations between clinicopathologic data and HES1 expression were analyzed using Chi-square test or Fisher's exact test. Kaplan-Meier survival curves were preformed to estimate OS and DFS. A value of P<0.05 was considered statistically significant.
Results
Correlation between HES1 level and clinicopathological variables
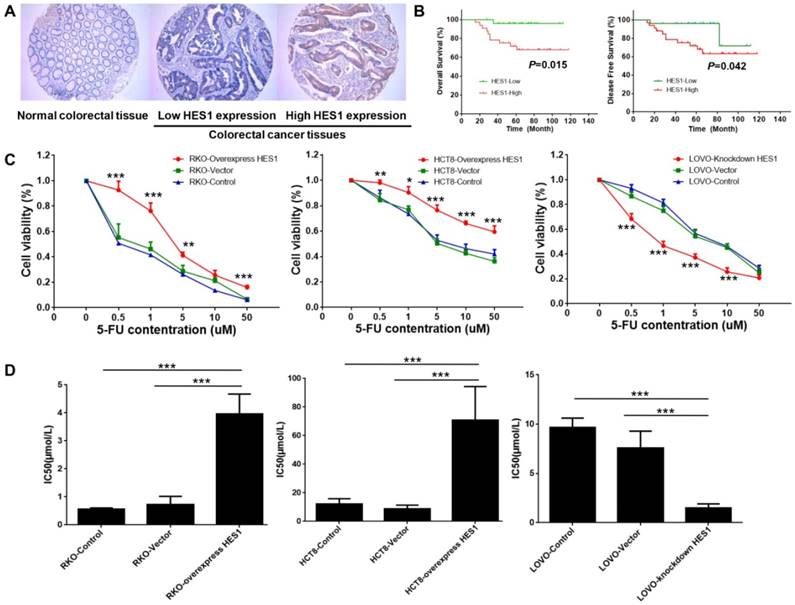
To study the clinical significance of HES1 expression, 121 CRC samples in stage II/III (n=121) patients who received 5-Fu based adjuvant chemotherapy were examined by tissue microarray. HES1 protein was mainly located in cancer cell cytoplasm (Fig.1A). Patients were divided into high and low HES1 expression groups, using the median expression index (2.95) as the cutpoint. Clinic-pathological characteristics and correlation with HES1 expression were shown in Table 1. Correlation analysis revealed that HES1 expression was significantly correlated with CEA level (P=0.038), not with age, gender, CA199 level, tumor type, location, stage or recurrence. For stage II patients only, HES1 expression was correlated with CA199 level (P=0.016) and recurrence rate (P=0.022) (Table 2).
HES1 prompted chemoresistance in CRC (A) Representative images of HES1 staining in normal colorectal tissues and CRC (Magnification: ×200). (B) Kaplan-Meier survival curves for over-all survival (left) and disease-free survival (right) of stage II CRC patients who received 5-Fu based adjuvant chemotherapy. (C) Cell viability of RKO, HCT8, LOVO in different 5-Fu concentrations. (D) IC50 of RKO, HCT8, LOVO in different 5-Fu concentrations. The bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Correlation between expression of HES1 and clinicopathological variables
| Variables | All cases | HES1 expression | P value† | |
|---|---|---|---|---|
| Low | High | |||
| No. of patients | 121 | 60 | 61 | - |
| Median HES1 expression index | 2.95 | 0.53 | 9.70 | - |
| Age | ||||
| <60/≥60 | 67/54 | 37/23 | 30/31 | 0.167 |
| Gender | ||||
| Male/Female | 76/45 | 40/20 | 36/25 | 0.384 |
| CEA, ng/ml | ||||
| <5/≥5 | 74/41 | 32/26 | 42/15 | 0.038* |
| CA199, U/ml | ||||
| <37.5/≥37.5 | 79/31 | 39/17 | 40/14 | 0.606 |
| Stage | ||||
| II/III | 58/63 | 25/35 | 33/28 | 0.171 |
| Histological type | ||||
| Adenocarcinoma/Mucinous | 106/14 | 55/5 | 51/9 | 0.255 |
| Location | ||||
| Colon/Rectum | 51/69 | 26/34 | 25/35 | 0.853 |
| Recurrence | ||||
| Yes/No | 28/93 | 10/50 | 18/43 | 0.094 |
† The P value was calculated by Chi square test.
* P<0.05.
Correlation between expression of HES1 and clinicopathological variables in stage II patients
| Variables | All cases | HES1 expression | P value† | |
|---|---|---|---|---|
| Low | High | |||
| No. of patients | 58 | 25 | 33 | - |
| Age | ||||
| <60/≥60 | 26/32 | 13/12 | 13/20 | 0.339 |
| Gender | ||||
| Male/Female | 36/22 | 15/10 | 21/12 | 0.777 |
| CEA, ng/ml | ||||
| <5/≥5 | 35/20 | 13/9 | 22/11 | 0.199 |
| CA199, U/ml | ||||
| <37.5/≥37.5 | 38/15 | 17/7 | 21/8 | 0.016* |
| Histological type | ||||
| Adenocarcinoma/Mucinous | 51/7 | 23/2 | 28/5 | 0.687‡ |
| T stage | ||||
| T3/T4 | 54/4 | 25/0 | 29/4 | 0.126‡ |
| Location | ||||
| Colon/Rectum | 20/38 | 8/17 | 12/21 | 0.729 |
| Recurrence | ||||
| Yes/No | 13/45 | 2/23 | 11/22 | 0.022* |
† The P value was calculated by Chi square test .
‡ The P value was calculated by Fisher's exact test.
* P<0.05.
Kaplan-Meier analysis revealed that stage II patients with higher HES1 expression level had poor OS (P=0.015) and DFS (P=0.042) (Fig. 1B).
Over-expression of HES1 induce chemoresistance in CRC cells
Chemoresistance is a key obstacle to the efficacy of CRC treatment and may result in recurrence. To determine the potential role of HES1 in chemoresistance, stable over-expression and inhibition of HES1 gene were established in colon cancer cell lines including RKO, HCT8 and LOVO, which were then exposed to different concentrations of 5-Fu treatment in vitro. CCK8 test showed that HES1 over-expression significantly promoted cell viability of RKO and HCT8 cells, whereas HES1 inhibition significantly decreased cell viability of LOVO cells (Fig.1C). The IC50 of RKO (P=0.016 vs vehicle, P=0.001 vs control) and HCT8 cells (P<0.001 vs vehicle, P<0.001 vs control) were significantly increased by HES1 over-expression. However, HES1 inhibition resulted in a significant decreased IC50 in LOVO cells (P<0.001 vs vehicle, P<0.001 vs control) (Fig.1D).
Over-expression of HES1 induce EMT in CRC cells
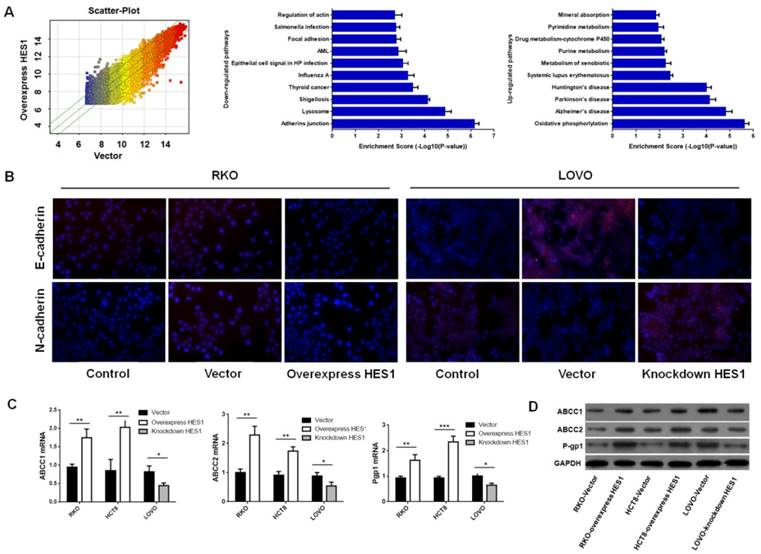
To determine the mechanisms of HES1 mediated CRC chemoresistance, RKO cell lines that stable over-expressed wild type HES1 and mutant HES1 gene were stablished. Changes in two cell lines were determined by whole-genome cDNA microarray. As shown in Fig.2A, several pathways were changed by HES1 over-expression. Briefly, pathways with drug metabolism were up-regulated, and pathways with adhering junction, focal adhesion and actin cytoskeleton were markedly down-regulated. 2668 genes were up-regulated after HES1 over-expression, while 1304 genes were down-regulated. Genes with most significant changes were shown in Table 3.
HES1 induced EMT and ABC transporters proteins of CRC cells (A) cDNA microarray of HES1 over-expression in RKO. Pathway enrichment analysis on the distinguished expressed genes. Enrichment score values are shown as-log10 (P value). (B) Immunofluorescence staining for E-cadherin and N-cadherin in RKO and LOVO. (C) qPCR assay of ABCC1, ABCC2 and P-gp1 in RKO, HCT8 and LOVO. Western blot analysis of ABCC1, ABCC2 and P-gp1 in RKO, HCT8 and LOVO. The bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Genes with most significant expression changes by overexpression HES1 in cDNA microarray
| NCBI Gene ID | Gene name | Fold change† | Description |
|---|---|---|---|
| NM 3598 | IL13RA2 | 7.34±0.47 | Interleukin 13 receptor, alpha 2 |
| NM 4582 | MUC1 | 6.71±1.02 | Mucin 1, cell surface associated |
| NM1396 | CRIP1 | 5.25±0.73 | Cysteine-rich protein 1 (intestinal) |
| NM 1000 | CDH2 | 5.19±0.24 | Cadherin 2, type 1, N-cadherin |
| NM19900 | ABCC1 | 4.62±0.13 | ATP-binding cassette, sub-family C, member 1 |
| AF541977 | ABCC4 | 3.98±0.44 | ATP-binding cassette, sub-family C, member 4 |
| NM1606 | ABCA2 | 3.62±0.81 | ATP-binding cassette, sub-family A, member 2 |
| NM33 | ABCD1 | 3.25±0.63 | ATP-binding cassette, sub-family D (ALD), member 1 |
| NM6624 | FSCN1 | -17.31±2.99 | Fascin homolog 1, actin-bundling protein |
| NM26012 | NELF | -14.45±0.68 | Nasal embryonic LHRH factor |
| NM5829 | PXN | -12.88±1.98 | Paxillin |
| NM 4134 | MAP4 | -8.24±1.67 | Microtubule-associated protein 4 |
† Data represent as means ± SEM for duplicate samples.
Considering the changes in adhesion and actin cytoskeleton pathways, we hypothesized HES1 over-expression might induce EMT. To verify this idea, we examined the expression of two critical EMT markers, E-cadherin and N-cadherin, in RKO and LOVO cell lines. Immunofluorescence assay revealed that HES1 over-expression increased the level of N-cadherin, and decreased E-cadherin in RKO cells. The opposite results were found in LOVO cells after HES1 inhibition (Fig.2B). Thus, HES1 over-expression might induce EMT in CRC cells.
Over-expression of HES1 induce ABC transporter genes in CRC cells
Since several members of ATP-binding cassette transporter family (ABC) are up-regulated by HES1 over-expression in cDNA microarray, we reasoned that ABC transporter proteins could be more commonly induced by HES1, and rendered chemoresistance. To test this hypothesis, we examined expression of ABCC1, ABCC2 and P-gp1 (three critical molecules in drug metabolism) in CRC cell lines mentioned above. QPCR and western-blot analysis showed that HES1 over-expression increased ABCC1, ABCC2 and P-gp1 expression in RKO and HCT8 cells, and HES1 inhibition of in LOVO cells showed opposite results (Fig.2C, 2D). Thus, these data suggested that HES1 might prompt chemoresistance by inducing ABC transporter proteins in CRC cells.
Discussion
Previous studies have revealed that Notch signaling pathway involved in CRC chemoresistance. However, as an important downstream transcriptional factor in Notch signaling pathway, the role of HES1 in CRC chemoresistance is still unclear. In this study, we investigated the clinical significance of HES1 expression in stage II/III CRC patients who received adjuvant chemotherapy, and demonstrated its role of chemoresistance in vitro.
Notch signaling pathway plays important role in the differentiation balance of intestinal crypts and carcinogenesis in CRC. Several studies showed HES1 is over-expressed in colorectal cancer [8, 18, 19], and its prognostic value in CRC has also been investigated [18, 20]. However, there is no study showed its role in CRC chemotherapy. In this study, we found stage II CRC patients with high HES1 expression had higher recurrence rate and poor prognosis (OS and DFS) after 5-Fu based adjuvant chemotherapy. This might be the result from the chemoresistance induced by HES1 over-expression. This correlation was not found in stage III patients. To our knowledge, this is the first time establishing a correlation between HES1 expression and CRC recurrence after 5-Fu based adjuvant chemotherapy.
Notch signaling was found to participate in chemoresistance in numerous cancers and inhibiting the Notch pathway could enhance chemosensitivity [9, 21-23]. Whether HES1 is involved in colorectal chemoresistance is unclear. What is more, HES1 could be activated by other upstream pathways besides Notch pathways [11-13], and HES1 is considered as a marker of colon CSCs which also might contribute to chemoresistance [16]. Thus, the role of HES1 in CRC chemoresistance is unpredictable. In this study, we found HES1 could promote chemoresistance of CRC and targeted inhibited HES1 could enhance chemosensitivity in vitro.
Epithelial-mesenchymal transition (EMT) has been shown to play a crucial role in chemoresistance and tumor recurrence [24-26]. Increasing evidences suggest EMT-associated transcription factors are involved in chemoresistance in different cancers [27-29]. In colorectal cancer, EMT could prompt chemoresistance to oxaliplatin by upregulating P-gp expression [30]. Notch signaling could promote chemoresistance via EMT in other type of cancer [31, 32]. In the present study, cDNA microarray profiling and western blot analysis demonstrated that over-expression of HES1 could down-regulate E-cadherin and up-regulate N-cadherin. Thus, HES1 might prompt chemoresistance by the induction of EMT.
ATP-binding cassette transporter (ABC) transporters involved in chemoresistance by decreasing cellular drug uptake and accumulation, and are considered as a major cause for chemotherapy failure. Over-expression of the ABCC1 transporter confers resistance to a wide range of anticancer drugs [33, 34]. In breast cancer, ABCC3 transporter is confirmed involving in drug resistance to chemotherapy [35]. The transfection of human embryonic kidney cells with the ABCC10 gene conferred resistance to various anticancer drugs including paclitaxel, docetaxel, vincristine, gemcitabine [36-39]. In this study, we found expression of several members of ABC transporters was increased after HES1 over-expression by cDNA microarray profiling, and western blot confirmed P-gp1, ABCC1 and ABCC2 are up-regulated by over-expression of HES1. Therefore, up-regulation of ABC transporters might be one of the mechanisms of HES1 induced chemoresistance.
In conclusion, our study showed that HES1 was an unfavorable factor for recurrence in stage II CRC patients who received adjuvant chemotherapy, and HES1 could promote CRC chemoresistance via induction of EMT and ABC transporters. Thus, HES1 might be a novel therapeutic strategy in CRC treatment.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (No.81402019, 81672436); Scientific Research Project of Guangzhou Municipal Colleges and Universities (No.1201630159); Special funds for public welfare research and capacity building in Guangdong Province (No.2014A020212333); Science and Technology Program of Guangzhou (No.201400000001-4); Startup Foundation for Doctor of Philosophy of Guangzhou Medical University (No.2015C21).
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goldberg RM. Intensive surveillance after stage II or III colorectal cancer: is it worth it? Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:330-1
2. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770-6
3. Gridley T. Notch signaling and inherited disease syndromes. Human molecular genetics. 2003;12:R9-13
4. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-33
5. Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nature reviews Cancer. 2003;3:756-67
6. Groeneweg JW, DiGloria CM, Yuan J, Richardson WS, Growdon WB, Sathyanarayanan S. et al. Inhibition of notch signaling in combination with Paclitaxel reduces platinum-resistant ovarian tumor growth. Frontiers in oncology. 2014;4:171
7. Nwabo Kamdje AH, Bassi G, Pacelli L, Malpeli G, Amati E, Nichele I. et al. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood cancer journal. 2012;2:e73
8. Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB. et al. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer research. 2009;69:573-82
9. Lee JY, Song SY, Park JY. Notch pathway activation is associated with pancreatic cancer treatment failure. Pancreatology: official journal of the International Association of Pancreatology. 2014;14:48-53
10. Schreck KC, Taylor P, Marchionni L, Gopalakrishnan V, Bar EE, Gaiano N. et al. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:6060-70
11. Ingram WJ, McCue KI, Tran TH, Hallahan AR, Wainwright BJ. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene. 2008;27:1489-500
12. Stockhausen MT, Sjolund J, Axelson H. Regulation of the Notch target gene Hes-1 by TGFalpha induced Ras/MAPK signaling in human neuroblastoma cells. Experimental cell research. 2005;310:218-28
13. Curry CL, Reed LL, Golde TE, Miele L, Nickoloff BJ, Foreman KE. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene. 2005;24:6333-44
14. Roy S, Majumdar AP. Signaling in colon cancer stem cells. Journal of molecular signaling. 2012;7:11
15. Gao F, Zhang Y, Wang S, Liu Y, Zheng L, Yang J. et al. Hes1 is involved in the self-renewal and tumourigenicity of stem-like cancer cells in colon cancer. Scientific reports. 2014;4:3963
16. Gerger A, Zhang W, Yang D, Bohanes P, Ning Y, Winder T. et al. Common cancer stem cell gene variants predict colon cancer recurrence. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6934-43
17. Wu XR, He XS, Chen YF, Yuan RX, Zeng Y, Lian L. et al. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. Journal of surgical oncology. 2012;106:130-7
18. Reedijk M, Odorcic S, Zhang H, Chetty R, Tennert C, Dickson BC. et al. Activation of Notch signaling in human colon adenocarcinoma. International journal of oncology. 2008;33:1223-9
19. Katoh M, Katoh M. Notch signaling in gastrointestinal tract (review). International journal of oncology. 2007;30:247-51
20. Candy PA, Phillips MR, Redfern AD, Colley SM, Davidson JA, Stuart LM. et al. Notch-induced transcription factors are predictive of survival and 5-fluorouracil response in colorectal cancer patients. British journal of cancer. 2013;109:1023-30
21. Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer research. 2006;66:4715-24
22. Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer research. 2006;66:1517-25
23. Nair P, Somasundaram K, Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. Journal of virology. 2003;77:7106-12
24. Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A. et al. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2008;11:123-51
25. Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC. et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141:279-91 91 e1-5
26. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature reviews Cancer. 2009;9:265-73
27. Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Current pharmaceutical design. 2015;21:1301-10
28. Sui H, Zhu L, Deng W, Li Q. Epithelial-mesenchymal transition and drug resistance: role, molecular mechanisms, and therapeutic strategies. Oncology research and treatment. 2014;37:584-9
29. Shang Y, Cai X, Fan D. Roles of epithelial-mesenchymal transition in cancer drug resistance. Current cancer drug targets. 2013;13:915-29
30. Deng JJ, Zhang W, Xu XM, Zhang F, Tao WP, Ye JJ. et al. Twist mediates an aggressive phenotype in human colorectal cancer cells. International journal of oncology. 2016;48:1117-24
31. Kang M, Jiang B, Xu B, Lu W, Guo Q, Xie Q. et al. Delta like ligand 4 induces impaired chemo-drug delivery and enhanced chemoresistance in pancreatic cancer. Cancer letters. 2013;330:11-21
32. Hindriksen S, Bijlsma MF. Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors. Cancers. 2012;4:989-1035
33. Anreddy N, Gupta P, Kathawala RJ, Patel A, Wurpel JN, Chen ZS. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules. 2014;19:13848-77
34. Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chinese journal of cancer. 2012;31:58-72
35. Balaji SA, Udupa N, Chamallamudi MR, Gupta V, Rangarajan A. Role of the Drug Transporter ABCC3 in Breast Cancer Chemoresistance. PloS one. 2016;11:e0155013
36. Bessho Y, Oguri T, Ozasa H, Uemura T, Sakamoto H, Miyazaki M. et al. ABCC10/MRP7 is associated with vinorelbine resistance in non-small cell lung cancer. Oncology reports. 2009;21:263-8
37. Chen ZS, Hopper-Borge E, Belinsky MG, Shchaveleva I, Kotova E, Kruh GD. Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10). Molecular pharmacology. 2003;63:351-8
38. Malofeeva EV, Domanitskaya N, Gudima M, Hopper-Borge EA. Modulation of the ATPase and transport activities of broad-acting multidrug resistance factor ABCC10 (MRP7). Cancer research. 2012;72:6457-67
39. Sun YL, Chen JJ, Kumar P, Chen K, Sodani K, Patel A. et al. Reversal of MRP7 (ABCC10)-mediated multidrug resistance by tariquidar. PloS one. 2013;8:e55576
Author contact
![]() Corresponding authors: Jia Ke, M.D. Ph.D. Email Address: 278494686com. Department of Colorectal Surgery, The Sixth Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510655, Guangdong Province, P.R. China. Tel: +86 020 38254159. Pro.Chuyuan Hong. Email Address: 1750699409com. Department of Gastrointestinal Surgery, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou 510260, Guangdong Province, P.R. China. Tel: +86 020 34153215.
Corresponding authors: Jia Ke, M.D. Ph.D. Email Address: 278494686com. Department of Colorectal Surgery, The Sixth Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510655, Guangdong Province, P.R. China. Tel: +86 020 38254159. Pro.Chuyuan Hong. Email Address: 1750699409com. Department of Gastrointestinal Surgery, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou 510260, Guangdong Province, P.R. China. Tel: +86 020 34153215.