Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(15):3049-3061. doi:10.7150/jca.19594 This issue Cite
Research Paper
Eukaryotic Translation Initiation Factor 3b is both a Promising Prognostic Biomarker and a Potential Therapeutic Target for Patients with Clear Cell Renal Cell Carcinoma
Yuanwei Zang1, Xiang Zhang1, Lei Yan1, Gangli Gu1, Dawei Li1, Yongzhen Zhang1, Liang Fang1, Shanshan Fu2, Juchao Ren1 ![]() , Zhonghua Xu1
, Zhonghua Xu1 ![]()
1. Department of Urology, Qilu Hospital, Shandong University, 107 Wenhua West Road, Jinan 250012, China;
2. Department of Gastroenterology, Shandong Provincial Qianfoshan Hospital, 16766 Jingshi Road, Jinan 250014, China.
Received 2017-2-9; Accepted 2017-8-6; Published 2017-9-2
Abstract
Eukaryotic translation initiation factors (eIFs) constitute a new class of therapeutic cancer targets. EIF3b is the major scaffold protein of eIF3 (the largest core of eIFs). We sought to define the role played by and the mechanism of action of eIF3b in patients with clear cell renal cell carcinoma (ccRCC). We found that high-level eIF3b expression in tumors was not only associated with an aggressive tumor phenotype, but was also independently prognostic for patients with ccRCC. Knockdown of eIF3b impaired the action of the Akt pathway, thus inhibiting cell proliferation by disrupting the cell cycle and triggering apoptosis. Furthermore, the epithelial-to-mesenchymal transition was impaired after eIF3b depletion, via suppression of cell migration and invasion. Additionally, eIF3b knockdown significantly inhibited the growth of subcutaneous xenografts in mice. Together, these data show that eIF3b is both a promising prognostic biomarker and a potential therapeutic target for patients with ccRCC.
Keywords: eIF3b, clear cell renal cell carcinoma, Akt pathway, β-catenin pathway, EMT, cell proliferation, cell invasion, cell migration.
Introduction
In 2017, it is predicted that 63,990 patients will be diagnosed with kidney cancer in the USA and 14,400 inpatients will die [1]. Renal cell carcinoma (RCC) is the predominant form of kidney cancer, accounting for 2-3% of all adult malignancies [2]. Clear cell RCC (ccRCC) is the most common (75%) and most lethal subtype of RCC [3]. Although recent surgical developments have improved the overall survival of patients with ccRCC, the long-term prognosis remains unsatisfactory. Therefore, there is an urgent need for a better understanding of the molecular mechanisms underlying ccRCC development.
In recent years, deregulated protein synthesis and degradation have been shown to contribute to cancer development and progression [4, 5]. Translation initiation is the critical rate-limiting process in the synthesis of most proteins [6]. Research has shown that inhibition of eukaryotic translation initiation factors (eIFs) is key in terms of reducing tumor initiation and progression, and improving prognosis; eIFs are promising therapeutic targets [4, 5].
Of the 12 known eIFs, the multi-subunit complex known as eukaryotic translation initiation factor 3 (eIF3) is the largest, with 13 putative subunits [7]. Of the 13 subunits, eIF3b forms a central core to which the other components bind with varying affinities [6, 8]. After binding to the 40S ribosomal subunit [9], eIF3 serves as a platform that recruits other protein components to form the 43S pre-initiation complex [10]. Furthermore, eIF3b interacts with eIF4G, resulting in formation of the 48S pre-initiation complex, which recognizes the AUG start codon [11]. The contribution made by the eIF3 subunits eIf3a, eIf3b, eIF3c, eIF3e, eIF3h, eIF3i, and eIF3m to malignant transformation and progression have been defined over the past few years [12-17]. These factors are potentially prognostic biomarkers of several types of cancer, are easy to measure, and may form the basis of a new class of cancer therapeutics. Within the eIF3 complex, subunit b (eIF3b) is a major scaffold protein with which the other core subunits interact [18].
Earlier research showed that eIF3b played an important role in colon cancer [13], glioblastoma [12], esophageal squamous carcinoma [19], and bladder cancer [20]. However, the role of eIF3b in ccRCC remains unclear. In the present study, we show that eIF3b is both a promising prognostic biomarker and a potential therapeutic target in patients with ccRCC.
Materials and Methods
Patients and specimens
A total of 82 cancer specimens and tumor-adjacent renal tissues were obtained from patients with pathologically confirmed ccRCC who underwent radical or partial nephrectomy in 2009 or 2010 at the Department of Urology, Shandong University Qilu Hospital, Jinan, China. No patient underwent preoperative radiotherapy, chemotherapy, or immunotherapy. All specimens were re-evaluated using the TNM staging system [21]; the nuclear grades were scored using the Fuhrman criteria [22]. Patients with incomplete follow-up records were excluded. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed on random tissue samples obtained during radical, partial, or cytoreductive nephrectomy; the tissues were stored in liquid nitrogen prior to use. The samples included 27 normal kidney (NK) tissues taken >2 cm distant from tumors; 30 adjacent NK tissues (ANKs) taken <1 cm distant from tumors; 25 non-metastatic tumor tissues; 5 pairs of primary tumor tissues and metastatic tumor tissues obtained from patients with metastatic ccRCC. Written informed consent was obtained from all patients. Our study was approved by the Ethics Board of Qilu Hospital and was performed in accordance with all relevant principles of the Declaration of Helsinki.
Cell culture
All cell lines were obtained from the American Type Culture Collection (ATCC, USA). A498 human kidney cancer cells were cultured in Eagle's Minimum Essential Medium (ATCC, USA) with 10% (v/v) fetal bovine serum (FBS). KRC-Y and 786-o human kidney cancer cells, and HK-2 proximal tubular cells, were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Hyclone, USA) with 10% (v/v) FBS. CAKI-1 and CAKI-2 kidney cancer cells were cultured in McCoy's Modified 5a Medium (Gibco, USA) with 10% (v/v) FBS. All cells were incubated at 37°C under 5% (v/v) CO2.
Small interfering RNA (siRNA) transfection
SiRNA was transfected with the aid of Lipofectamine® RNAiMAX (catalog no.13778-150; Invitrogen, USA) according to the manufacturer's instructions. The siRNA targeting eIF3b (designed by Invitrogen, USA) was 5′- GUCCAAAGCCUCAAAGGAAdTdT-3′. We also used a negative control siRNA.
RNA preparation and reverse transcription PCR
Total RNA was extracted from frozen tissues and cells using an RNeasy® Mini Kit (catalog no. 74104; Qiagen, USA). cDNA was synthesized from 2-µg amounts of total RNA using a ReverTra Ace® qPCR RT Kit (catalog no. FSQ-101; Toyobo, Japan) according to the manufacturer's instructions. All cDNA was stored at -20°C prior to analysis.
qRT-PCR
Primers were synthesized by Biosune (Jinan, China). EIF3b expression levels were quantified using a LightCycler® 2.0 (Roche, USA); we employed the R = 2-ΔΔCT method. All experiments were performed in triplicate and the data were presented as means ± standard deviation (SD). PCR was performed with the aid of the SYBR® Green Real-Time PCR Master Mix (catalog no. QPK-201; Toyobo, Japan). The primers were: eIF3b forward: 5′-CGGTGCCTTAGCGTTTGTG-3′; reverse: 5′-CGGTCCTTGTTGTTCTTCTGC-3′; α-tubulin forward: 5′-CATGTACGTTGCTATCCAGGC-3′; reverse: 5′-CTCCTTAATGTCACGCACGAT-3′.
Western blotting
For Western blotting, siRNA-transfected cells were harvested after 72 h of culture into RIPA Lysis Buffer (Beyotime, China) and PMSF (Phenylmethanesulfonyl fluoride, Beyotime) at a 100:1 (v/w) ratio. Protein concentrations were measured using a BCA Protein Quantitative Kit (DBI Bioscience, Germany). We used an anti-eIF3B antibody (catalog no. ab124778; Abcam, USA).
Immunohistochemistry and immunofluorescence
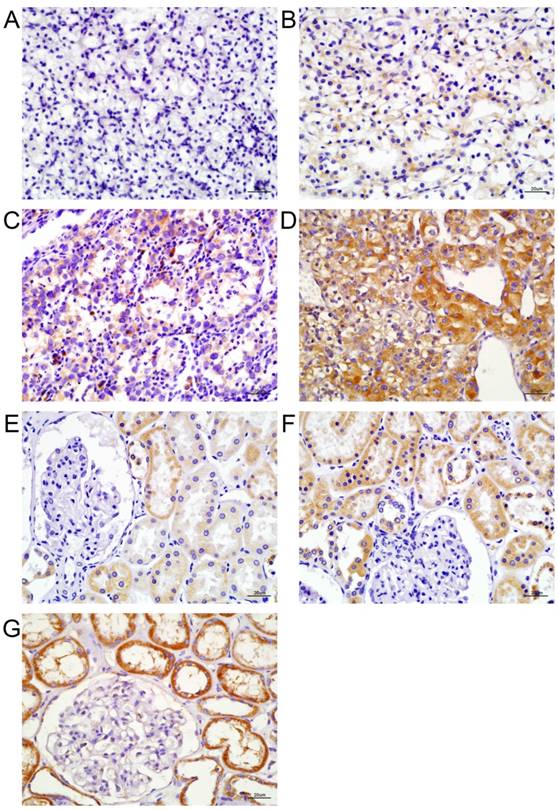
FFPE (Formalin Fixed and Paraffin Embedded) samples were retrieved from the Department of Pathology of Qillu hospital and cut into 5-µm-thick sections. Final staining scores were determined by the staining intensity (0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining) and the percentage of positive cells (scored 0, 0-10%; 1, 10-25%; 2, 26-50%; 3, 51-75%; and 4, 76-100%). The final staining score was the sum of the staining intensity and the percentage of positive cells, and was further graded as follows: 0-1, (-); 2-3, (+); 4-5, (++); and 6-7, (+++) (Figure 1A and 1B). On this basis, the tumor eIF3b expression level was rated as either negative (-) or positive (+, ++, or +++). The expression of eIF3b in ANK tissue was graded as low (+ and ++) or high (+++). Nuclear status was graded by three observers using the Fuhrman system. For immunofluorescence measurements, cells were cultured on chambered slides for 72 h after siRNA transfection and an anti-eIF3B antibody (catalog no. sc-271539; Santa Cruz Biochemicals, USA) was added. Images were acquired using an Olympus IX81 microscope (Olympus, Japan).
eIF3b expression levels in tumors and ANK tissues. A-D: The extent of immunohistochemical staining for eIF3b in ccRCC tissues, graded as -, +, ++, or +++. E-G: The extent of immunohistochemical staining for eIF3b in ANK tissues, graded as +, ++, or +++.
Cell counting kit 8 (CCK-8) assay and cell proliferation
To prepare standard curves, A498 and CAKI-2 cells were seeded into 96-well plates at cell densities of 1000, 2000, 4000, 6000, 8000, and 10000 per well. Optical densities (ODs) at 450 nm were read with the aid of a Varioskan Flash (Thermo Scientific, Finland) after incubation at 37°C for 1.5 h with 2-(4-indophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2-tetrazolium monosodium salt (the CCK-8 reagent; Bestbio, China). Next, logarithmically growing siRNA-transfected cells were seeded into 96-well plates at 2,300 and 1,200 cells/well, respectively, and incubated for 0, 8, 16, 24, 32, 40, 48, 56, 64 and 72 h. At various times, samples were taken and incubated with the CCK-8 reagent for 1.5 h at 37°C and the ODs were measured. The averages of the values obtained in five independent experiments were used to generate cell proliferation curves.
Scratch migration assay
A498 and CAKI-2 cell monolayers were transfected with negative-control or eIF3b-targeting siRNA for 48 h, scratched with a 100-µL pipette tip, washed twice with phosphate buffer saline (PBS) to remove detached cells, and incubated further in growth medium without FBS to ensure that the cells did not proliferate extensively and that most distributional changes would reflect cell migration. Wound closure (reflected by cell migration into the initially cell-free scratch area) was measured after 0, 8, 16, and 24 h. All experiments were performed in triplicate.
Transwell migration and invasion assays
We used Corning Matrigel® Invasion Chambers (8-μM pore size; BD Biosciences, USA). Cells (5.0 × 104/500 μL serum-starved medium) were added to the upper chambers and complete medium to the bottom wells. After 16 h, 5% (w/v) glutaraldehyde was added to both chambers, followed by 0.1% (w/v) crystal violet. The upper faces of the filters were then wiped with cotton swabs. Images of five 200× fields were captured from each membrane and the numbers of invading cells were counted. In the invasion assay, the upper faces of the membranes were coated with 60-µL amounts of a 1:6 (w/v) dilution of Matrigel®. After incubation at 37°C for 1 h, the chambers were used in the invasion assay. The subsequent steps were the same as described above.
Cell cycle analysis and measurement of apoptosis
Seventy-two hours after transfection, cells were harvested and subjected to flow cytometry, for evaluation of cell cycle status using a Bestbio (China) kit (catalog no. BB-4104-50T) and measurement of apoptosis status using a BD (USA) kit (catalog no. 556547). We employed a BD FACSCaliburTM cytometer. Cell cycle status was evaluated using Modifit LT 4.1 software (Verity Software House, USA) and apoptosis status analyzed with the aid of FCS version V3 software (De Novo, USA).
Lentivirus transfection
A lentivirus-mediated eIF3b siRNA-delivery system was constructed by Genechem, China. The GV248 vector contained the elements hU6-MCS-ubiquitin-EGFP-IRES-puromycin; the clone was transfected with the aid of Enhanced Infection Solution (Genechem, China) with 5 µg/mL polybrene. After 12 h of incubation, the transfection medium were changed to normal culture medium with 10% (v/v) FBS.
Colony formation
Lentivirus-transfected cells were seeded at 100 cells/well into 6-well culture dishes, incubated for 8 d, and fixed and stained with 0.1% (w/v) crystal violet. Colonies containing > 50 cells were counted. All experiments were performed in triplicate.
Subcutaneous xenografts in nude mice
Tumor xenografts were established by injecting 5 × 106 A498-LV-eIF3b-RNAi cells in 200-µL amounts of PBS into the right flanks of 6-week-old female BALB/c nude mice (n = 5 per group; Biomedical Research Institution of Nanjing University, China). The two greatest tumor diameters were measured every 4 days with calipers and tumor volumes were calculated as V = L × l2 × 0.5, where L and l represent the larger and smaller diameters. All animal care followed our institutional guidelines. Mice were sacrificed before the volume of the largest tumor attained 1,000 mm3. All animal experiments followed the dictates of the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the work was approved by the Committee on the Use and Care of Animals of Shandong University.
Statistical Analysis
Statistical analyses were performed with the aid of SPSS version 20 (IBM, USA) and GraphPad Prism® version 5.01 (GraphPad Software, USA). The immunohistochemically measured eIF3b levels and the clinicopathological parameters were compared using chi-squared and Fisher's exact tests. Kaplan-Meier analysis followed by log-rank testing was employed in the univariate analysis. Multivariate analyses were performed using Cox's proportional hazards regression model. A p-value < 0.05 was considered to indicate statistical significance. Migration and invasion levels were compared using the Student's t-test. Real-time PCR data were compared with the aid of the Mann-Whitney U test.
Results
Patient characteristics
The 82 patients with ccRCC had a mean age of 57 years and included 55 males (66.7%) and 27 females (33.3%). Twenty patients (24.7%) died during the follow-up period. The estimated 5-year overall survival (OS) was 84.1%. The numbers of patients with TNM grades I, II, III, and IV were 64, 7, 7, and 4, respectively. The numbers of patients with Fuhrman nuclear grades 1, 2, 3, and 4 were 7, 25, 28, and 22, respectively. The median follow-up time was 76 months. Table 1 lists the details.
Correlations between tumor eIF3b expression levels and the clinicopathological variables of patients with ccRCC (n = 82)
| Variables | No. of patients | eIF3b expression (tumor) | P value | |
|---|---|---|---|---|
| eIF3b negative n=37 | eIF3b positive n=45 | |||
| Age | ||||
| ≥57 years | 41 | 15 | 26 | 0.12 |
| <57 years | 41 | 22 | 19 | |
| Gender | ||||
| Male | 55 | 25 | 30 | 0.931 |
| Female | 27 | 12 | 15 | |
| T stage | ||||
| T1-2 | 74 | 36 | 38 | 0.067 |
| T3-4 | 8 | 1 | 7 | |
| N stage | ||||
| N0 | 78 | 37 | 41 | 0.123 |
| N1 | 4 | 0 | 4 | |
| M stage | ||||
| M0 | 76 | 36 | 40 | 0.215 |
| M1 | 6 | 1 | 5 | |
| TNM grade | ||||
| I - II | 71 | 35 | 36 | 0.1 |
| III - IV | 11 | 2 | 9 | |
| Furhman nulcear grade | ||||
| 1-2 | 32 | 19 | 13 | 0.044 |
| 3-4 | 50 | 18 | 32 | |
| eIF3b expression (ANK) | ||||
| Low expression (+, ++) | 30 | 15 | 15 | 0.5 |
| High expression (+++) | 52 | 22 | 30 | |
eIF3b, eukaryotic translation initiation factor 3 b; ccRCC , clear cell renal cell carcinoma; ANK, adjacent normal kidney.
EIF3b expression and association thereof with clinicopathological variables
The cytoplasm and nuclei of both cancer and renal tubular cells stained positively for eIF3b (Figure 1). In tumors, the staining level correlated with the Fuhrman nuclear grade (p = 0.044; Table 1). Notably, the eIF3b expression levels in ANK tissues were much higher than those in tumors. All ANK tissues (n = 82) stained positively; 30 exhibited low expression (+, ++) and 52 exhibited high expression (+++) (Table 2). eIF3b expression in ANK tissue was associated with the tumor T stage (p = 0.046; Table 2). eIF3b downregulation in tumor tissue was also confirmed by qRT-PCR of fresh samples (Figure 2A). eIF3b expression was significantly higher in NK tissue (p = 0.038) and ANK tissue (p = 0.04) than in tumors. Additionally, eIF3b was expressed at a significantly higher level in metastatic than in non-metastatic tumors (p = 0.0262), indicating that eIF3b contributes to tumor progression.
Correlations between the eIF3b expression levels in adjacent normal kidney (ANK) tissues and the clinicopathological variables of patients with ccRCC (n = 82)
| Variables | No. of patients | eIF3b expression (ANK) | P value | |
|---|---|---|---|---|
| Low (+, ++) n=30 | High (+++) n=52 | |||
| Age | ||||
| ≥57 years | 41 | 15 | 26 | 1 |
| <57 years | 41 | 15 | 26 | |
| Gender | ||||
| Male | 55 | 20 | 35 | 0.953 |
| Female | 27 | 10 | 17 | |
| T stage | ||||
| T1-2 | 74 | 24 | 50 | 0.046 |
| T3-4 | 8 | 6 | 2 | |
| N stage | ||||
| N0 | 78 | 28 | 50 | 0.621 |
| N1 | 4 | 2 | 2 | |
| M stage | ||||
| M0 | 76 | 26 | 50 | 0.185 |
| M1 | 6 | 4 | 2 | |
| TNM grade | ||||
| I - II | 71 | 23 | 48 | 0.088 |
| III - IV | 11 | 7 | 4 | |
| Furhman Nulcear grade | ||||
| 1-2 | 32 | 9 | 23 | 0.245 |
| 3-4 | 50 | 21 | 29 | |
A: qRT-PCR of fresh samples revealed that eIF3b mRNA expression was downregulated in cancer tissues (p = 0.0137, n = 20) and was significantly greater in metastatic than in non-metastatic tumors (p = 0.0262). B: Kaplan-Meier analysis of the effect of the tumor eIF3b level on OS. C: Kaplan-Meier analysis and log-rank testing of the effect of the TNM grade on OS. D: Kaplan-Meier analysis and log-rank testing of the effect of the nuclear Fuhrman grade on OS. E: Kaplan-Meier analysis of the effect of the ANK eIF3b level on OS. F: The tumor eIF3b expression correlated with the nuclear grade. G: Kaplan-Meier analysis of the effect of the eIF3b level (four categories) on OS. H: Kaplan-Meier analysis of eIF3b level in both tumor and adjacent normal (four categories) on OS.
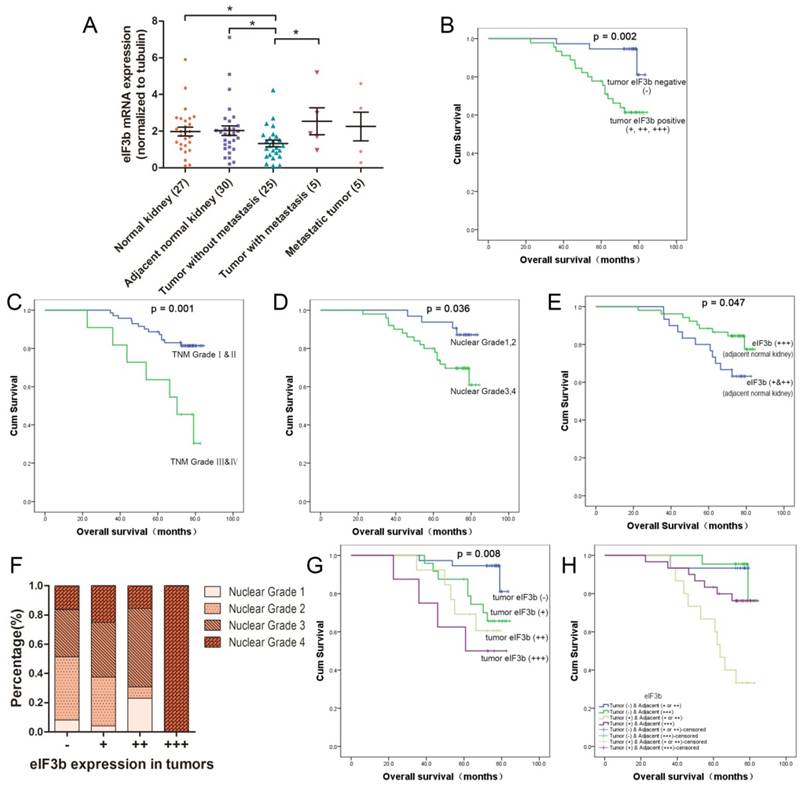
The tumor eIF3b expression level was independently prognostic for patients with ccRCC
Patients with tumors positive for eIF3b (+, ++, +++) (p = 0.002; Table 3, Figure 2B), high TNM grades (p = 0.001; Table 3, Figure 2C), advanced Fuhrman nuclear grades (p = 0.036; Table 3, Figure 2D), and low-level eIF3b expression in ANK tissue (p = 0.047; Table 3, Figure 2E) had significantly poorer prognoses. Furthermore, when tumor eIF3b expression was divided into four categories (-, +, ++, +++), the correlation analysis showed that such expression was significantly (positively) associated with the nuclear grade (Figure 2F). Kaplan-Meier analysis indicated that the greater the level of tumor eIF3b expression, the poorer the OS (p = 0.008; Figure 2G). If we divided all the patients, according to eIF3b expression, into four groups including tumor (-) & adjacent (+ or ++), tumor (-) & adjacent (+++), tumor (+) & adjacent (+ or ++) and tumor (+) & adjacent (+++) (Supplementary Figure 2). Kaplan-Meier analysis indicated that patients with tumor eIF3b (+) and ANK (+ or ++) experienced worse OS compared to the other three groups. (Figure 2H). The multivariate analysis showed that the TNM grade (p = 0.023; Table 3) and the eIF3b tumor expression level (p = 0.019; Table 3) were independently prognostic for patients with ccRCC.
Results of the univariate and multivariate analyses
| Variables | Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|---|
| P value | HR | 95%CI | P value | ||
| Age | 0.934 | ||||
| Gender | 0.285 | ||||
| TNM grading | 0.001 | 2.982 | 1.166 | 7.624 | 0.023 |
| Furhman Nuclear Grade | 0.036 | 2.328 | 0.771 | 7.03 | 0.134 |
| eIF3b expression In ANK | 0.047 | 0.542 | 0.184 | 1.597 | 0.266 |
| eIF3b expression In tumor | 0.002 | 4.488 | 1.283 | 15.704 | 0.019 |
HR, hazard ratio; CI, confidence interval.
Depletion of eIF3b inhibited cell proliferation, migration, and invasion
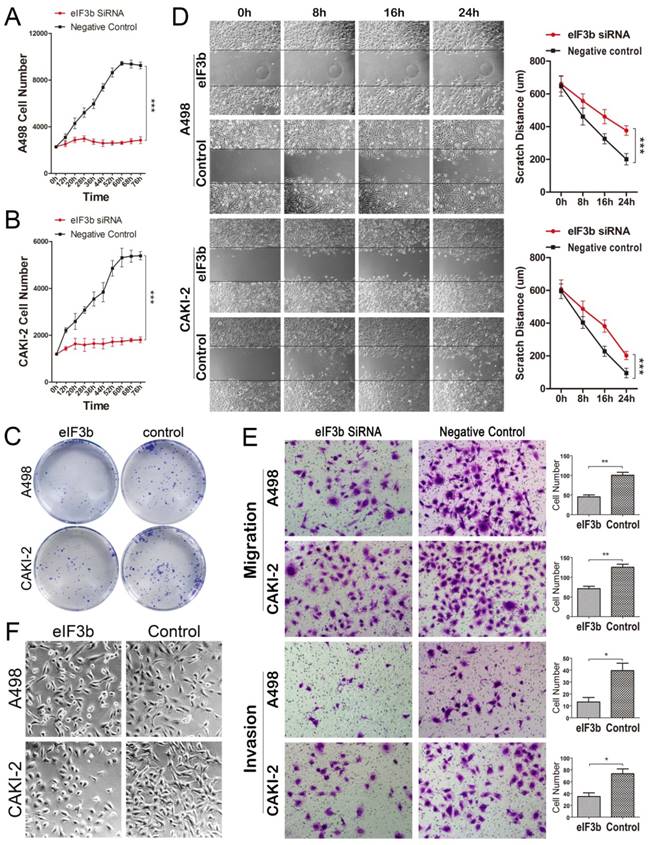
A498 and CAKI-2 cells (which express high levels of eIF3b) were used to conduct in vitro experiments (Supplementary Figure 1A). Cell proliferation was significantly inhibited after eIF3b knockdown (Figure 3A and 3B; Supplementary Figure 1B). Additionally, cell colony numbers fell after eIF3b depletion (Figure 3C). Furthermore, migration capacity was significantly impaired after eIF3b knockdown (Figure 3D). In the transwell assay, eIF3b depletion reduced the number of cells traversing the membrane (compared with control cells) (Figure 3E). Therefore, eIF3b depletion impaired both cell migration and invasion. Notably, eIF3b depletion caused the cells to become smaller and rounded, suggesting that both migration and adhesion were impaired compared with control cells (Figure 3F).
eIF3b knockdown inhibited cell proliferation, migration, and invasion. A-B: eIF3b knockdown inhibited the proliferation of A498 (A) and CAKI-2 (B) cells. C: EIF3b-depleted cells formed fewer colonies. D: The scratch migration assay indicated that A498 and CAKI-2 migration was inhibited after eIF3b knockdown. E: The transwell assay also revealed that migration was impaired (cell numbers, eIF3b siRNA vs. negative control: A498 cells, 45.3 ± 4.9 vs. 100.3 ± 7.3 [p < 0.01]; CAKI-2 cells, 71.0 ± 6.1 vs. 125.3 ± 7.8 [p < 0.01]), as was the invasive capacity (cell numbers, eIF3b siRNA vs. negative control: A498 cells, 13.3 ± 3.8 vs. 39.7 ± 6.2 [p < 0.05]; CAKI-2 cells, 35.0 ± 6.1 vs. 73.3 ± 8.1 [p < 0.05]), upon eIF3b depletion. F: Changes in cellular morphology after eIF3b depletion.
eIF3b depletion triggered G1/S and G2/M arrest
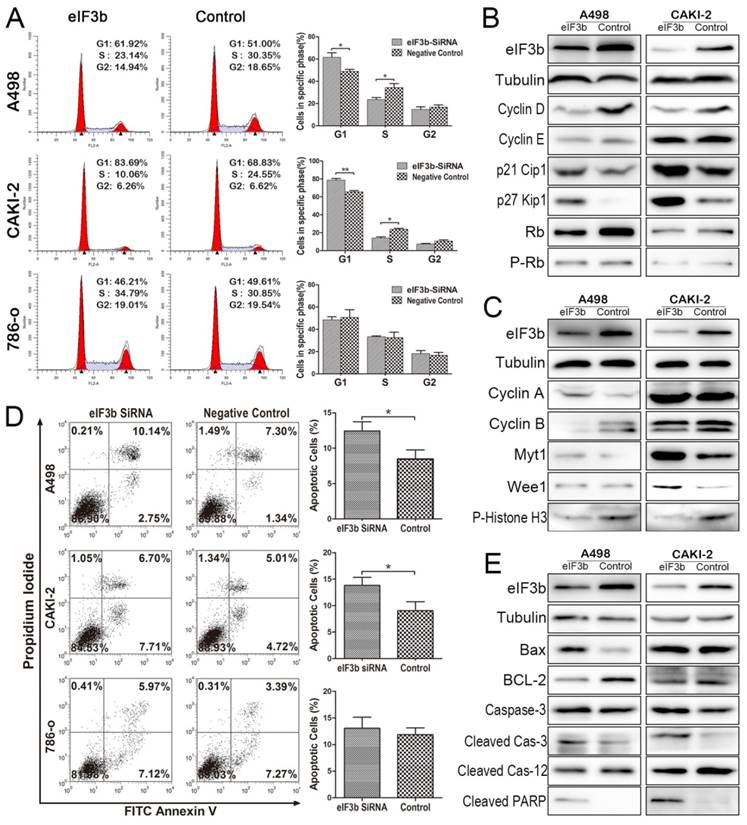
We explored the effects of eIF3b depletion on the cell cycle. The proportion of cells in S-phase was lower in eIF3b-depleted cells (Figure 4A) and the proportion in the G1 phase was higher (Figure 4A) than that in the negative control. Western blotting showed that the G1/S arrest was caused by eIF3b depletion (Figure 4B). During the G1/S transition, cyclins D and E combine with cyclin-dependent kinases (CDK) to form the cyclin/CDK complexes required for the transition. The complexes phosphorylate the retinoblastoma protein (Rb), releasing the E2F transcription factor that activates expression of G1/S progression genes. We found that the levels of both cyclins D and E decreased after eIF3b knockdown. Cyclins D and E, Rb, and the inactivated form of Rb (p-Rb) were downregulated after eIF3b knockdown. In addition, the levels of p27 Kip1 and p21 Cip1, inhibitors of the cyclin/CDK complexes, significantly increased after eIF3b knockdown. Interestingly, Western blotting of G2/M-related proteins indicated that the G2/M transition was also inhibited after eIF3b knockdown (Figure 4C). Cyclin A, which accumulates steadily during the G2 phase and is abruptly destroyed at mitosis, was upregulated after eIF3b depletion. Cyclin B is required for the G2/M transition; we found that the cyclin B level fell. Also, the levels of Myt1 and Wee1 increased after eIF3b depletion; these proteins inhibit cell entry into mitosis. The observed decrease in histone H3 phosphorylation indicated that chromosome condensation was reduced by eIF3b depletion; fewer cells were in the mitotic phase.
Changes in the cell cycle and apoptosis after eIF3b depletion. A: Cell cycle flow cytometry identified G1/S arrest (with more G1-phase cells and fewer S-phase cells). B: Western blotting of G1/S-related proteins verified the G1/S arrest. C: Changes in G2/M-related protein levels indicated that G2/S arrest was also present after eIF3b depletion. D: Cell apoptosis flow cytometry revealed that apoptosis increased after eIF3b depletion. E: Western blotting confirmed the apoptotic changes.
Knockdown of eIF3b promoted apoptosis of renal cancer cells
As eIF3b depletion reduced cell proliferation in vitro, we explored whether apoptosis was involved. The levels of apoptosis increased slightly after eIF3b depletion (Figure 4D). Further analysis (Figure 4E) showed that, after eIF3b knockdown, the pro-apoptotic factors Bax, caspase-3, and the activated form thereof (cleaved caspase-3) were all upregulated and the pro-survival factor Bcl-2 was downregulated. More importantly, cleavage of poly-ADP-ribose polymerase (c-PARP), a marker of apoptosis, was also increased. Therefore, apoptosis increased after eIF3b knockdown. However, interestingly, knockdown reduced the level of cleaved caspase-12, which is required for endoplasmic reticulum-stress-induced apoptosis (Figure 4E).
Impairment of the β-catenin pathway inhibited the epithelial-to-mesenchymal transition (EMT) after eIF3b depletion
The EMT is a key event in tumor invasion and metastasis, including RCC [23]. We found that the epithelial marker E-cadherin was upregulated and the levels of repressors thereof (Slug and Snail) were downregulated after eIF3b depletion. Additionally, the mesenchymal markers N-cadherin and vimentin were downregulated. Therefore, the EMT was inhibited by eIF3b depletion (Figure 5A). The β-catenin pathway is involved in regulation of the EMT [24]. We found that β-catenin expression was significantly downregulated after eIF3b depletion (Figure 5A). The level of cyclin D1 (Figure 4B), a target of β-catenin, was also downregulated after eIF3b depletion. Together, our data showed that eIF3b depletion inactivated the β-catenin pathway and inhibited the EMT of ccRCC.
Pathways affected by eIF3b knockdown. A: eIF3b depletion inhibited EMT progression by inactivating the β-catenin pathway. B: The Akt pathway was inhibited after eIF3b depletion.
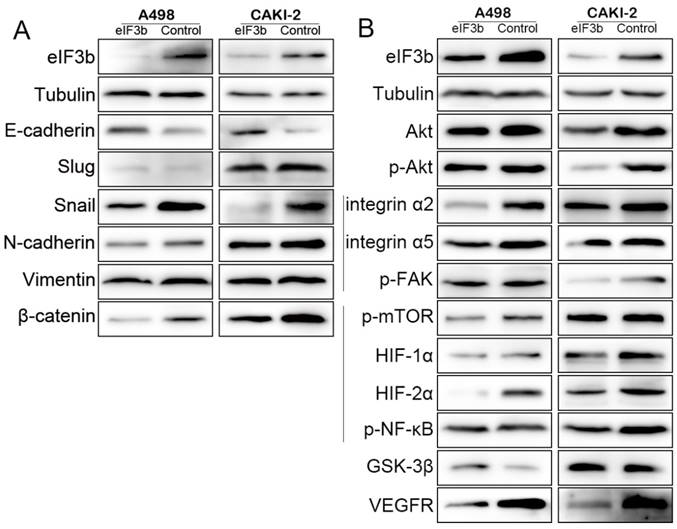
The Akt pathway was impaired after eIF3b depletion
The serine/threonine kinase Akt (also termed protein kinase B or PKB) has attracted a great deal of attention because Akt plays critical roles in the regulation of many cellular functions including metabolism, growth, proliferation, survival, transcription, and protein synthesis [25-29]. We found that eIF3b depletion was not associated with any significant change in Akt levels; however, the level of the activated form, p-Akt, fell in parallel with the extent of eIF3b depletion, indicating that the Akt signaling pathway was involved in such depletion.
The significant morphological changes in cells after eIF3b depletion (Figure 3F) suggested that the integrin pathway might be affected; integrin links the extracellular matrix to the intracellular cytoskeleton to facilitate focal adhesion [30]. After eIF3b knockdown, the levels of integrins α2 and α5 fell significantly, as did the level of the upstream phosphorylated Focal adhesion kinase (p-FAK) protein, suggesting that integrin/FAK/Akt signaling was inhibited (Figure 5B).
Akt plays a critical role in cell growth by directly phosphorylating the mechanistic target of rapamycin (mTOR) [31]. We found that p-mTOR was downregulated after eIF3b knockdown, indicating that the Akt/mTOR pathway was impaired (Figure 5B). Furthermore, the observed downregulation of HIF-1α, HIF-2α, and p-NF-κB after eIF3b depletion may indicate that the Akt/mTOR/HIF [29] and Akt/mTOR/NF-κB [28] pathways were also downregulated (Figure 5B), compromising cell proliferation and inducing apoptosis. The observed reduction in HIF-1α and HIF-2α levels suggested that the cells would be less able to endure hypoxia, reducing cell survival. Vascular Endothelial Growth Factor Receptor (VEGFR), which is often targeted in patients with RCC, was also downregulated (Figure 5B).
Akt promotes cell survival by inhibiting apoptosis via phosphorylation (inactivation) of several proteins [32], including Bcl-2 and Bax (Figure 4E). Apart from the roles played in survival and apoptosis, the Akt pathway is also involved in cell cycle regulation, preventing GSK-3β-mediated phosphorylation and degradation of cyclin D1 [25, 33] and negatively regulating the actions of p27 Kip1 [26] and p21 Waf1/Cip1 [27]. We found that GSk-3β and cyclin D1 were upregulated and p27 Kip1 and p21 Cip1 were downregulated after eIF3b depletion (Figures 4B and 5B), indicating that the Akt/GSK-3β pathway was involved in cell cycle regulation of A498 and CAKI-2 cells.
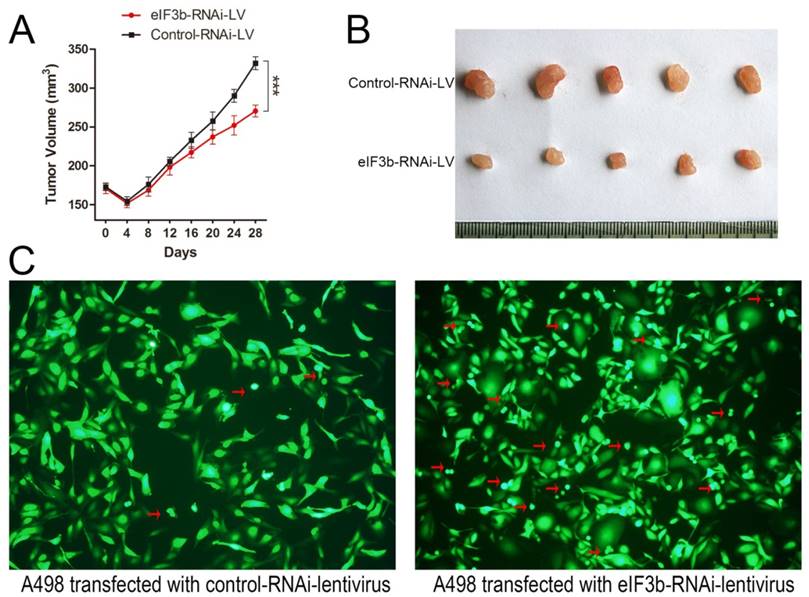
EIF3b depletion inhibited the growth of subcutaneous xenografts in mice
To further investigate the roles played by eIF3b and to validate eIF3b as a potential therapeutic target in vivo, we injected nude mice with A498 cells transfected with the eIF3b-RNAi-containing lentivirus (Supplementary Figure 1C). Control cells yielded could grow into significantly larger tumors than did eIF3b-RNAi-lentivirus transfected cells (p < 0.001; Figure 6A and 6B). Therefore, eIF3b knockdown inhibited the growth of subcutaneous xenografts in mice. And about 2 months after transfection, cells transfected with eIF3b-RNAi lentivirus was found in a worse situation with more cell debris than control cells (Figure 6C).
The animal experiment. A: eIF3b knockdown inhibited the growth of subcutaneous RCC xenografts in mice. B: Xenograft samples after 28 d of growth. C: A498 cells transfected with the eIF3b-RNAi lentivirus (left: eIF3b expression was significantly reduced; right: the transfected cells were abnormal in shape with much more cell debris than control cells) (red arrow: cell debris).
Discussion
Recent studies have shown that certain components of several eIFs can serve as potential prognostic biomarkers and therapeutic targets for several cancers. The relevant components include eIF4E, eIF4A, eIF2α, eIF3a, eIF4B, eIF4F, and eIF5A [4, 34-42]. However, the role of eIF3b in RCC was previously unclear.
We found that higher-level tumor eIF3b expression was associated with a more aggressive phenotype and, more importantly, that the tumor eIF3b expression level was independently prognostic of ccRCC progression. This carcinogenic role of eIF3b was confirmed by in vitro experiment that eIF3b knockdown inhibited tumor cell proliferation, migration, and invasion.
We then showed that eIF3b depletion inhibited cell proliferation by compromising the cell cycle and inducing cell apoptosis. The G1/S arrest after eIF3b depletion has also been noted in bladder cancer [20], glioblastoma [12], and esophageal squamous carcinoma [19]. Wang et al. found that reduction in overall protein synthesis by eIF3b depletion lead to the smaller size of cells which cannot pass the major size regulatory “R or restriction point” checkpoint, which is also a G1/S arrest reason [20]. We also found, in vitro, that the G2/M transition was impaired after eIF3b depletion. However, such impairment may be weak; it was not apparent on flow cytometry. In addition, apoptosis increased after eIF3b depletion. Similar pro-apoptotic effects have been reported in colon cancer [13], glioblastoma [12], and esophageal squamous carcinoma [19]. Interestingly, ER-stress-induced apoptosis seemed to be downregulated after eIF3b depletion. However, the overall effect of eIF3b depletion was pro-apoptotic in ccRCC.
Cell proliferation, migration, and invasion were reduced after eIF3b knockdown. The EMT is a key event in renal tumor invasion and metastasis [23]. We found that the EMT was impaired after eIF3b knockdown, which was characterized by upregulation of E-cadherin and downregulation of N-cadherin, vimentin, and repressors of E-cadherin. We also found that inhibition of the β-catenin pathway contributed to the EMT impairment after eIF3b depletion.
eIF3b knockdown inhibited many activities of the Akt pathway network in RCC cells, including the integrin/FAK/Akt, Akt/mTOR/HIF/VEGF, Akt/mTOR/NF-κB, Akt/Bcl-2/Bax, and Akt/GSK-3β pathways. Inhibition of the integrin/FAK/Akt pathway changed the cellular morphology and impaired adhesion. Inhibition of the Akt/mTOR/HIF/VEGF and Akt/mTOR/NF-κB pathways compromised cell proliferation and increased apoptosis. Akt pathway inhibition also induced apoptosis via downregulation of Bcl-2 and Bax. Inhibition of the Akt/GSK-3β pathway contributed to G1/S arrest.
Dysregulation of protein synthesis has been implicated in the impaired oncogenicity after eIF3b depletion. The inherent “translatability” of different mRNAs varies widely [43, 44]. “Weak” mRNAs encoding proteins involved in cell proliferation are translated after activation of the protein synthesis apparatus [14, 45]. Not all mRNAs are translated to an equal extent, as we also found. mRNA encoding oncogenic proteins seem to be poor competitors (compared with other mRNAs) of the translational complex. Therefore, after eIF3b depletion, the translation of oncogenic proteins in the EMT and Akt signaling pathways is significantly inhibited. In other words, these pathways are stimulated when the level of overall protein synthesis is high (e.g., in tumors expressing high levels of eIF3b). Activation of protein synthesis triggers overproduction of oncogenic proteins and rapid cell growth; downregulation of protein synthesis preferentially impairs the expression of oncogenic proteins, restoring controls on proliferation. This explains why patients with high levels of tumor eIF3b experience poor OS.
Many clinical analyses and in vitro experiments have indicated that tumor eIF3b plays a role in tumor progression. EIF3b is highly expressed in certain tumors [12, 13, 19, 20]. It is thus of considerable interest that we found eIF3b to be downregulated in ccRCC, as evident both immunohistochemically and upon qRT-PCR. Furthermore, we found that high-level eIF3b expression in ANK tissue correlated with longer OS and a less aggressive tumor phenotype; eIF3b thus seems to play a totally different role in normal kidney cells. It is well-known that, transforming growth factor beta (TGF-β) plays different roles in cancer initiation and progression, both suppressing and promoting such progression [46]. We suggest that eIF3b may, like TGF-β, play different roles in kidney cancer. In tumor cells, eIF3b contributes to tumor progression but in non-tumor cells, it plays a protective role in patients with ccRCC. Further in vitro research and study of a larger sample of tumor and normal tissues are required.
eIF3b knockdown inhibited the growth of subcutaneous tumors in mice, confirming eIF3b as a potential therapeutic target for patients with ccRCC. As the high expression in normal kidney tissues, the impact on normal kidney tissues cannot be ignored and it is difficult to design specific therapeutic agents to target this protein. However, after we uncover the role of eIF3b in normal kidney in the future, it is possible to apply this protein to the clinic. Nowadays, RNA-based therapeutics, such as small-interfering (siRNA), microRNAs, antisense oligonucleotides (ASOs), CRISPR-Cas9, have great potential to target a large part of the currently undruggable genes to treat cancers [47]. Besides, recent advances has helped to overcome the lipid bilayer to deliver the RNA into cells cytoplasm and nucleus and enabled RNA-based therapeutics directed to other cancer targets to progress into clinical trials for cancer [48, 49]. There is also research showing that intratumoral administration of ASOs can antagonize pancreatic cancer growth in vivo [50], which points out a promising way of for clinic application research of RNA-based therapeutics targeting eIF3b.
There were certain limitations to our study. We failed to explore the reason why eIF3b expression is higher in ANK than that in tumors. Regulation of involved protein in cancer progression is very complicated. Methylation is probably one of them. A research on breast cancer showed that the status of methylation of genes in tumor is not exactly the same as the adjacent normal [51]. The difference of methylation of eIF3b gene may contribute to the discrepancy of eIF3b protein expression in tumor compared to adjacent tissue. So more work need to be done to uncover the methylation and gene status of ANK and NK tissue.
Together, our results suggest that high-level expression of eIF3b in cancer cells may be essential for expression of the transformed phenotype, and that tumor eIF3b expression is independently prognostic for patients with ccRCC. Furthermore, in vitro eIF3b knockdown effectively impaired the Akt pathway, inhibiting cell proliferation by disrupting the cell cycle, and triggering apoptosis. eIF3b depletion also impaired the EMT, suppressing cell migration and invasion. More importantly, the in vivo work showed that eIF3b may be a useful therapeutic target for patients with ccRCC. More work is needed to understand what role eIF3b plays in non-tumor kidney cell and how eIF3b interacts with other eIFs in terms of cancer development.
Supplementary Material
Supplementary figures.
Abbreviations
eIFs, eukaryotic translation initiation factors; ccRCC, clear cell Renal Cell Carcinoma; OS, Overall Survival; EMT, Epithelial-mesenchymal transition; Normal Kidney, NK; ANK, adjacent normal kidney.
Acknowledgements
The study was supported by National Natural Science foundation of China (No. 81372335; No. 81402108; No. 81400696; No. 81502213). China Postdoctoral Science Foundation (No. 2014M551915).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017
2. Chow WH, Devesa SS. Contemporary epidemiology of renal cell cancer. Cancer J. 2008;14:288-301
3. Hakimi AA, Pham CG, Hsieh JJ. A clear picture of renal cell carcinoma. Nat Genet. 2013;45:849-850
4. Hagner PR, Schneider A, Gartenhaus RB. Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood. 2010;115:2127-2135
5. De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189-3199
6. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113-127
7. Hinnebusch AG. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem Sci. 2006;31:553-562
8. Roobol A, Roobol J, Carden MJ, Smith ME, Hershey JW, Bastide A, Knight JR, Willis AE, Smales CM. The chaperonin CCT interacts with and mediates the correct folding and activity of three subunits of translation initiation factor eIF3: b, i and h. Biochem J. 2014;458:213-224
9. Kolupaeva VG, Unbehaun A, Lomakin IB, Hellen CU, Pestova TV. Binding of eukaryotic initiation factor 3 to ribosomal 40S subunits and its role in ribosomal dissociation and anti-association. RNA. 2005;11:470-486
10. Valasek L, Nielsen KH, Hinnebusch AG. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 2002;21:5886-5898
11. Dong Z, Zhang JT. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit Rev Oncol Hematol. 2006;59:169-180
12. Liang H, Ding X, Zhou C, Zhang Y, Xu M, Zhang C, Xu L. Knockdown of eukaryotic translation initiation factors 3B (EIF3B) inhibits proliferation and promotes apoptosis in glioblastoma cells. Neurol Sci. 2012;33:1057-1062
13. Wang Z, Chen J, Sun J, Cui Z, Wu H. RNA interference-mediated silencing of eukaryotic translation initiation factor 3, subunit B (EIF3B) gene expression inhibits proliferation of colon cancer cells. World J Surg Oncol. 2012;10:119
14. Zhu Q, Qiao GL, Zeng XC, Li Y, Yan JJ, Duan R, Du ZY. Elevated expression of eukaryotic translation initiation factor 3H is associated with proliferation, invasion and tumorigenicity in human hepatocellular carcinoma. Oncotarget. 2016;7:49888-49901
15. Emmanuel R, Weinstein S, Landesman-Milo D, Peer D. eIF3c: a potential therapeutic target for cancer. Cancer Lett. 2013;336:158-166
16. Qi J, Dong Z, Liu J, Zhang JT. EIF3i promotes colon oncogenesis by regulating COX-2 protein synthesis and beta-catenin activation. Oncogene. 2014;33:4156-4163
17. Goh SH, Hong SH, Hong SH, Lee BC, Ju MH, Jeong JS, Cho YR, Kim IH, Lee YS. eIF3m expression influences the regulation of tumorigenesis-related genes in human colon cancer. Oncogene. 2011;30:398-409
18. Liu Y, Neumann P, Kuhle B, Monecke T, Schell S, Chari A, Ficner R. Translation initiation factor eIF3b contains a nine-bladed beta-propeller and interacts with the 40S ribosomal subunit. Structure. 2014;22:923-930
19. Xu F, Xu CZ, Gu J, Liu X, Liu R, Huang E, Yuan Y, Zhao G, Jiang J, Xu C, Chu Y, Lu C, Ge D. Eukaryotic translation initiation factor 3B accelerates the progression of esophageal squamous cell carcinoma by activating beta-catenin signaling pathway. Oncotarget. 2016;7:43401-43411
20. Wang H, Ru Y, Sanchez-Carbayo M, Wang X, Kieft JS, Theodorescu D. Translation initiation factor eIF3b expression in human cancer and its role in tumor growth and lung colonization. Clin Cancer Res. 2013;19:2850-2860
21. Park MS, Chung KY, Kim KD, Yang WI, Chung JH, Kim YS, Chang J, Kim JH, Kim SK, Kim SK. Prognosis of thymic epithelial tumors according to the new World Health Organization histologic classification. Ann Thorac Surg. 2004;78:992-997 discussion 997-998
22. Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6:655-663
23. He H, Magi-Galluzzi C. Epithelial-to-mesenchymal transition in renal neoplasms. Adv Anat Pathol. 2014;21:174-180
24. Wang DX, Zou YJ, Zhuang XB, Chen SX, Lin Y, Li WL, Lin JJ, Lin ZQ. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3beta/beta-catenin signaling pathways. Acta Pharmacol Sin. 2016
25. Chen X, Zhang L, Zheng S, Zhang T, Li M, Zhang X, Zeng Z, McCrae MA, Zhao J, Zhuang H, Lu F. Hepatitis B Virus X Protein Stabilizes Cyclin D1 and Increases Cyclin D1 Nuclear Accumulation through ERK-Mediated Inactivation of GSK-3beta. Cancer Prev Res (Phila). 2015;8:455-463
26. Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem. 2000;275:39223-39230
27. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245-252
28. Lee KB, Byun HJ, Park SH, Park CY, Lee SH, Rho SB. CYR61 controls p53 and NF-kappaB expression through PI3K/Akt/mTOR pathways in carboplatin-induced ovarian cancer cells. Cancer Lett. 2012;315:86-95
29. Shafee N, Kaluz S, Ru N, Stanbridge EJ. PI3K/Akt activity has variable cell-specific effects on expression of HIF target genes, CA9 and VEGF, in human cancer cell lines. Cancer Lett. 2009;282:109-115
30. Schwartz MA. Integrin signaling revisited. Trends Cell Biol. 2001;11:466-470
31. Yamada Y, Kohashi K, Fushimi F, Takahashi Y, Setsu N, Endo M, Yamamoto H, Tokunaga S, Iwamoto Y, Oda Y. Activation of the Akt-mTOR pathway and receptor tyrosine kinase in patients with solitary fibrous tumors. Cancer. 2014;120:864-876
32. Kim HJ, Oh JE, Kim SW, Chun YJ, Kim MY. Ceramide induces p38 MAPK-dependent apoptosis and Bax translocation via inhibition of Akt in HL-60 cells. Cancer Lett. 2008;260:88-95
33. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-3511
34. Bramham CR, Jensen KB, Proud CG. Tuning Specific Translation in Cancer Metastasis and Synaptic Memory: Control at the MNK-eIF4E Axis. Trends Biochem Sci. 2016;41:847-858
35. Cencic R, Pelletier J. Hippuristanol - A potent steroid inhibitor of eukaryotic initiation factor 4A. Translation (Austin). 2016;4:e1137381
36. Yang SS, Gao Y, Wang DY, Xia BR, Liu YD, Qin Y, Ning XM, Li GY, Hao LX, Xiao M, Zhang YY. Overexpression of eukaryotic initiation factor 5A2 (EIF5A2) is associated with cancer progression and poor prognosis in patients with early-stage cervical cancer. Histopathology. 2016;69:276-287
37. Han KS, Li N, Raven PA, Fazli L, Frees S, Ettinger S, Park KC, Hong SJ, Gleave ME, So AI. Inhibition of endoplasmic reticulum chaperone protein glucose-regulated protein 78 potentiates anti-angiogenic therapy in renal cell carcinoma through inactivation of the PERK/eIF2alpha pathway. Oncotarget. 2015;6:34818-34830
38. Madden JM, Mueller KL, Bollig-Fischer A, Stemmer P, Mattingly RR, Boerner JL. Abrogating phosphorylation of eIF4B is required for EGFR and mTOR inhibitor synergy in triple-negative breast cancer. Breast Cancer Res Treat. 2014;147:283-293
39. Robert F, Roman W, Bramoulle A, Fellmann C, Roulston A, Shustik C, Porco JA Jr, Shore GC, Sebag M, Pelletier J. Translation initiation factor eIF4F modifies the dexamethasone response in multiple myeloma. Proc Natl Acad Sci U S A. 2014;111:13421-13426
40. Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene. 2004;23:3790-3801
41. Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254-266
42. Moon HS, Kim B, Gwak H, Suh DH, Song YS. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2alpha) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol Carcinog. 2016;55:346-356
43. Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68:631-634
44. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109-113
45. Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res. 2010;16:4914-4920
46. Tirado-Rodriguez B, Ortega E, Segura-Medina P, Huerta-Yepez S. TGF- beta: an important mediator of allergic disease and a molecule with dual activity in cancer development. J Immunol Res. 2014;2014:318481
47. Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222-229
48. Tamm I, Dorken B, Hartmann G. Antisense therapy in oncology: new hope for an old idea? Lancet. 2001;358:489-497
49. Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Curr Opin Oncol. 2014;26:584-589
50. Fox RG, Lytle NK, Jaquish DV, Park FD, Ito T, Bajaj J, Koechlein CS, Zimdahl B, Yano M, Kopp JL, Kritzik M, Sicklick JK, Sander M, Grandgenett PM, Hollingsworth MA, Shibata S, Pizzo D, Valasek MA, Sasik R, Scadeng M, Okano H, Kim Y, MacLeod AR, Lowy AM, Reya T. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534:407-411
51. Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM, Centeno B, Weber F, Leu YW, Shapiro CL, Eng C, Yeatman TJ, Huang TH. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res. 2006;12:6626-6636
Author contact
![]() Corresponding authors: Zhonghua Xu. E-mail: xuzhonghua1963com. Tel: +86 18560086000 Juchao Ren. E-mail: rjchao1983com. Tel: +86 18560083898
Corresponding authors: Zhonghua Xu. E-mail: xuzhonghua1963com. Tel: +86 18560086000 Juchao Ren. E-mail: rjchao1983com. Tel: +86 18560083898