Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(15):2933-2943. doi:10.7150/jca.20319 This issue Cite
Research Paper
Anti-Melanoma Activities of Haspin Inhibitor CHR-6494 Deployed as a Single Agent or in a Synergistic Combination with MEK Inhibitor
Lili Han1, Peiling Wang1, Yang Sun1, Sijing Liu1, Jun Dai1, 2, 3 ![]()
1. School of Pharmaceutical Science and Technology, Tianjin University, Tianjin, 300072 P. R. China;
2. Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin, Madison, WI 53705 USA;
3. Cutaneous Biology Research Center, Massachusetts General Hospital, Charlestown, MA 02129 USA.
Received 2017-3-29; Accepted 2017-5-28; Published 2017-8-25
Abstract
Background: Melanoma is a heterogeneous malignancy that presents an immense challenge in therapeutic development. Recent approaches targeting the oncogenic MAP kinase pathways have shown tremendous improvement in the overall survival of patients with advanced melanoma. However, there is still an urgent need for identification of new strategies to overcome drug resistances and to improve therapeutic efficacy. Haspin (Haploid Germ Cell-Specific Nuclear Protein Kinase) belongs to a selected group of mitotic kinases and is required for normal mitosis progression. In contrast to inhibitors of other mitotic kinases, anti-tumor potential of haspin inhibitors has not been well explored. Herein, we aim to examine effects of CHR-6494, a small molecule inhibitor of haspin, in melanoma cells.
Methods: Anti-tumor activities of the haspin inhibitor CHR-6494 were tested in a number of melanoma cell lines either as a single agent or in combination with the MEK inhibitor Trametinib (GSK1120212). Experiments are based on: 1) Cell viability determined by the crystal violet staining assay; 2) apoptotic responses measured by the caspase 3/7 activity assay and western blot analysis for the level of cleaved PARP (Poly ADP-Ribose Polymerase); 3) cell cycle analysis conducted using flow cytometry; and 4) cell migratory ability assessed by the scratch assay and the transwell migration assay.
Results: We have found that CHR-6494 alone elicits a dose dependent inhibitory effect on the viability of several melanoma cell lines. This growth inhibition is accompanied by an increase in apoptotic responses. More importantly, CHR-6494 appears to synergize with the MEK inhibitor Trametinib in suppressing cell growth and enhancing apoptosis in both wild type and BRAFV600E mutant melanoma cell lines. Administering of these two small molecules as a combination is also capable of suppressing cell migration to a greater extent than the individual agent.
Conclusion: These results suggest that haspin can be considered as a viable anti-melanoma target, and that concomitant inhibition of haspin and MEK activities with small molecules could represent a novel therapeutic strategy with improved efficacy for treatment of melanoma.
Keywords: melanoma, mitotic kinases, haspin, MEK, inhibitor, targeted therapy, combined therapy.
Introduction
Melanoma is one of the most aggressive and drug-resistant human cancers. Development of melanoma is largely dependent upon the oncogenic activation of the mitogen-activated protein kinase (MAPK) pathway composed of the receptor tyrosine kinase (RTK)-NRAS-BRAF-MEK-ERK [1, 2]. Gain-of-function mutations of NRAS and BRAF genes are detected in about 15-30% and 60% of all melanomas, respectively. With the onset of discovering small molecule based inhibitors of BRAF as well as its downstream kinases MEK and ERK, the overall survival rate of patients with advanced melanoma has been improved tremendously in the past decade [3]. Despite such advancement, deploying BRAF inhibitors as single agents in chemotherapy still confront a number of challenges such as lack of complete responses and relapse through drug resistance [4]. Therefore, it remains imperative to pursue new and more effective therapeutics for treatment of melanoma [5].
Cancer cells in general exhibit an increased rate of proliferation relative to normal cells with mitosis as a centrally critical step. Vinca alkaloids and taxanes have become conventional chemotherapeutic drugs that target mitosis by disturbing microtubule dynamics [6-8]. Despite their therapeutic success for an impressive range of tumor types, a major concern in employing these anti-mitotic compounds has been their toxicity to normal cell types such as neurons [9]. Recently, a selected group of protein kinases has been identified as important orchestrators for mitotic progressions [10]. This select group includes cyclin dependent kinases (Cdks) [11, 12], Polo-like-kinases [13], and Aurora kinases [14, 15]. It has been widely recognized that mitotic kinases represent attractive targets for developing cancer therapeutics [16, 17]. Activities of mitotic kinases could be selectively modulated using small molecule based inhibitors [18], which could potentially have fewer side effects and be more selective in attacking proliferative cells than the conventional microtubule poisons [19]. Recent studies have revealed elevated expression levels of Aurora A, Aurora B, and the polo-like kinase 1 (Plk1), in clinical melanoma samples [20, 21], and small molecule inhibitors of these kinases have shown promising potential in delaying melanoma progression either as single-agent or in combination with existing therapeutic drugs, such as MEK inhibitors [22-24].
Haspin (Haploid Germ Cell-Specific Nuclear Protein Kinase) is a recently identified mitotic kinase that phosphorylates histone H3 at threonine 3 [25]. This specific phosphorylation serves as a docking site for the chromosome passenger complex at the centromere during mitosis and is critical for centromeric functions of Aurora B [26]. Depletion of haspin by small interfering RNAs (siRNAs) leads to chromosome misalignment, precocious loss of cohesion between sister chromatids, and formation of multipolar spindles [27, 28]. Haspin is among a few proteins that are considered as atypical eukaryotic protein kinases, due to the lack of the conserved ATP/Mg2+ binding motif Asp-Phe-Gly (DFG) in its activation segment and the divergent structure in his kinase domain [29]. Such structural distinction has rendered it possible to design more selective or specific inhibitors of haspin and to reduce side effects potentially due to concomitant activities against kinases with a more conserved activation segment [30].
A number of small molecule based inhibitors targeting the ATP binding site of haspin has been discovered via high throughput screening [31-34]. Recently, bisubstrate inhibitors of haspin have also been designed by conjugating the ATP-site targeting aromatic fragment with a peptide that mimics the N-terminus of histone H3 tail [35, 36]. Among known haspin inhibitors, compound CHR-6494 has shown anti-proliferative effects on a number of cancer cell lines including cervical, colon, and breast cancer cells [33]. The mode of action is likely the mitotic catastrophe associated with chromosome misalignment and formation of multipolar spindles, and ex vivo assays also reveal anti-angiogenesis activities of CHR-6494. However, to the best of our knowledge, activities of CHR-6494 have not been examined in melanoma cell lines, and the potential of haspin as an anti-mitotic target to melanoma treatment has not been explored. We wish to report herein antitumor potential of the haspin inhibitor CHR-6496 as a single agent and in combination with other oncogene targeting drugs in melanoma treatment.
Methods
Chemicals and reagents
CHR-6494 was purchased from Sigma-Aldrich (St Louis, Missouri, USA). GSK1120212 (Trametinib) and PLX4032 were from Selleck Chemicals (Houston, Texas, USA).
Cell culture
Human melanoma cell lines were cultured in DMEM (Corning Incorporated, Corning, New York, USA) or RPMI1640 (Life Technologies, Shanghai, China) supplemented with 10% (v/v) fetal bovine serum (FBS) (Capricorn Scientific) and penicillin (100 U/mL)/ streptomycin (100 ug/mL) (Solarbio, Beijing, China) [37]. Human dermal fibroblasts (HDFs) were cultured in DMEM medium supplemented with 10% FBS and penicillin/ streptomycin. All cells were maintained at 37° C with 5% CO2.
Cell viability assay
Cells were seeded as triplicates in 96-well culture plates (Corning) at a density of 4,000 cells/well. After 24 h of attachment, cells were treated with CHR-6494, a MEK inhibitor, or vehicle (DMSO) at various concentrations. At the end of treatment, cells were fixed with 4% paraformaldehyde/PBS at room temperature for 10 min, and stained with 0.05% crystal violet diluted in PBS at room temperature for 20 min. After washing these plates with tap water and air dried, the dye was solubilized with 30% acetic acid and the absorbance was measured at 590 nm using the Tecan Infinite 200 Pro Microplate Reader. Cell viability of the treated group was normalized to the vehicle control. IC50 values were determined using the Prism 7.0 Software (GraphPad Software Inc., LaJolla, California, USA). Degrees of synergy between two compounds were assessed numerically using Combination Index (CI) and Dose-Reduction Index (DRI), which were calculated based on the Chou-Talalay method [38] employing the CalcuSyn Software (Biosoft, Cambridge, UK). CI < 1, synergy; CI = 1, additive; CI > 1, antagonism; DRI = 1, no dose-reduction; DRI > 1, favorable dose-reduction, DRI > 1, unfavorable dose-reduction.
Western blot analysis
Whole cell lysates for immunoblotting were prepared with the 0.5 X Laemmli Sample Buffer. The volume of buffer was adjusted according to the cell number. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, Darmstadt, Germany). The following antibodies were used for immunoblotting: Anti-phospho-Histone H3 (Thr3) Antibody, (clone JY325, EMD Millipore, Darmstadt, Germany), cleaved PARP (Cell Signaling, Danvers, MA, USA), and α-tubulin (Sigma-Aldrich, St. Louis, MO).
Caspase 3/7 activity-based apoptosis assay
Cells were seeded in a six-well plate (2.5x105 cells/well). After attachment for 24 h, cells were treated with compounds at various concentrations. At the end of treatment, cells were detached and cell pellets were lysed with 300 μL of 1X cell lysis buffer (CWBIO, Beijing, China). Protein concentrations were determined using the BCA Protein Assay Kit (CWBIO, Beijing, China). Caspase 3/7 activity was measured with the Caspase-Glo 3/7 Assay kit (Promega, Madison, Wisconsin, USA) according to manufacturer's instruction, using the Tecan Infinite 200 Pro Microplate Reader. The Caspase-Glo 3/7 activity was normalized to the protein concentration.
Cell cycle analysis
MeWo or MDA-MB-435 cells were plated in 60-mm dishes at a density of 8.5 x105/dish. After treatment with haspin or MEK inhibitors for 48 h, cells were fixed in ice-cold 70% ethanol, and stained with 50 μg/mL propidium iodide and 100 U/mL RNAse A (Leagene, Beijing, China) diluted in PBS at 25°C for 1 h. DNA content analysis was conducted on a FACSCalibur flow cytometer (Becton Dickinson, San Diego, CA).
Scratch assay
MDA-MB-435 cells were grown to confluence in 60-mm dishes. After serum starvation for 18 h, cells were treated with mitomycin C (10 μg/mL) for 2 h to prevent cell proliferation during the later migration process. The cell-free wound areas were generated on monolayers using a 200-μL pipette tip. After washing with PBS twice, the marked area of each wound was photographed under Nikon Eclipse Ti-S Inverted phase-contrast microscope. Cells were re-fed with DMEM medium containing 5% FBS as well as various compounds or DMSO (vehicle). After incubating for 48 h, the marked wound area was re-photographed. The wound closure area (cell migration) was analyzed using the Image J Software.
Transwell migration assay
Cell migration assays were performed using 24-well Co-star transwell inserts (8 μm pore; Corning). MDA-MB-435 cells were treated with various compounds or DMSO (vehicle) for 3 days. After which, an equal amount of cells (1.2 × 105) were seeded into the upper chamber of a transwell inserted in serum-free medium. The lower chamber of the transwell was filled with medium containing 10% FBS as chemo-attractant. After incubating for 18 h, cells that have migrated to the bottom side of the transwell membrane were fixed with 4% PFA/PBS followed by staining with 0.1% crystal violet at room temperature for 20 min. The migrated cells were viewed with Nikon Eclipse Ti-S inverted microscope and counted from 5 different fields to get an average sum of cells.
Statistics
All statistical evaluations were carried out using the GraphPad Prism 7.0 Software. Data were analyzed by Student's t-test for comparison between two groups or two-way ANNOVA for comparison between multiple groups. Combined data were obtained from 3 independent experiments and presented as mean-fold over control ± S.E.M. P-values < 0.05 were considered significant.
Results
CHR-6494 inhibits histone H3 phosphorylation at Thr3 and exhibits anti-proliferative and pro-apoptotic effects as a single agent in melanoma cell lines
We first examined sensitivities of a panel of melanoma cell lines to the haspin inhibitor CHR-6494 and the MEK inhibitor GSK1120212/Trametinib (referred to as MEKi) as single agents. CHR-6494 showed a dose-dependent inhibitory effect on cell viability in all melanoma cell lines tested, which included BRAFV600E mutants, NRAS mutants, and wild type cells. The respective IC50 values ranged between 396 nM and 1229 nM (Fig. 1A). While most melanoma cell lines were responsive to MEKi with IC50 values between 1.1 nM and 3.6 nM, the BRAFV600E mutant RPMI-7951 and the wild type COLO-792 cells were resistant to MEKi even at 50 nM, which was the highest concentration dosage employed (Fig. 1B). In contrast to other BRAF mutant cell lines, RPMI-7951 was also resistant to BRAF inhibitors PLX4032 (data not shown) and PLX4702 [37]. Our subsequent studies focused on anti-tumor potential of CHR-6494 as a single agent against these two specific melanoma cell lines that are non-responsive to BRAF and MEK inhibitors.
CHR-6494 decreased cell viability and increased apoptosis in melanoma cells. Melanoma cells (as indicated) were plated as triplicates in 96-well plates, and treated with CHR-6494, MEK inhibitor GSK1120212 (MEKi), or DMSO (vehicle control) at various concentrations for 72 h. (A-C) Cell viability was determined with crystal violet staining assay. The y-axis represents mean-percentage of viable cells ± S.E.M, N=3 independent experiments. (D) Western blot analysis using an antibody against histone H3 Thr3 phosphorylation (H3T3P). (E) Caspase 3/7 activity measured using the Caspase 3/7 Glo Assay Kit. Respective values are normalized to protein concentrations and presented as mean-fold over control ± S.E.M. *, p < 0.05, **, p < 0.01, N=3. (F) Western blot analysis for the cleaved form of PARP.
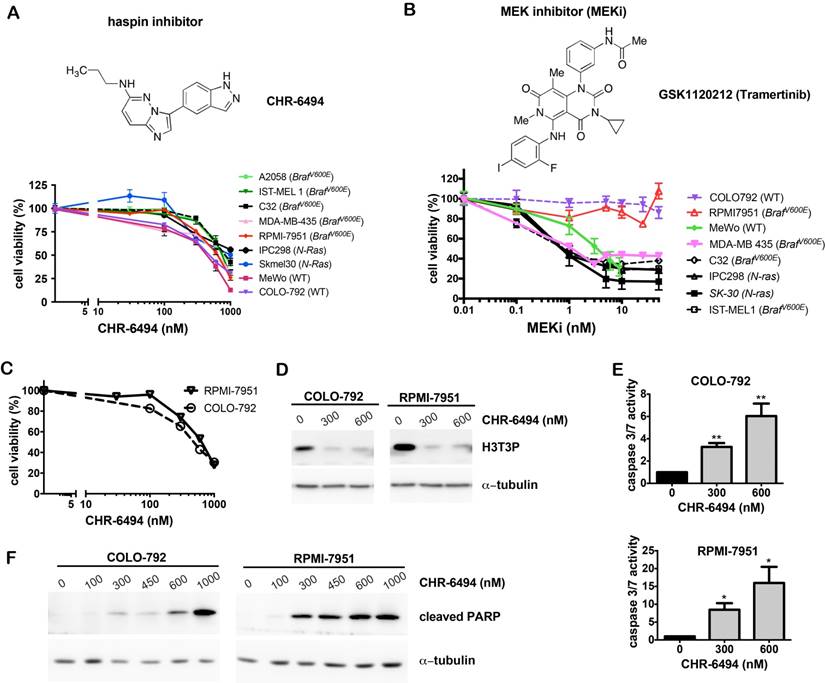
As shown in Fig. 1C, treatment of CHR-6494 suppressed the viability of COLO-792 and RPMI-7951 cells in a dose-dependent manner with IC50 values of 497 nM and 628 nM, respectively. Western blot analysis showed that CHR-6494 markedly reduced the level of histone H3 Thr3-phosphorylation (H3T3P), thereby revealing for the first time its inhibitory effect on haspin activity in melanoma cells (Fig. 1D). Moreover, when using 300 nM and 600 nM of CHR-6494, caspase 3/7 activity was increased by 3- and 6-fold, respectively in COLO-792 cells, and to 8.5- and 16-fold in RPMI-7951 cells (Fig. 1E). The pro-apoptotic effect of CHR-6494 was further confirmed by increased level of caspase 3-cleaved form of PARP (cPARP) as measured by western blot analysis (Fig. 1F). These results indicate that treatment with the haspin inhibitor as a single agent could reduce cell viability and induce apoptosis in both oncogene mutant and wild type melanoma cells, particularly those that are resistant to BRAF and MEK inhibitors.
Haspin and MEK inhibitors synergistically inhibit viability of melanoma cells
We next determined the combined effect on melanoma cells when employing both CHR-6494 and the MEK inhibitor GSK1120212 (Trametinib) simultaneously. Both the BRAFV600E mutant MDA-MB-435 and the wild type MeWo melanoma cell lines were responsive to treatment with either compound as a single agent. As shown in Fig. 2A, CHR-6494 inhibited viabilities of MeWo and MDA-MB-435 cells in a dose dependent manner with respective IC50 values of 396 nM and 611 nM, whereas normal dermal fibroblasts (HDFs) were less sensitive with an IC50 > 1μM (Fig. 2A). In MeWo cells, the IC50 value for single MEKi treatment was approximately 3.6 nM (Fig 1B). Dual treatment by combining CHR-6494 (100 nM, 300 nM, or 600 nM) with MEKi (0.3 nM, 1 nM, or 3 nM) elicited an enhancement in cytotoxicity (Fig. 2B). For example, treatment with CHR-6494 (300 nM) and MEKi (1 nM) as single agents reduced cell viability to 64.4% and 72.8% of the control, respectively. On the other hand, dual treatment by combining both inhibitors led to an enhanced effect of reducing cell viability to only 22.6% of the control (Fig. 2B).
CHR-6494 and MEK inhibitor synergistically reduced cell viability of MeWo and MDA-MB-435 melanoma cell lines. MeWo and MDA-MB-435 melanoma cells or human dermal fibroblasts (HDFs) were treated with CHR-6494, MEK inhibitor GSK1120212 (MEKi), or the combination of two compounds at various concentrations for 5 d. Cell viabilities were measured by crystal violet staining assay in: (A) Cells treated with CHR-6494 as single agent; (B) MeWo melanoma cells. (C) MDA-MB-435 melanoma cells; (D) HDFs. Respective values are presented as mean-percentage of control ± S.E.M. *, p < 0.05, **, p < 0.01 ***, p < 0.001, N=3. (E) The degree of synergy between CHR-6494 and MEKi was assessed numerically employing Combination Index (CI) and Dose Reduction Index (DRI), which were calculated based on the Chou-Talalay method using the CalcuSyn Software. CI < 1, synergy; CI = 1, additive; CI > 1, antagonism. DRI = 1, no dose-reduction; DRI > 1, favorable dose-reduction; DRI < 1, unfavorable dose-reduction.
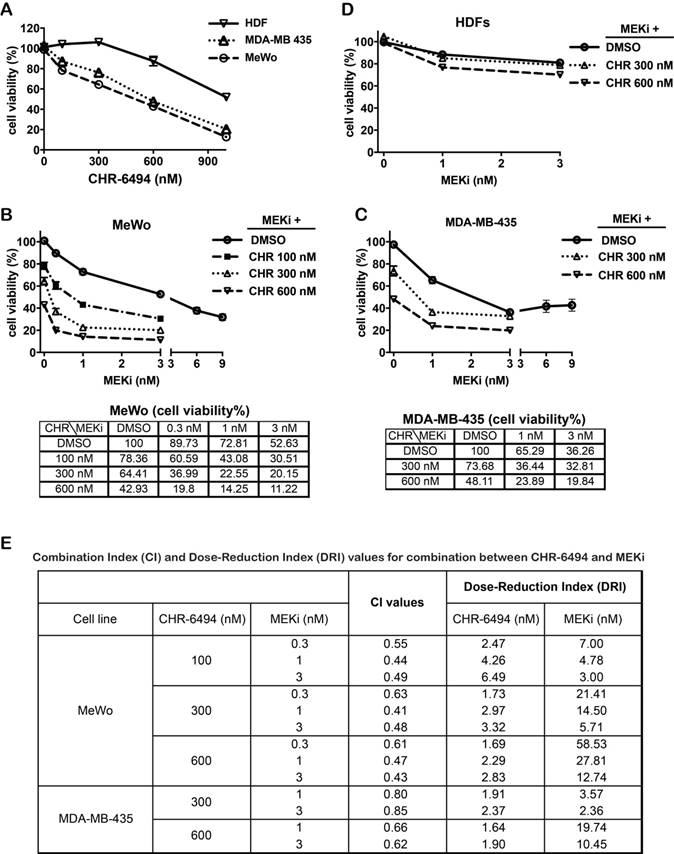
In comparison with MeWo cells, MDA-MB-435 cells were more sensitive to MEKi as a single agent (IC50 = 2.6 nM) (Fig. 1B). An enhanced cytotoxicity was also detected when CHR-6494 (300 nM or 600 nM) was combined with MEKi (1 nM or 3 nM) in this cell line (Fig. 2C). In contrast, normal human dermal fibroblasts (HDFs) were relative insensitive to MEKi, and more importantly, MEKi induced growth inhibition was only slightly enhanced when it was combined with 600 nM of CHR-6494 (Fig. 2D). Degrees of synergy between CHR-6494 and MEKi in melanoma cells were further assessed through Combination Index (CI) and the reciprocal Dose-Reduction Index (DRI), using the Chou-Talalay method [38, 39]. While CI < 1 is often used as an indication of synergy, DRI quantifies how many folds in dosages single drugs can be reduced to achieve the synergistic effect at a given level. DRI > 1 represents a situation of favorable dose-reduction. As shown in Fig. 2E, CI < 1 and DR > 1 values were achieved via all combination options tested in our studies, thereby confirming the synergistic effects between CHR-6494 and MEKi in the two melanoma cell lines.
Co-inhibition of haspin and MEK enhances apoptosis in melanoma cells
Our next goal was to determine whether the observed synergism between haspin and MEK inhibitors on cell viability was associated with enhanced apoptosis. For both MeWo and MDA-MB-435 cells, treatment with CHR-6494 (300 nM or 600 nM) as a single agent markedly increased the expression level of cPARP, and MEKi alone only showed a marginal effect at 3 nM (Fig. 3A). The intensity of cPARP signal was significantly enhanced when employing CHR-6494 and MEKi in combination (Fig. 3A). In contrast, the cPARP expression was not detected in HDFs when treated with CHR-6494 and MEKi either as single agents or as dual inhibitors (Fig. 3B).
Combination between CHR-6494 and the MEK inhibitor enhanced apoptosis in MeWo and MDA-MB-435 melanoma cell lines. MeWo and MDA-MB-435 melanoma cells or HDFs were treated with CHR-6494, MEK inhibitor GSK1120212 (MEKi), or the combination of two compounds at various concentrations for 5 days. (A-B) Cells were harvested for western blot analysis using an antibody against the cleaved form of PARP. (C) Caspase 3/7 activity was measured using the Caspase 3/7 Glo Assay Kit and normalized to protein concentrations. Respective values are presented as mean-fold over control ± S.E.M. *, p < 0.05, **, p < 0.01 ***, p < 0.001, N=3.
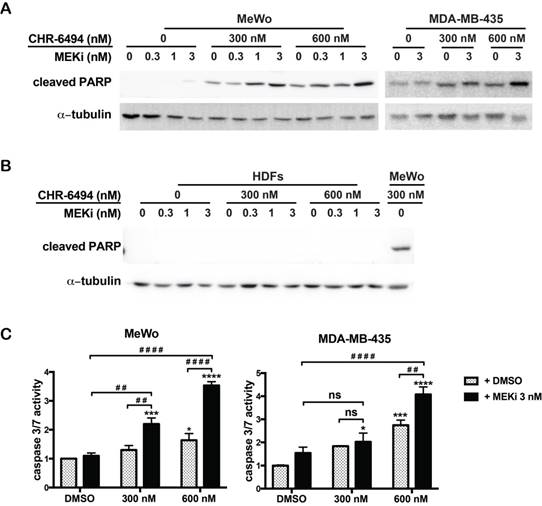
Consistently, when used as a single agent, MEKi (3 nM) showed no significant impact on the caspase 3/7 activity, while CHR-6494 alone at 600 nM increased this activity to 1.6- and 2.7-fold in MeWo and MDA-MB-435 cells, respectively (Fig. 3C). Caspase 3/7 activity was significantly increased following dual treatment of these two compounds (Fig. 3C). These results support the notion that the synergistic effect of haspin and MEKi inhibition on cell viability is associated with increased apoptotic responses in melanoma cells.
Haspin and MEK inhibitors modulate cell cycle progression independently by arresting melanoma cells at different phases
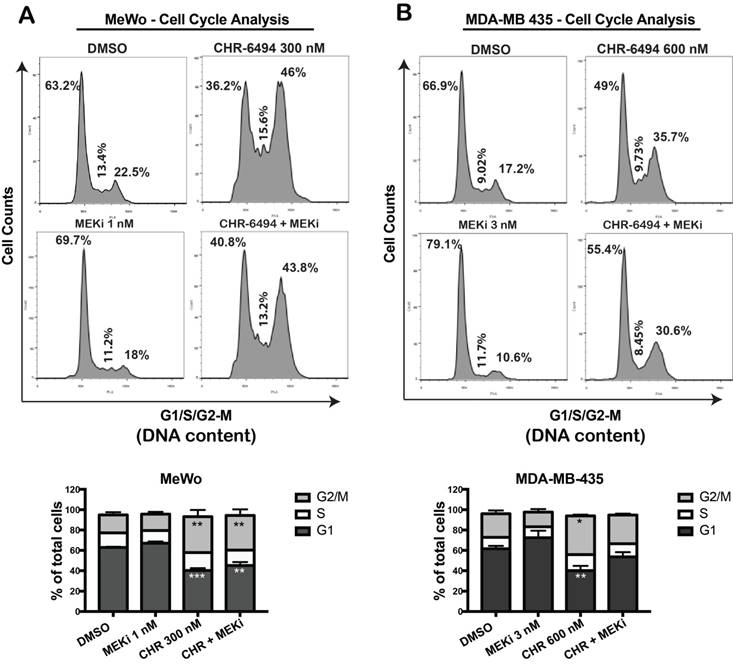
We utilized flow cytometry approach to determine whether the observed synergistic effect of haspin and MEKi inhibitors were associated with a perturbed cell cycle progression. In MeWo cells, while single treatment with 1 nM of MEKi caused a slight increase in G1 population, CHR-6494 at 300 nM led to a significant cell cycle arrest at G2/M phase (Fig. 4A). Combined treatment led to a cell cycle profile comparable to that of cells treated with CHR-6494 alone, although the magnitude of G2/M arrest was decreased (Fig. 4A). Similarly in MDA-MB-435 cells, a significant G2/M arrest was achieved with 600 nM of CHR-6494, whereas a moderate increase in G1 population was observed in cells treated with 3 nM of MEKi (Fig. 4B). Under dual inhibitions, the level of G2/M arrest induced by CHR-6494 alone was reduced, with more cells shifting to G1 phase (Fig. 4B). These results indicate that inhibitions of haspin and MEK likely interfere cell cycle progression independently, thereby rendering cells arrested at different phases.
CHR-6494 and MEKi led to cell cycle arrest at different phases. MeWo (A) and MDA-MB-435 (B) melanoma cells were treated with CHR-6494, MEK inhibitor GSK1120212 (MEKi), or the combination of two compounds at various concentrations for 48 h. Cells were fixed with cold 70% ethanol and stained with propidium iodide. DNA content was measured by flow cytometry. Representative results are shown in top panels. In the bottom panels, data are presented as mean percentage of total cells ± S.E.M. *, p < 0.05, **, p < 0.01 ***, p < 0.001, N=3 independent experiments.
Haspin and MEK inhibitors cooperatively suppress migration of melanoma cells
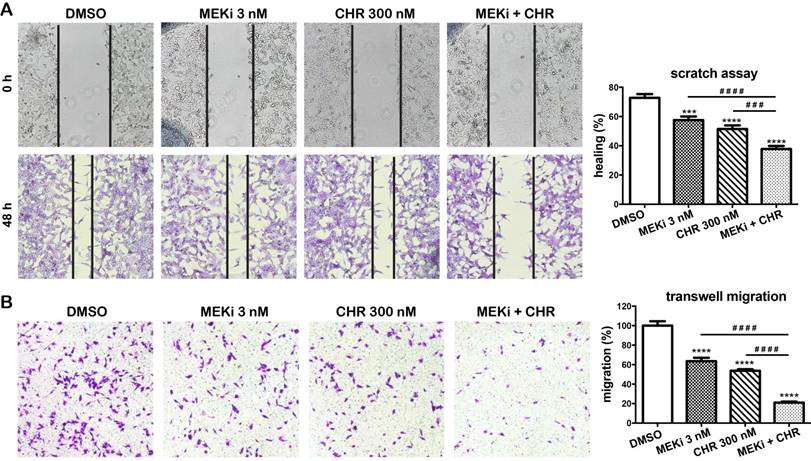
We next investigate effects of haspin inhibitor alone or in combination with MEKi on migration in MDA-MB-435 melanoma cells. In a two-dimensional scratch assay, after 48 h, only 57.6% and 51.5% of the gap were closed in cells treated with 3 nM of MEKi and 300 nM of CHR-9494, respectively, whereas 72.8% of the original gap was closed in the vehicle (DMSO) treated control cells (Fig. 5A). In contrast, 37.9% of original gap was closed when both compounds are utilized in a dual treatment (Fig. 5A). The migratory ability of cells was also examined by the transwell migration assay using Boyden chambers. MDA-MB-435 cells were treated with CHR-6494 and MEKi as single agents or in combination for 72 h before the same number of cells were loaded into the upper chamber of transwells and examined 18 h later. As shown in Figure 5B, the number of cells that migrated to the bottom side of the membrane was reduced to 66% and 54% of the control with individual treatment using either 3 nM of MEKi or 300 nM of CHR-9494, respectively. In contrast, the migrated cell number was decreased to 21% of the control after dual treatment using both CHR-6494 and MEKi (Fig. 5B). These results indicate that simultaneous inhibition of haspin and MEK activities may elicit a much greater potential in suppressing migratory ability of melanoma cells.
Co-treatment with CHR-6494 and MEKi enhanced the inhibitory effects on cell migration in MDA-MB-435 melanoma cell lines. (A) Scratch assay (wound closure assay). Cell monolayers were scratched and wounds at the marked areas were photographed at 0 h. After 48 h incubation with various compounds or DMSO, cells were fixed and stained with 0.1% crystal violet. Cells from the same wound areas were photographed. Photographs shown are representative of three independent experiments. The value is presented as mean-percentage of migration ± S.E.M. ***, p < 0.001, ****, p < 0.0001, N=3. (B) Transwell migration assay. MDA-MB-435 cells were treated with a respective compound at various concentrations for 72 h. Surviving cells (1.2x105) were seeded onto the upper chamber of transwell inserts as described in Materials and Methods. Cells were then incubated with 3 nM of MEKi (GSK-1120212), 300 nM of CHR-6494, or both compounds, for 18 h at 37 °C. The number of migrated cells was counted from 5 fields per experiment. Data shown are the mean percentage of control ± S.E.M. from three independent experiments, **** P < 0.0001.
Discussion
Melanoma is a heterogeneous malignancy that is one of the most aggressive tumors resistant to treatment when becoming metastatic, and thus, presents an immense challenge in the development of therapeutic approaches. Aberrant activations of the MAP kinase pathway have been associated with an array of human cancers including melanoma [1]. Therapeutics targeting against BRAF as well as its downstream kinases MEK and ERK have shown great potential in prolonging the life span of patients with advanced melanoma. However, the common occurrence of drug resistance to single-agent chemotherapeutic treatment accentuates the need to continue discovering alternative or new strategies for anti-melanoma [4].
In this study, we found evidence of utilizing CHR-6494, a small molecule based inhibitor of the mitotic kinase haspin, to treat melanoma cell lines including both wild type and BRAF mutant cells. As a single agent, CHR-6494 was shown to cause a dose dependent growth inhibitory and pro-apoptotic effect in COLO-792 and RPMI-7951 cells, which are cells not responsive to the MEK inhibitor GSK1120212 (Trametinib). Intriguingly, a combination of haspin and MEK inhibitions revealed a synergistic effect on the viability of both wild type (MeWo) and the BRAFV600E mutant (MDA-MB-435) cell lines. Most importantly, the dual treatment also enhanced apoptotic responses and elicited a greater inhibitory effect on cell migrations, and overall, these effects were not observed in the normal human dermal fibroblasts. Therefore, our findings suggest that haspin could serve as a novel target for melanoma treatment either via a single-agent or in combination therapy, and provides an alternative approach for drug-resistant melanoma cells.
Mitosis has been widely recognized as an attractive target for cancer therapeutic development because it is an essential step for cell proliferations. While none of the commonly used antimitotic agents has led to improved survival, the newly developed solvent-free and albumin-bound form of Paclitaxel (Nab-paclitaxel) has shown beneficial effect in prolonging overall survival of chemotherapy-naïve patients with metastatic melanoma in an ongoing phase III trial [40]. Furthermore, Aurora B inhibitor barsertib-HQPA has been shown to exert an anti-proliferative effect on metastatic melanoma cells regardless of their BRAF mutational status [41]. Therefore, by identifying novel inhibitors of mitotic kinases or structurally modifying existing spindle poisons, anti-mitosis represents an attractive strategy for melanoma treatment. Our results have particularly pinpointed the great potential of using small molecule based inhibitors of the mitotic kinase haspin as a single-agent or in combinational therapy for treatment of melanoma.
It should be noted that MEK inhibitors have shown a synergistic effect with several anti-mitotic agents. In lung and ovarian cancer cell lines with constitutive activation of the ERK signaling pathways, MEK inhibitor U0126 selectively enhances the apoptotic responses induced by the microtubule inhibitor paclitaxel [42]. The increased cell death by MEK/paclitaxel combination is associated with the cooperative up-regulation of the pro-apoptotic protein Bim and down-regulation of the anti-apoptotic protein Mcl-1 [43]. MEK/ERK inhibition could also sensitize neuroblastoma cells to the inhibitors of centromere-associated protein E (CENP E), a kinesin motor protein that is critical for chromosome attachment to mitotic spindles [44]. Moreover, combined inhibition of MEK and Plk1 elicits synergistic antitumor activity specifically in NRAS mutant melanoma [24]. In our study, the synergy found between the inhibitors of MEK and the other mitotic kinase haspin in both wild type and BRAF mutant melanoma cells further support the beneficial potential of simultaneously targeting mitosis and ERK pathways in melanoma therapy.
Despite the synergistic effect found between anti-mitotic drugs and MEK inhibitors, the underlying mechanism remains unclear. At this moment, our cell cycle analysis showed that inhibitions of haspin and MEK as a single agent led to cell cycle arrests at G2/M and G1, respectively, but degrees of both arrests were down regulated when cells were treated with these two compounds as a combination. Therefore, it is unlikely that the enhanced growth inhibitory and apoptotic response by dual-inhibition are due to enhanced activation of either G2/M or G1 checkpoints. Instead, it is possible that single inhibition of haspin and MEK could independently activate G2/M and G1 checkpoints, thereby resulting an improved efficiency to impede cell cycle progression and to initiate apoptotic responses.
Lastly, it has been suggested that growth factors secreted in the tumor microenvironment can rescue cancer cells from kinase inhibitions. When the A431 epidermoid cancer cells are treated with the epithelial growth factor receptor (EGFR) inhibitor Gefitinib, haspin is the most up-regulated kinase followed by fibroblast growth factors (FGF2) stimulation, thereby implying that haspin could be part of the molecular mechanism in the Gefitinib resistance [45]. Moreover, FGF2 rescued cells were highly sensitive to inhibition of haspin, and dual inhibitions of EGFR and haspin activities showed an enhanced anti-tumor efficacy than the single treatment [45]. Their results further support the notion that haspin could be an ideal target to overcome resistance to drugs targeting growth factors and their downstream pro-survival pathways in different types of cancer cells including melanoma.
Conclusion
Our study has demonstrated that haspin, a structurally atypical mitotic kinase, could be a viable target for anti-melanoma therapy and possesses the potential for developing combination therapy using small molecule based drugs. Given its unique protein structure, selective or specific inhibitors of haspin could be envisioned and developed to overcome off-target effects common for inhibitors of other known kinases. Further studies will focus on whether haspin inhibitors could enhance the growth arrest and apoptosis in combination with other inhibitors of MAPK pathways such as BRAF inhibitors, and whether these strategies could be applied to all melanoma and other cancer cell types.
Abbreviations
Haspin: Haploid Germ Cell-Specific Nuclear Protein Kinase; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; MEK: MAPK/ERK kinase; Cdks: cyclin dependent kinases; Plk1: polo-like kinase 1; siRNA: small interfering RNA; H3T3P: histone H3 Thr3 phosphorylation; CI: Combination Index; PARP: Poly ADP-Ribose Polymerase; DRI: Dose Reduction Index; cPARP: caspase 3-cleaved form of PARP; HDFs: human dermal fibroblasts.
Acknowledgements
Authors are grateful for generous funding from School of Pharmaceutical Science and Technologies at Tianjin University as well as from School of Pharmacy at University of Wisconsin-Madison. JD also acknowledges partial support of this work by The National Institutes of Health (K01AR062132). Authors would like to thank Professor David E. Fisher of Cutaneous Biology Research Center at Massachusetts General Hospital and Professor Richard P. Hsung of School of Pharmacy at University of Wisconsin-Madison for invaluable discussions.
Authors' Contributions
JD and LH conceived and designed the experiments. LH, PW, YS, and SL performed the experiment. JD and LH wrote the manuscript. All authors wrote and approved the final version of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. 2012;12:349-61
2. Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M. et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5:1701-52
3. Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132:854-63
4. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297-304
5. Flaherty KT. Targeting metastatic melanoma. Annu Rev Med. 2012;63:171-83
6. Marzo I, Naval J. Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem Pharmacol. 2013;86:703-10
7. Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat. 2007;10:162-81
8. van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015;76:1101-12
9. Gornstein E, Schwarz TL. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology. 2014:76 Pt A: 175-83
10. Ma HT, Poon RY. How protein kinases co-ordinate mitosis in animal cells. Biochem J. 2011;435:17-31
11. Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K. et al. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859-64
12. Malumbres M, Pevarello P, Barbacid M, Bischoff JR. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci. 2008;29:16-21
13. Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther. 2016;15:1427-35
14. Hrabakova R, Kollareddy M, Tyleckova J, Halada P, Hajduch M, Gadher SJ. et al. Cancer cell resistance to aurora kinase inhibitors: identification of novel targets for cancer therapy. J Proteome Res. 2013;12:455-69
15. Taylor S, Peters JM. Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol. 2008;20:77-84
16. Penna LS, Henriques JA, Bonatto D. Anti-mitotic agents: Are they emerging molecules for cancer treatment?. Pharmacol Ther. 2017
17. Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell. 2015;60:524-36
18. Bayliss R, Fry A, Haq T, Yeoh S. On the molecular mechanisms of mitotic kinase activation. Open Biol. 2012;2:120136
19. Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012;19:369-77
20. Cholewa BD, Ndiaye MA, Huang W, Liu X, Ahmad N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017;385:179-87
21. Wang X, Moschos SJ, Becker D. Functional analysis and molecular targeting of aurora kinases a and B in advanced melanoma. Genes Cancer. 2010;1:952-63
22. Fletcher GC, Brokx RD, Denny TA, Hembrough TA, Plum SM, Fogler WE. et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol Cancer Ther. 2011;10:126-37
23. Xie L, Meyskens FL Jr. The pan-Aurora kinase inhibitor, PHA-739358, induces apoptosis and inhibits migration in melanoma cell lines. Melanoma Res. 2013;23:102-13
24. Posch C, Cholewa BD, Vujic I, Sanlorenzo M, Ma J, Kim ST. et al. Combined Inhibition of MEK and Plk1 Has Synergistic Antitumor Activity in NRAS Mutant Melanoma. J Invest Dermatol. 2015;135:2475-83
25. Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19:472-88
26. Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT. et al. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science. 2010;330:231-5
27. Dai J, Sullivan BA, Higgins JM. Regulation of mitotic chromosome cohesion by Haspin and Aurora B. Dev Cell. 2006;11:741-50
28. Dai J, Kateneva AV, Higgins JM. Studies of haspin-depleted cells reveal that spindle-pole integrity in mitosis requires chromosome cohesion. J Cell Sci. 2009;122:4168-76
29. Higgins JM. Haspin-like proteins: a new family of evolutionarily conserved putative eukaryotic protein kinases. Protein Sci. 2001;10:1677-84
30. Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW. et al. Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci U S A. 2009;106:20198-203
31. Patnaik D, Jun X, Glicksman MA, Cuny GD, Stein RL, Higgins JM. Identification of small molecule inhibitors of the mitotic kinase haspin by high-throughput screening using a homogeneous time-resolved fluorescence resonance energy transfer assay. J Biomol Screen. 2008;13:1025-34
32. De Antoni A, Maffini S, Knapp S, Musacchio A, Santaguida S. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J Cell Biol. 2012;199:269-84
33. Huertas D, Soler M, Moreto J, Villanueva A, Martinez A, Vidal A. et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene. 2012;31:1408-18
34. Wang F, Ulyanova NP, Daum JR, Patnaik D, Kateneva AV, Gorbsky GJ. et al. Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J Cell Biol. 2012;199:251-68
35. Kestav K, Lavogina D, Raidaru G, Chaikuad A, Knapp S, Uri A. Bisubstrate inhibitor approach for targeting mitotic kinase Haspin. Bioconjug Chem. 2015;26:225-34
36. Kestav K, Viht K, Konovalov A, Enkvist E, Uri A, Lavogina D. Slowly on, Slowly off: Bisubstrate-Analogue Conjugates of 5-Iodotubercidin and Histone H3 Peptide Targeting Protein Kinase Haspin. Chembiochem. 2017
37. Friedman AA, Amzallag A, Pruteanu-Malinici I, Baniya S, Cooper ZA, Piris A. et al. Landscape of Targeted Anti-Cancer Drug Synergies in Melanoma Identifies a Novel BRAF-VEGFR/PDGFR Combination Treatment. PLoS One. 2015;10:e0140310
38. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
39. Cuccarese MF, Singh A, Amiji M, O'Doherty GA. A novel use of gentamicin in the ROS-mediated sensitization of NCI-H460 lung cancer cells to various anticancer agents. ACS Chem Biol. 2013;8:2771-7
40. Hersh EM, Del Vecchio M, Brown MP, Kefford R, Loquai C, Testori A. et al. A randomized, controlled phase III trial of nab-Paclitaxel versus dacarbazine in chemotherapy-naive patients with metastatic melanoma. Ann Oncol. 2015;26:2267-74
41. Porcelli L, Guida G, Quatrale AE, Cocco T, Sidella L, Maida I. et al. Aurora kinase B inhibition reduces the proliferation of metastatic melanoma cells and enhances the response to chemotherapy. J Transl Med. 2015;13:26
42. MacKeigan JP, Collins TS, Ting JP. MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem. 2000;275:38953-6
43. Kawabata T, Tanimura S, Asai K, Kawasaki R, Matsumaru Y, Kohno M. Up-regulation of pro-apoptotic protein Bim and down-regulation of anti-apoptotic protein Mcl-1 cooperatively mediate enhanced tumor cell death induced by the combination of ERK kinase (MEK) inhibitor and microtubule inhibitor. J Biol Chem. 2012;287:10289-300
44. Mayes PA, Degenhardt YY, Wood A, Toporovskya Y, Diskin SJ, Haglund E. et al. Mitogen-activated protein kinase (MEK/ERK) inhibition sensitizes cancer cells to centromere-associated protein E inhibition. Int J Cancer. 2013;132:E149-57
45. Iwamoto E, Ueta N, Matsui Y, Kamijo K, Kuga T, Saito Y. et al. ERK Plays a Role in Chromosome Alignment and Participates in M-Phase Progression. J Cell Biochem. 2016;117:1340-51
Author contact
![]() Corresponding author: Jun Dai, PhD, Assistant Professor, Division of Pharmaceutical Sciences, School of Pharmacy, 777 Highland Avenue, University of Wisconsin, Madison, WI 53705 USA Tel: 608-890-1063 Fax: 608-262-5345 Email: jdai32edu
Corresponding author: Jun Dai, PhD, Assistant Professor, Division of Pharmaceutical Sciences, School of Pharmacy, 777 Highland Avenue, University of Wisconsin, Madison, WI 53705 USA Tel: 608-890-1063 Fax: 608-262-5345 Email: jdai32edu