Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(15):3037-3048. doi:10.7150/jca.19315 This issue Cite
Research Paper
MiR-340 Inhibits Triple-Negative Breast Cancer Progression by Reversing EZH2 Mediated miRNAs Dysregulated Expressions
Zhendong Shi1, 2, 3* ![]() , Yang Li1, 2, 3*, Xiaomin Qian4*, Yunhui Hu1, 2, 3, Jingjing Liu1, 2, 3, Sheng Zhang1, 2, 3, Jin Zhang1, 2, 3
, Yang Li1, 2, 3*, Xiaomin Qian4*, Yunhui Hu1, 2, 3, Jingjing Liu1, 2, 3, Sheng Zhang1, 2, 3, Jin Zhang1, 2, 3 ![]()
1. 3rd Department of Breast Cancer, China Tianjin Breast Cancer Prevention, Treatment and Research center, Tianjin Medical University Cancer Institute and Hospital;
2. Key laboratory of breast cancer prevention and therapy of ministry of education;
3. Key laboratory of cancer prevention and therapy, Tianjin, PR China;
4. School of Laboratory Medicine, Tianjin Medical University, Tianjin, PR China.
* These authors contributed equally to this work.
Received 2017-1-23; Accepted 2017-5-29; Published 2017-9-2
Abstract
The anti-tumor efficacy of miR-340 has been recently characterized in cancers. However, the underlying mechanisms of miR-340 inhibited cell growth and invasion in triple-negative breast cancer (TNBC) have not been well elucidated. In this study, we found that miR-340 expression was negatively correlated with EZH2 (Enhancer of zeste homolog 2) expression in TNBC tissues and cell lines. Subsequent luciferase reporter assay confirmed that EZH2 was a novel molecule target of miR-340. Upregulated miR-340 levels by mimics transfection significantly inhibited the MDA-MB-231 and MDA-MB-468 breast cancer cells proliferation, invasion and migration, and induced more cell apoptosis. Meanwhile, miR-340 inhibited the tumor growth in an orthotopic MDA-MB-231 breast cancer mouse model. Furthermore, we found the reduced EZH2 expression by miR-340 mimics transfection decreased the DNMT1, H3K27me3, β-catenin and P-STAT3 expressions, which ultimately resulted in miR-21 activity blockage and miR-200a/b expression upregulation. The results of rescue experiments further confirmed that miR-340 inhibited triple-negative breast cancer progression through targeting EZH2. Taken together, our results identified miR-340 as a tumor suppressor in TNBC, moreover, an EZH2 medicated regulatory loop was established. Post-transcriptional suppression of EZH2 expression not only blocked STAT3 mediated miR-21 trans-activation, but also reversed the miR-200a/b silencing via reducing DNMT1 and H3K27me3 expressions. MiR-21 inhibition and miR-200a/b expression triggered by miR-340 ultimately cooperated in the TNBC progression.
Keywords: miR-340, EZH2, breast cancer, progression, miRNAs.
Background
Breast cancer is the most frequent carcinoma and the second most common cause of cancer-related mortality in women [1]. The cancer progression involves a complex cascade of events, including cancer cell proliferation, migration, local invasion, angiogenesis, and survival in the circulatory system and then settling down at a distant site to form a metastatic tumor [2]. Therefore, understanding of the underlying molecular mechanisms and networks governing breast cancer progression and metastasis may provide a rationale for developing more effective cancer therapies.
Recently, miRNA, a non-coding RNA molecule that composed of ~22 nucleotides, has been proved playing significant role in the progression of tumors, including breast cancer, glioma, lung cancer and so on [3-5]. It can regulate gene expression at the post transcriptional level by binding to the 3′ untranslated region (UTR) of target mRNAs, Which leads to cleavage of the target mRNA by the Ago2 ribonuclease in the RNA induced silencing complex (RISC) or inhibition of translation [6]. MiRNAs upregulation or deregulation was found to have oncogenic potential. For example, the miR-21 miRNA cluster cooperates with the oncogene PI3K to induce tumors in a mouse model [7] and deregulation of the oncogene RAS by loss of regulation through the let-7 family of miRNAs might contribute to carcinogenesis [8]. Recent studies reported that MiR-340 could function as a tumor suppressor via inhibiting tumor cell proliferation and inducing apoptosis by targeting multiple negative regulators of p27 in non-small cell lung cancer [9], and inhibited glioblastoma cell proliferation by suppressing CDK6, cyclin-D1 and cyclin-D2 [10]. In addition, miR-340 could also suppress breast cancer cell migration and invasion through targeting of oncoprotein c-Met [11]. Although multiple regulatory mechanisms are implicated in the anti-tumor of miR-340, the role of miRNA in mediating breast cancer progression has only recently been investigated and still remains largely elusive.
EZH2, a methyl transferase and component of PRC2 (the polycomb repressive complex 2), is primarily responsible for H3K27 methylation, which mediate target genes transcriptional silencing [12]. In current reported studies, the common discovery was that EZH2 expression was elevated in cancer tissues compared to adjacent normal tissues, being the highest expression in the most advanced stages of cancer, and correlated with poor prognosis in patients [13]. EZH2 expression was also found to correlate with breast cancer aggressiveness and acting as an independent predictor of recurrence and survival [14]. A set of genes and pathways associated with cell proliferation and invasion are regulated by EZH2. Such as RUNX3, RAD51, and CDKN1C (p57KIP2), which regulate cell proliferation, and FOXC1 and CDH1 (E-cadherin) have been linked to metastasis [15]. There are also reports indicated that enhanced expression of EZH2 in breast tumor initiating cells results in accumulation of genomic abnormalities that mediate deregulated signaling (RAF1-ERK-β-catenin) promoting cell expansion and cancer progression [16]. Furthermore, our previous study demonstrated that knockdown of EZH2 not only impacted H3K27 trimethylation but also reduced DNMT1 presence on the miR-200b/a/429 promoter which dramatically suppressed cancer progression in vitro and in vivo [17].
Therefore, based on EZH2 transcriptional regulation and epigenetic gene silencing role in cancer, and microRNAs (miRNAs) function as post-transcriptional regulators of cognate target gene expression, in this study, we first indicted that tumor-suppressor activity of miR-340 in TNBC via altering a series of miRNAs expressions by reversing EZH2 expression.
Materials and Method
Tissue samples
A total of 62 primary TNBC tissues and corresponding adjacent normal tissues from patients with breast cancer were collected from the Department of Breast Cancer, Tianjin Medical University Cancer Institute and Hospital during the period from 2013 to 2014. The collection of the patient's sample was approved by Tianjin Medical University Cancer Institute and Hospital. Patients without radiotherapy and chemotherapy before surgery were considered for this study.
Cell culture and transfection
Human breast cancer cell lines MCF-7, T47D, Cal51, MDA-MB-231, MDA-MB-468 were obtained from ATCC (the American Type Culture Collection, Manassas, VA, USA). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Hyclone) supplemented with 10% Fetal bovine serum (FBS, Hyclone) and were incubated at 37°C in a 5% CO2 atmosphere. The cell transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
RNA extraction and Real-time PCR assay
Total RNA extraction was carried out using Total RNA isolation kit (Bioteke Corporation, Beijing, China). For MicroRNA, we used miRcute miRNA first-strand cDNA synthesis kit and miRcute miRNA qPCR detection kit (SYBR Green) (Tiangen, Beijing, China) to detect the levels of miR-340 according to the manufacturer's protocol. U6 was as an internal control. Real-time PCR was performed on a CFX96 Real-Time System (Bio-Rad, USA) using the following primers:
MiR-340 stem loop primer: 5'-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGACGTGGATACGACAATCAG-3'; forward primer, 5'-GCGGTTATAAAGCAATGAGA-3'; reverse primer, 5'-GTGCGTGTCGTGGAGTCG-3'; U6 forward primer, 5'-GCTTCGGCAGCACATATACTAAAAT-3'; reverse primer, 5'-CGCTTCACGAATTTGCGT. For mRNA, we put 2 μg of total RNA to reverse-transcribed with BioTeke super RT Kit (Bioteke Corporation, Beijing, China) to synthesize cDNA samples. 2 ml of cDNA product were used to polymerase chain reaction (PCR) amplification with Go Tag qPCR Master Mix (Promega, USA) on a thermal cycler using the following primers.
Cell cycle and colony formation analysis
Cell cycle distribution of different groups was analyzed by propidium iodide (PI) staining and flow cytometryas previously reported [18]. For the colony formation analysis, the treated cells were plated in 6-well plates at 200 cells per well and grown for 2 weeks. Afterwards, the cells were washed with PBS for three times, fixed in methanol, and stained with 0.5% crystal violet (Sigma, St. Louis, MO, USA). All the assays were repeated at least three times.
Apoptosis and transwell invasion assay
The cell apoptosis was evaluated by flow cytometry analysis. Briefly, after treatment with miR-340 mimics, inhibitor or the NC for 48h, the cells were collected and subjected to an annexin V/PI stain using an annexin V-FITC Apoptosis Detection Kit (BioVision, Palo Alto, CA). Then the fluorescence was measured by flow cytometry using a FACS flow cytometer (Becton Dickinson, San Jose, CA). Cell invasion abilities were examined using a 24-well matrigel-coated transwell inserts (BD Biosciences, Bedford, MA, USA). Cells with treatment were transferred on the top of matrigel-coated invasion chambers with serum-free DMEM and were fixed in methanol and stained with 0.1% crystal violet later. The average numbers of cells per field were determined by counting the cells in four random fields per well. All tests were performed in triplicate.
Immunofluorescence and In Situ Hybridization (FISH)
Immunofluoresence staining was conducted using antibodies against E-cadherin, β-catenin, and MMP-9 (1:100 dilutions; Cell Signaling Technology), and the cells were visualized using Fluo View Confocal Laser Scanning Microscopes-FV1000 (Olympus, Tokyo, Japan) and analyzed by IPP5.1 (Olympus). In-Situ Hybridization kit was purchased from Wuhan Boster Biological Technology, Ltd. Sequences of oligonucleotide probes are as shown: has-miR-340 5'-AATCAGTCTCATTGCTTTATAA-3'; hsa-miR-200a 5'-ACA TCG TTA CCA GACAGT GTT A-3'; hsa-miR-200b 5'-TCATCATTACCAGGCAGTATTA-3'; hsa-miR-21 5'-TCAACATCAGTCTGATAAGCTA-3'. Antisense locked nucleic acid (LNA) modified oligonucleotide probes: LNA-miR-340, LNA-miR-200a, LNA-miR-200b and LNA-miR-21 hybridization solution were used for detection as previously described [19].
Histology and immunohistochemistry (IHC)
Immunohistochemistry analysis was performed as previously described [20]. Briefly, the sections were incubated with primary antibodies (1:100 dilutions) overnight at 4℃, followed by a biotin-labeled secondary antibody (1:100 dilutions) for 1h at 37℃ and then ABC peroxidase and 3,30-diaminobenzidine were added, hematoxylin was counterstained and visualized using a light microscope. Staining results were scored by two pathologists. The quantity and intensity scores were calculated such that a final score of 0-1 indicated negative expression (-), 2-3 indicated weak expression (+), 4-5 indicated moderate expression (++), and 6 indicated strong expression (+++). Cases with score discrepancies were discussed to reach an agreement.
Protein extraction and western blot
The breast cells transfected with miR-340 mimics, inhibitor or the NC was cultured for 48h. Then each total proteins and nuclear proteins were isolated from cell lines using 1% of Nonidet P-40 lysis buffer. Western blotting assay was performed as previously described [20]. DNMT1, H3K27me3, β-catenin, p-STAT3 and EZH2 were used as the detected antibodies. GAPDH was selected as a housekeeping gene. The protein was detected using a Super Signal protein detection kit (Pierce, Rockford, IL).
Luciferase Reporter Assay
The EZH2 3′-UTR-Luc reporter was created by the ligation of EZH2 3′-UTR PCR products into the XbaI site of the pGL3 control vector (Promega). For the reporter assay, cells were cultured in 96-well plates and transfected with luciferase reporters, and wild type miR-340 sequences or mutant ones. After 48h incubation, luciferase activity was measured using a dual-luciferase reporter system (Promega). The Renilla luciferase activity was used as an internal control.
Nude mouse tumor xenograft model and treatment
BALB/c-A nude mice at 3-4 weeks of age were purchased from the Animal Center at the Cancer Institute at Chinese Academy of Medical Science (Beijing, PR China). Animal handling and experimental procedures were performed according to the regulations and internal biosafety and bioethics guidelines of Tianjin Medical University. The female nude mice were subcutaneously inoculated with MDA-MB-231 cells (1 × 10 7 cells/mouse) for the construction of the tumor xenograft model. When tumors reached a volume of about 100mm3, the animals were randomized divided into three groups and injected 200pM miR-340 mimics, miR-340 inhibitor and NC respectively every 2 days for 21 days. Tumor lengths and widths were measured every 3 days using a digital caliper, and tumor volumes were calculated using the equation volume(mm3) = length × width2/2. At the end of the experiment on day 21, the mice were sacrificed, tumor weight was measured, and tumors were used for examination as well as HE stains.
Statistical Analysis
Statistics were performed using the SPSS Graduate Pack, version 11.0, statistical software (SPSS). Descriptive statistics, including mean and SE and one-way analysis of variance, were used to determine statistically significant differences. P <0.05 was considered to be statistically significant.
Results
MiR-340 was downregulated in TNBC and inversely correlated with the EZH2 expression
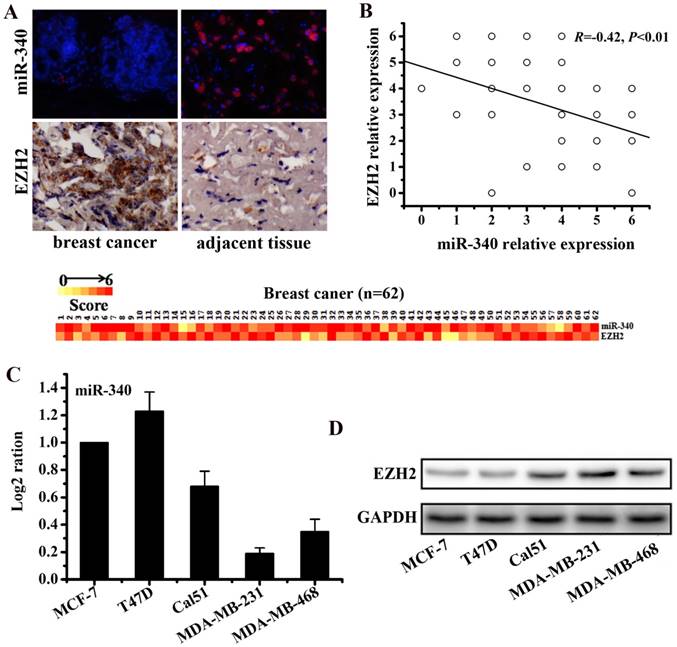
To detect the expressions of miR-340 and EZH2 in TNBC tissues, 62 pairs of breast cancer tissues and adjacent normal tissues were used for IHC and FISH assay. The results revealed that miR-340 expression was reduced in cancer tissues compared with the adjacent normal tissues, whereas EZH2 was over-expressed in cancer tissues (Fig. 1A). Furthermore, the expression level of miR-340 was inversely correlated with the EZH2 expression (R=-0.42, P<0.01, Fig. 1B). Meanwhile, Real-time PCR assay established that miR-340 was lowly expressed in TNBC cell lines (Cal51, MDA-MB-231 and MDA-MB-468), compared with hormone-receptor positive cells (T47D and MCF-7) (Fig. 1C). However, EZH2 were overexpressed mainly in TNBC cell lines, especially in MDA-MB-231 and MDA-MB-468 cells (Fig. 1D). Based on these results, a negative correlation between miR-340 and EZH2 expression might also be existed in TNBC cell lines.
Differential expressions of miR-340 and EZH2 assayed respectively by FISH and IHC in cancer and adjacent normal tissues of 62 TNBC samples. (A) MiR-340 expression was reduced in cancer tissues compared with adjacent normal tissues, whereas EZH2 was over-expressed in cancer tissues. The heat map represents gene expression on a linear scale, with a maximum immunoreactive score of 6 to highlight differences within this range. (B) Inverse correlation (R=-0.42, P<0.01) of miR-340 and EZH2 relative expression in cancer and normal tissues. A trend line is provided in each plot, which represented the 'best fit,' as determined by simple linear regression. (C) MiR-340 was lowly expressed in TNBC cells (Cal-51, MDA-MB-231 and MDA-MB-468) in comparison with hormone receptor positive cells (MCF-7 and T47D). (D) However, EZH2 was over-expressed in TNBC cells, especially in MDA-MB-231 and MDA-MB-468.
EZH2 is a novel target of miR-340
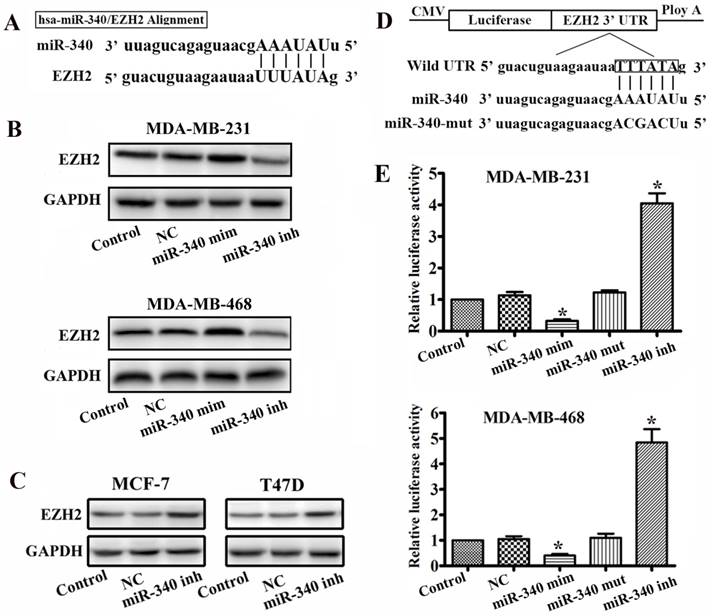
By using the miRanda and mirRVR algorithms, we speculated EZH2 as a candidate target for miR-340. Sequence analysis revealed that the seed region of miR-340 could form complementary base pairs with the 3'UTR of EZH2 mRNAs (Fig. 2A). To test whether EZH2 expression was regulated by miR-340, we used miR-340 mimics and inhibitor to change the miR-340 endogenous levels in MDA-MB-231 and MDA-MB-468 cells. There was an increased in the EZH2 protein expression levels 48h after transfection with the miR-340 inhibitor, whereas EZH2 expresses were reduced after miR-340 mimics transfection (Fig. 2B). Meanwhile, miR-340 inhibitor also upregulated EZH2 expression levels in MCF-7 and T47D (Fig. 2C). To demonstrate a direct interaction between the 3'UTR of EZH2 and miR-340, we inserted the 3'UTR region of human EZH2, which contains the putative miR-340 recognition sites and flanking sequences downstream of a renilla luciferase reporter gene, into a pGL3 vector (Fig. 2D). The miR-340 inhibitor and mimics were transfected into TNBC cells along with the reporter gene. The reporter gene activity was significantly increased in miR-340 inhibitor transfected cells, whereas reduced in mimics transfected group (Fig. 2D). It has been shown that the base pairing of miRNA seeding sequences to the target genes is essential for the binding of miRNA to its target mRNA, repressing target gene expression. To test whether the predicted miR-340 targeting sites in the EZH2 3' UTR are critical for the repression of EZH2 expression by miR-340, point mutations disrupting the base-pairing of the predicted miR-340 targeting sites were introduced into the miR-340 sequence. Mutation of the miR-340 targeting sites abolished the repression (Fig. 2E). These results suggest that miR-340 represses EZH2 expression through the predicted targeting sites in the EZH2 3'UTR.
EZH2 is a target of miR-340. (A) Diagram of seed sequence of miR-340 matched the 3'UTR of the EZH2 gene. (B) EZH2 expression was detected by western blot after miR-340 mimics and miR-340-inhibitor transfection. (C) EZH2 expression was assayed by western blot after miR-340 inhibitor transfection in MCF-7 and T47D cells. (D) Schematic diagram of EZH2 3'UTR containing reporter constructs. (E) Luciferase reporter assays in breast cancer cells, following cotransfection with the 3'UTR EZH2 and miR-340 mimics, miR-340-inhibitor, and mutant miR-340 sequence as indicated. The data represent the fold change in expression (means ± SE) of three replicates. Data represent mean ± SD of three replicates. *P < 0.05.
Up-regulation of miR-340 inhibits the tumorigenic properties of TNBC cells
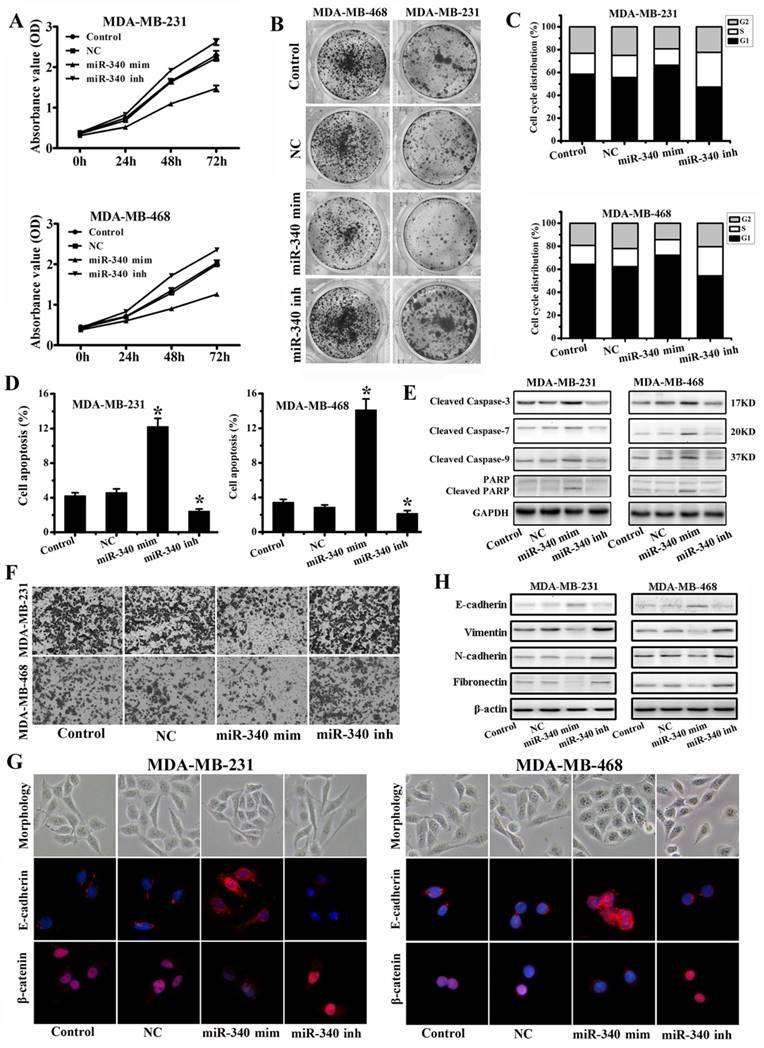
To assess the tumor suppressive role of miR-340, we elevated miR-340 levels respectively in MDA-MB-231 and MDA-MB-468 followed by functional assays. MTT (Fig. 3A) and Colony formation (Fig. 3B) assays revealed that overexpression of miR-340 can significantly inhibit TNBC cells proliferation, whereas miR-340 inhibitor transfection stimulated the cells growth. Cell cycle analysis showed that miR-340 elevated expression resulted in cell arrest in G0/G1 phase, however, miR-340 inhibitor transfection reduced the G0/G1 phase (Fig. 3C). Furthermore, we measured apoptosis and Cleaved Caspases and PARP expressions in Control and transfected cells by annexin-V-PI assay and Western blot. It was observed, in both TNBC cells, that the average apoptotic cell fractions (Fig. 3D) and Cleaved Caspase-3, Cleaved Caspase-7, Cleaved Caspase-9, and Cleaved PARP (Fig. 3E) were significantly increased on miR-340 re-expression group compared with Control, NC and inhibitor transfection group. To assess the role of miR-340 in cell invasion, transwell assay and epithelial-mesenchymal transition (EMT) associated markers expression levels were detected. MiR-340 mimics transfection decreased the invasion of cancer cells, whereas miR-340 inhibitor transfection enhanced invasion, compared with Control and NC (Fig. 3F). In addition, miR-340 mimics treatment induced a close cell-cell contact epithelial-like morphology, however, cells acquired spindle and fibroblastoid shapes with increased cell gaps after miR-340 inhibitor treatment. Confocal microscopy detection showed that E-cadherin expression was increased after miR-340 mimics transfection, meanwhile, the nuclear β-catenin expression was significantly down-regulated. However, miR-340 inhibitor promoted the β-catenin nuclear translocation (Fig. 3G), which was associated with the EMT. Furthermore, western blot assay indicated that miR-340 mimics increased E-cadherin expression and decreased Vimentin, N-cadherin and Fibronectin expressions, whereas miR-340 inhibitor reduced E-cadherin expression and upregulated Vimentin, N-cadherin and Fibronectin levels compared with Control and NC (Fig. 3H). Together, these data demonstrate that miR-340 is potent at suppressing breast cancer cell growth and invasion.
Enforced expression of miR-340 inhibits growth and invasion, and induces cell apoptosis in breast cancer cells in vitro. (A) Effect of miR-340 on cell proliferation was measured by MTT assay after NC, mimics or inhibitor transfection in MDA-MB-231 and MDA-MB-468 cells. (B) Colony formation, (C) cell cycle distribution, and (D) apoptosis were detected after NC, mimics and inhibitor transfection. (E) Cleaved caspase-3, Cleaved caspase-7, Cleaved caspase-9 and Cleaved PARP were detected by western blot. (F) Cell invasion and migration activity was assayed by transwell. (G) Cell morphology was photographed and confocal microscopy was used to test the E-cadherin and β-catenin expressions after transfections. (H) E-cadherin, Vimentin, N-cadherin and Fibronectin were analyzed by western blot. Data represent mean ± SD of three replicates. *P < 0.05.
MiR-340 exhibits anti-tumor efficacy in an orthotopic mouse model
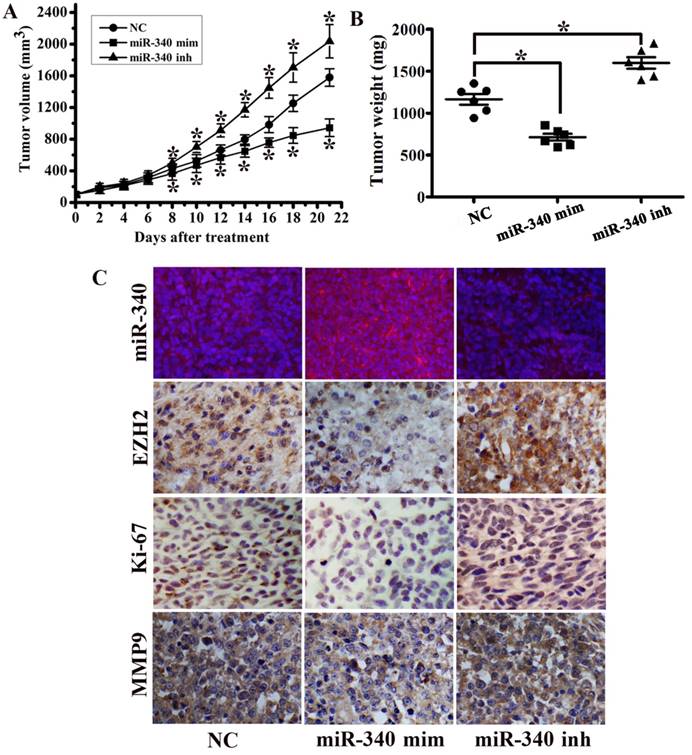
To investigate the potential impact of miR-340 on TNBC growth in vivo, a MDA-MB-231 orthotopic mouse model was used. MiR-340 mimics treated group displayed a significant growth reduction, whereas tumor growth was increased in miR-340 inhibitor treated group (Fig. 4A). Tumor mass were significantly greater in miR-340 inhibitor treated tumors compared with those treated with NC, and tumors from miR-340 mimics treated group achieved the smaller weights (Fig. 4B). FISH analysis confirmed increased miR-340 expression in mimics treated group, but reduced in inhibitor treated group. Moreover, miR-340 mimics treated mouse models displayed decreased expressions of EZH2, Ki-67 and MMP9 compared with NC and inhibitor treated group (Fig. 4C). These data indicated that miR-340 re-introduction in vivo exhibited tumor suppressive efficacy similar in vitro.
MiR-340 suppressed tumor growth in an orthotopic breast cancer mouse model. (A) MDA-MB-231 subcutaneous tumors were treated with NC, miR-340 mimics and miR-340 inhibitor. Growth curves of tumor volumes in NC, mimics and inhibitor group (n=8 in each group). (B) Tumors were resected and tumor mass was determined 21 days following implantation. Data indicate decreased tumor mass miR-340 mimics-treated tumors compared with NC and miR-340-inhibitor treatment. (C) MiR-340, EZH2, Ki-67 and MMP9 expressions were detected by IHC and FISH. Data represent mean ± SD of three replicates. *P < 0.05.
MiR-340 reversed EZH2 mediated miR-21 transactivation and miR-200a/b silencing
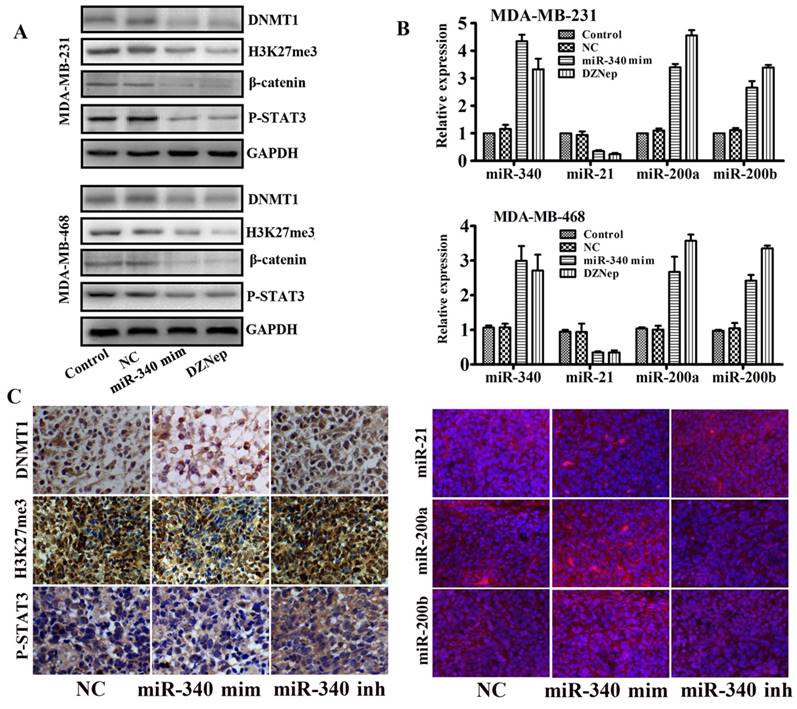
As our previous studies reported, EZH2 cooperated with β-catenin and STAT3 and participated in glioma progression [21]. Meanwhile, there were binding sites of P-STAT3 located in miR-21 gene promoter [22]. Therefore, we supposed that miR-21 might be trans-activated by EZH2. In addition, we also found that EZH2 mediated methylation silences the miR-200a/b gene and promoted the glioma and gastric cancer progression [17]. Base on above studies, we assayed whether miR-340 mediated miR-21 and miR-200a/b expressions and inhibited the TNBC progression by targeting EZH2. Western blot assay indicated that miR-340 mimics downregulated the DNMT1, H3K24me3, β-catenin and P-STAT3 expressions as the same as the EZH2 inhibitor DZNep treatment (Fig. 5A). Moreover, both miR-340 mimics and DZNep treatment elevated miR-340 expressions, and inhibited the miR-21 expression and induced miR-200a/b levels as detected by Real-time PCR (Fig. 5B). In addition, IHC and FISH detection revealed that DNMT1, H3K27me3, P-STAT3 and miR-21 expressions were significantly reduced in tissues from miR-340 mimics treated group, whereas miR-200a/b expression levels were increased (Fig. 5C).Taken together, we concluded that miR-340 inhibited the TNBC progression partly due to the reversion of EZH2 mediated a series of miRNAs activities, such as miR-21 and miR-200a/b.
EZH2 downstream target genes and miRNAs expressions were detected after miR-340-mimics treatment. (A) Western blot detection for DNMT1, H3K27me3, β-catenin and P-STAT3 expressions after miR-340-mimics and DZNep treatment. (B) Real-time PCR assay for miR-340, miR-21, miR-200a and miR-200b expressions after miR-340 mimics and DZNep treatment. (C) Expressions of DNMT1, H3K27me3, P-STAT3, miR-21, miR-200a and miR-200b were detected in MDA-MB-231 mouse model tissues. Data represent mean ± SD of three replicates. *P < 0.05.
MiR-340 inhibited triple-negative breast cancer progression through targeting EZH2
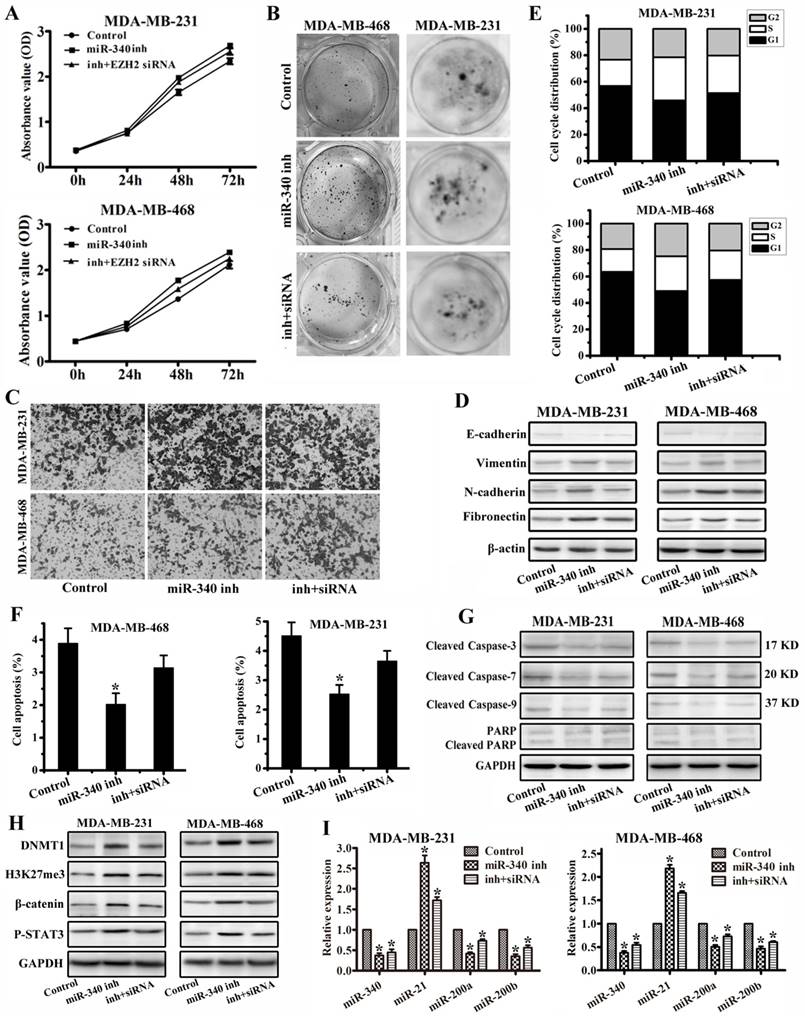
To further verify miR-340 functions through EZH2, we performed rescued experiments with miR-340 inhibitor and EZH2 siRNA transfection together. Compared with single miR-340 inhibitor treatment group, combined use of miR-340 inhibitor and EZH2 siRNA weakened the cancer cell proliferation and migration activity detected by MTT (Fig. 6A), colony formation (Fig. 6B) and transwell (Fig. 6C) assays, and increased E-cadherin expression and reduced Vimentin, N-cadherin and Fibronectin expressions (Fig. 6D). Meanwhile, miR-340 inhibitor and EZH2 siRNA transfection prolonged the G0/G1 phase (Fig. 6E), induced the more cell apoptosis (Fig. 6F), and increased Cleaved Caspase-3, Cleaved Caspase-7, Cleaved Caspase-9 and Cleaved PARP expressions (Fig. 6G) to some extent compared with miR-340 inhibitor treatment group. Furthermore, EZH2 siRNA transfection abolished miR-340 inhibitor effect on DNMT1, H3K27me3, β-catenin, P-STAT3, miR-21 upregulation and miR-200a/b reduction (Fig. 6 H, I). Taken together, these results further determined that miR-340 inhibited triple-negative breast cancer progression through targeting EZH2.
miR-340 inhibited triple-negative breast cancer progression through targeting EZH2. (A) MTT and (B) Colony formation were conducted to assay cell proliferation after single miR-340 inhibitor or miR-340 inhibitor combined with EZH2 siRNA treatment. (C) Cell invasion and migration activity was assayed by transwell. (D) E-cadherin, Vimentin, N-cadherin and Fibronectin were analyzed by western blot. (E) Cell cycle distribution and (F) apoptosis were detected after miR-340 inhibitor or miR-340 inhibitor combined with EZH2 siRNA transfection. (G) Cleaved caspase-3, Cleaved caspase-7, Cleaved caspase-9 and Cleaved PARP were detected by western blot. (H) Western blot detection for DNMT1, H3K27me3, β-catenin and P-STAT3 expressions and (I) real-time PCR assay for miR-340, miR-21, miR-200a and miR-200b expressions miR-340 inhibitor or miR-340 inhibitor combined with EZH2 siRNA transfection. Data represent mean ± SD of three replicates. *P < 0.05.
Discussion
Breast cancer, the most life-threatening disease for women, involves a complex cascade of events, including tumor cell increase, local and detached invasion, angiogenesis immune system suppression and so on [23]. Identification of the key molecules, genes and proteins that are essential for cancer cell survival is vital for understanding the underlying biology, as well as to design effective cancer therapeutics. Over the last decades, accumulated evidence has shown that miRNAs play a significant role in relevant molecular and cellular mechanisms governing breast tumorigenesis. With the small size and influence in a broad range of biological processes, miRNAs are very attractive therapeutic targets for breast cancer. For example, miR-125b, which targets erythropoietin (EPO) and its receptor (EPOR) as well as ERBB2, is found to be one of the most downregulated miRNAs in breast cancer [24]. Roopra A reported the results of miR-205 regulating HMGB3 and its ectopic expression significantly inhibited cell proliferation and promoted cell apoptosis in breast cancer [25]. Mei and colleagues reported miR-21 upregulation was associated with an increase of the anti-apoptosis protein BCL-2 and resulted in taxol resistance in breast cancer cells [26]. MiR-340 has been reported playing an important role in suppressing breast cancer invasion and metastasis [11]. Although abroving study has examined the role of miR-340 in breast cancer, the underlying molecular mechanism needs to be further identified.
Post-translational modifications of the N-terminal tails of core histones, including methylation, acetylation, ubiquitylation and phosphorylation, influence chromatin configuration that can modulate accessibility of transcription factors and transcriptional activity of nearby genes [27]. PRC1 (polycomb repressive complex 1) which catalyzes the mono ubiquitylation of histone H2A and PCR2 which contributes to the methylation of H3K27 are associated with chromatin condensation [28]. PRC2 complexes containing EZH2 are primarily responsible for H3K27 methylation. Experimental evidence suggests that H3K27 methylation is mainly achieved by EZH2 and influences proliferation and differentiation of stem cells and cancer cells [29]. Here we showed that EZH2 was the target of miR-340 by luciferase reporter assay and protein expression assay. In addition, miR-340 elavation results in the decreased expression of H3K27me3, β-catenin and p-STAT3, which are regulated by EZH2. In our previous studies, we have showed that DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes human gastric cancer and glioma progression [17]. Also there was a report that STAT3 bound to the miR-21 promoter region and transcribed its expression [22]. In this study, we verificated that upregulation of miR-340 inhibited miR-21 transactivation as well as increased miR-200a/b expression by decreased EZH2 expression. These results first indicated that miR-340 regulated miR-21 and miR-200a/b through targeting EZH2 in TNBC.
Taken together, in the present study, we identified EZH2 as a direct target of miR-340, which acted as a tumor suppressor in TNBC via the modulation of the expression of EZH2 not only in DNA methylation but also in transcriptional regulation process. MiR-340 upregulation was able to inhibit EZH2 expression and consequently decreased miR-21 and increased miR-200a/b expressions, which formed a miRNAs network pathway ultimately contribute to the malignant progression of TNBC. Our current data provided the functional significance of miR-340 target molecules, including EZH2, might provide a greater understanding of the complex mechanisms of TNBC development and progression.
Abbreviations
TNBC, triple-negative breast cancer; EZH2, enhancer of zeste homolog 2; PRC2, the polycomb repressive complex 2; FISH, Immunofluorescence and In Situ Hybridization; PI, propidium iodide
Acknowledgements
This study was supported by the National Natural Scientific Foundation (81502306), Tianjin Medical University Research Project (2014KYQ08), the Research Fund for the Doctoral Program of Tianjin Medical University Cancer Institute and Hospital (B1403) and the National Key Clinical Specialist Construction Programs of China.
Authors' contributions
Zhendong Shi performed most of the experiments, analyzed data, and wrote the manuscript. Yang Li and Yunhui Hu performed some experiments. Xiaomin Qian and Jingjing Liu reviewed and edited the manuscript. Sheng Zhang and Jin Zhang designed the experiments and edited the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-2917
2. Wang L, Wang J. MicroRNA-mediated breast cancer metastasis: from primary site to distant organs. Oncogene. 2012;31:2499-2511
3. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857-866
4. Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849-864
5. You JS, Jones PA. Cancer genetics and epigenetics. two sides of the same coin? Cancer Cell. 2012;22:9-20
6. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-531
7. Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849-864
8. Bussing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14:400-409
9. Fernandez S, Risolino M, Mandia N, Talotta F, Soini Y, Incoronato M, Condorelli G, Banfi S, Verde P. miR-340 inhibits tumor cell proliferation and induces apoptosis by targeting multiple negative regulators of p27 in non-small cell lung cancer. Oncogene. 2015;34:3240-3250
10. Li X, Gong X, Chen J, Zhang J, Sun J, Guo M. miR-340 inhibits glioblastoma cell proliferation by suppressing CDK6, cyclin-D1 and cyclin-D2. Biochem Biophys Res Commun. 2015;460:670-677
11. Wu ZS, Wu Q, Wang CQ, Wang XN, Huang J, Zhao JJ, Mao SS, Zhang GH, Xu XC, Zhang N. miR-340 inhibition of breast cancer cell migration and invasion through targeting of oncoprotein c-Met. Cancer. 2011;117:2842-2852
12. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM. et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871-874
13. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21-29
14. Collett K, Eide GE, Arnes J, Stefansson IM, Eide J, Braaten A, Aas T, Otte AP, Akslen LA. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin Cancer Res. 2006;12:1168-1174
15. Roh S, Park SY, Ko HS, Sohn JS, Cha EJ. EZH2 expression in invasive lobular carcinoma of the breast. World J Surg Oncol. 2013;11:299
16. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86-100
17. Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, Kang C, Zhang Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015;359:198-205
18. Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J, Yuan X, Kang C. Sequence-dependent synergistic inhibition of human glioma cell lines by combined temozolomide and miR-21 inhibitor gene therapy. Mol Pharm. 2012;9:2636-2645
19. Shi ZD, Qian XM, Zhang JX, Han L, Zhang KL, Chen LY, Zhou X, Zhang JN, Kang CS. BASI, a potent small molecular inhibitor, inhibits glioblastoma progression by targeting microRNA-mediated beta-catenin signaling. CNS Neurosci Ther. 2014;20:830-839
20. Shi Z, Qian X, Li L, Zhang J, Zhu S, Zhu J, Chen L, Zhang K, Han L, Yu S, Pu P, Jiang T, Kang C. Nuclear translocation of beta-catenin is essential for glioma cell survival. J Neuroimmune Pharmacol. 2012;7:892-903
21. Zhang J, Chen L, Han L, Shi Z, Pu P, Kang C. EZH2 is a negative prognostic factor and exhibits pro-oncogenic activity in glioblastoma. Cancer Lett. 2015;356:929-936
22. Han L, Yue X, Zhou X, Lan FM, You G, Zhang W, Zhang KL, Zhang CZ, Cheng JQ, Yu S, Pu P, Jiang T, Kang C. MicroRNA-21 expression is regulated by beta-catenin/STAT3 pathway and promotes glioma cell invasion by direct targeting RECK. CNS Neurosci Ther. 2012;18:573-583
23. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275-292
24. Ferracin M, Bassi C, Pedriali M, Pagotto S, D'Abundo L, Zagatti B, Corra F, Musa G, Callegari E, Lupini L, Volpato S, Querzoli P, Negrini M. miR-125b targets erythropoietin and its receptor and their expression correlates with metastatic potential and ERBB2/HER2 expression. Mol Cancer. 2013;12:130
25. Elgamal OA, Park JK, Gusev Y, Azevedo-Pouly AC, Jiang J, Roopra A, Schmittgen TD. Tumor suppressive function of mir-205 in breast cancer is linked to HMGB3 regulation. PLoS One. 2013;8:e76402
26. Mei M, Ren Y, Zhou X, Yuan XB, Han L, Wang GX, Jia Z, Pu PY, Kang CS, Yao Z. Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat. 2010;9:77-86
27. Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224-229
28. Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, Wutz A, Vidal M, Elderkin S, Brockdorff N. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664-678
29. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-349
Author contact
![]() Corresponding authors: Zhendong Shi or Jin Zhang, 3rd Department of Breast Cancer, China Tianjin Breast Cancer Prevention, Treatment and Research center, Tianjin Medical University Institute and Hospital. West Beihuanhu Road, He Xi District, Tianjin 300060, PR China. Tel/Fax: +86-22-23340123; Email: shizhendong3588com; Email: zhangjincom
Corresponding authors: Zhendong Shi or Jin Zhang, 3rd Department of Breast Cancer, China Tianjin Breast Cancer Prevention, Treatment and Research center, Tianjin Medical University Institute and Hospital. West Beihuanhu Road, He Xi District, Tianjin 300060, PR China. Tel/Fax: +86-22-23340123; Email: shizhendong3588com; Email: zhangjincom