Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3183-3189. doi:10.7150/jca.20015 This issue Cite
Research Paper
Synergy of purine-scaffold TLR7 agonist with doxorubicin on systemic inhibition of lymphoma in mouse model
Dong Gao1 ![]() , Wang Li1, Wanmin Wang2, Yongguang Cai3, Yuhuan Wang1, Xiaoling Luo1, Chih-Chang Wei1
, Wang Li1, Wanmin Wang2, Yongguang Cai3, Yuhuan Wang1, Xiaoling Luo1, Chih-Chang Wei1 ![]()
1. Shenzhen Hornetcorn Biotechnology Co., Ltd., Shenzhen, 518045, China;
2. Dalian Jinma Medical Technology Co., Ltd., Dalian, 116620, China;
3. The Fifth District of Chemotherapy, Department of Medical Oncology, Central Hospital of Guangdong Provincial Agricultural Reclamation, Zhanjiang, 524002, China.
Received 2017-3-10; Accepted 2017-8-30; Published 2017-9-15
Abstract
Chemo- and radio-therapy suffer from certain well-recognized drawbacks for lymphoma therapy. Passive immunotherapy with monoclonal antibody has improved outcome for patients with CD20+ B cell lymphoma, but not for T cell lymphoma. Therefore, novel treatment approaches are clearly required for T cell lymphoma. To date, the combined application targeting TLR7, 8 and 9 has established long-term antitumor immunity. We previously synthesized a purine-scaffold TLR7 agonist named GD5. Here, we report that the intratumoral administration of GD5 combined with doxorubicin (DOX), a conventional chemotherapeutic agent in T cell lymphoma. This combined treatment made mice to produce more cytokines in blood, and generate more potent cytotoxic T lymphocyte response, then result in effective eradication of both local and distant tumors in tumor-bearing mice. Our findings demonstrate the potential for enhancing the efficacy of the current standard DOX therapy through combination with TLR7 agonist GD5 to improve antitumor immune responses and provide durable remissions for T cell lymphoma.
Keywords: T cell Lymphoma, Combination therapy, Toll-like receptor 7, Doxorubicin, Innate immune
Introduction
Treatments for lymphoid malignancies, current chemotherapy and radiotherapy have serious drawbacks, and passive immunotherapy with monoclonal antibodies was limited to CD20+ B cell malignancies [1]. Furthermore, resistance and relapse are observed also after conventional treatment for many patients with B cell lymphoma [2,3]. Therefore, new therapeutic approaches are urgently required. Toll-like receptor (TLR) agonists [4], anti-CTLA4 [5,6] and anti-CD40 [7,8] have been demonstrated efficacy in preclinical and early phase clinical studies.
TLRs are evolutionarily conserved proteins that detect pathogen associated molecular patterns (PAMPs). They are critical for the innate immune response and are key regulators of both innate and adaptive immune responses. TLRs are constitutively expressed on immune cells, such as antigen-presenting dendritic cell (DC), macrophage; effector B, T; and natural killer cell (NK) [9-11]. TLRs signal regulates cytokine production, polarizing of CD4+ helper, CD8+ effector, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) [12-15].
TLR7 can be activated by natural ligands, such as viral guanosine and/or uridine-rich ssRNA, or by synthetic ligands, such as R848 (Resiquimod) [16], SM360320 (CL087) [17] and R837 (Imiquimod) [18]. TLR7 activation induces strong Th-biassed immune responses through the activation of pDC and mDC subsets, and thus elicits durable tumoricidal effect by supporting the activation and expansion of CD8+ T cells [19]. To date, the combined application targeting TLR7, 8 and 9 established long-term antitumor immunity [20]. A phase II clinical study in which a TLR9 agonist (CpG ODN1018 ISS) was used with rituximab for follicular lymphoma [21]. Systemic administration of an imidazoquinoline-scaffold TLR7 agonist (R848) combined with cyclophosphamide or radiation can prime a cytotoxic T cell response against lymphoma cells, and prime a memory immune response that may prevent recurrence of lymphoma [22]. Enhanced antitumor efficacy was also found when radio- and chemo-responses combined with the TLR9 agonist (CpG ODN1826) [23]. Thus, these cases encourage us to surmise that locally administered TLR7 agonist in combination with conventional chemotherapy would induce systemic antitumor immune responses.
In this study, we investigated intratumoral and intraperitoneal administration of the purine-scaffold TLR7 agonist GD5, 9-(4-carboxyphenyl)-8-hydroxy-2-(2-methoxyethoxy)-adenine, called 9e in our previous paper [24], in combination with chemotherapeutic agent doxorubicin (DOX) in mice T cell lymphoma graft model. Here we demonstrate that intratumoral administration of GD5 can enhance the therapeutic efficacy of DOX by generation of systemic immune response. This setting induced the release of proinflammatory cytokines, promoted the maturation of DC and activated T and B cells, resulting in eradication of both local and distant tumor in lymphoma-bearing murine model. This study provides evidence for translating principle to early phase clinical trial.
Materials and Methods
Mice
C57BL/6 mice were obtained from Guangdong Medical Laboratory Animal Center, China. Mice were bred at 22 ± 0.5 °C on a 12/12 h light-dark cycle from 7 a.m. to 7 p.m. All procedures and protocols were approved by the Institutional Animal Care and Use Committee.
Tumor Cell Lines
The EL4 T cell lymphoma cells were obtained from ATCC. Cell lines were grown in DMEM (Gibco) cell culture medium supplemented with 10% FBS and 100 U/ml penicillin/streptomycin at 37 ˚C in a humidified 5% CO2 atmosphere.
Tumor Models
Tumor models were established by injecting subcutaneously 1×106 EL4 cells at both sites, left and right sides, on the back of each mouse. Tumor size is reported as tumor volume (mm3) by measuring perpendicular diameters of the tumor and calculating as follows: (1/2) × tumor length × tumor width2 [25]; it is expressed as mean volume ± SEM of tumor volume for all mice of each experimental group.
Tumor Therapy
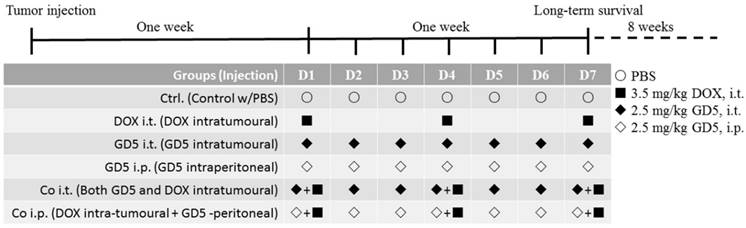
One week after the tumor injection, the tumor volume reached approximately 500-1,000 mm3, the mice were randomly assigned to the PBS-treated control arm or treated arm as Figure 1. DOX and GD5 stock were prepared in PBS at a concentration of 2 μg/μl, respectively. Tumor-bearing mice were treated with DOX, GD5 or both agents. For the monotherapy, GD5 was administered intratumorally or intraperitoneally at a dose of 2.5 mg/kg per mouse and repeated every day in the week. Or DOX, 3.5 mg/kg per model mouse, was applied on days 1, 4 and 7 of each course of treatment. For the combination therapy, mice received 3.5 mg/kg on days 1, 4 and 7, and simultaneously received GD5, intratumorally or intraperitoneally at a dose of 2.5 mg/kg every day in the week. Mice received equivalent PBS every day in the week as control. Tumor growth was measured daily, and long-term survival was evaluated.
The schematic diagram indicated the course schedule. Injected tumor cells subcutaneously at left and right side on the back of each mouse, 7 days later, pharmacotherapy was assigned as schematic diagram. For the monotherapy, GD5 was administered intratumorally or intraperitoneally at a dose of 2.5 mg/kg per mouse and repeated every day in the week, marked as GD5 i.t., ◆, and GD5 i.p., ◇. Or DOX, 3.5 mg/kg per model mouse, was applied on days 1, 4 and 7 of each course of treatment, marked as DOX i.t., ■. For the combination therapy, mice received 3.5 mg/kg on days 1, 4 and 7, and simultaneously received GD5, intratumorally or intraperitoneally at a dose of 2.5 mg/kg every day in the week, marked as Co i.t., or Co i.p.. Mice received equivalent PBS every day in the week as control. Tumor growth was measured daily, and long-term survival was evaluated.
Flow Cytometry and ELISA
One week after the tumor injection, the tumor-bearing mice were treated intraperitoneally with 2.5 mg/kg GD5 or 3.5 mg/kg DOX. After 2 and 4 h, the spleens were collected, and the splenocytes were prepared according to the manufacturer's instructions (BioLegend). Approximately 1 × 106 cells were stained with surface antibodies and analyzed by FACSCalibur flow cytometry (BD Biosciences). The antibodies including anti-CD11c-Alexa488, -CD86-PerCP/Cy5.5, -CD80-Alexa647, -CD3-Alexa488, -CD19-Alexa647 and -CD69-PerCP/Cy5.5 were purchased from BioLegend.
Sera samples were collected at 2, 4 or 24 h from the same administrated mice and stored at -20°C. ELISA was performed using a Ready-SET-Go!® ELISA kit (eBioscience).
Cytotoxicity Assay
After different therapies as described in Figure 1, long-term survival mice were sacrificed, and splenic lymphocytes were collected from each mouse using lymphocyte separation medium (Dakewe, Shenzhen, China), and were incubated with EL4 mouse T cell lymphoma cells at an effector: target cell ratio of 10:1. The cytotoxicity experiment was performed using the CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit according to the manufacturer's instructions.
Statistical Analysis
Statistical analysis and graph were performed using Prism software version 4.0c (GraphPad, San Diego, CA). Data (mean ± SE) were analyzed by one-way ANOVA and a value of p < 0.05 was considered statistically significant.
Results
GD5 Treatment Results in Immune Cell Activation and Cytokine Expression
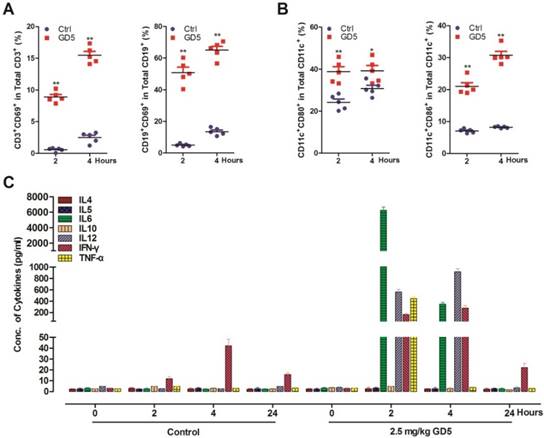
We have previously shown that GD5 directly activated TLR7 signal pathway [24]. Here, we prepared splenocytes from GD5-treated-mice (2.5 mg/kg) to investigate the immune activation effect of GD5 by flow cytometry. We found that GD5 significantly induced the expression of CD69, an early activation marker, in both CD19+ B cells and CD3+ T cells (Fig. 2A). The results also revealed that GD5 stimulated CD11c+ DCs to express the maturation and activation marker CD80 along with CD86, another earlier marker of the immune response (Fig. 2B). To the systemic induction of cytokines, 2 h later, the concentration of TNF-α increased to 440 pg/ml, IFN-γ to 151 pg/ml, IL-6 to 6,020 pg/ml and IL-12 to 580 pg/ml in the serum, in contrast, all of which were almost undetectable in the serum of control mice. Four hours later, the concentration of TNF-α was almost undetectable, and IL-6 decreased by 17.71-fold, Th1 cytokines IFN-γ increased to 1.87-fold and IL-12 to 1.62-fold (Fig. 2C).
Systemic activation of the immune system after administration of GD5. Tumor-bearing mice were treated intraperitoneally with 2.5 mg/kg GD5. After 2 and 4 h, splenocytes were harvested and stained with cell surface marker. The expression of CD3+CD69+ and CD19+CD69+ cells (A), CD11c+CD86+ and CD11c+CD80+ cells (B) were analyzed by flow cytometry. *p < 0.05, **p < 0.01, one-way ANOVA. Induction of systemic cytokines (C) were measured in the plasma. The serums were collected after 2, 4 and 24 h post GD5 treatment and analyzed by ELISA assay (5 mice/group).
Combination Therapy Improves Survival Time
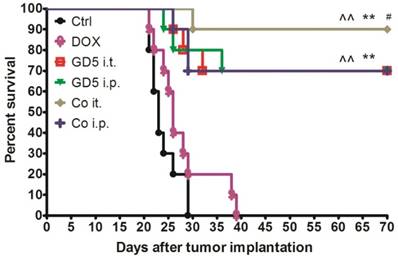
Eight-week-old mice were implanted with EL4 cells, and the therapy began when the tumor grew to 500-1,000 mm3 (day 1, D1). Tumor-bearing mice were randomly assigned to six treatment groups as shown in Figure 1. One course of treatment later, drug effectiveness and long-term survival were observed. Ten weeks later, the mice treated with GD5 have more survival than control or DOX treated groups (p<0.01, log-rank test). Combination of DOX with local administration of GD5 significantly improved survival compared with the two GD5 alone treatment groups (p<0.01, log-rank test). In the two combination groups, for GD5 administration, intratumoral group showed a notable increase in survival compared with intraperitoneal group (Fig. 3).
Survival curve for tumor-bearing mice dosed as graphical representation. Tumor-bearing mice were treated as described in Figure 1. Tumor-bearing mice were randomly assigned to 6 groups, PBS, GD5 i.t., GD5 i.p., DOX i.t., Co i.t., or Co i.p treatment group. Each treatment group contained 10 mice and are representative of at least 2 independent experiments. ^^p < 0.01, compared with ctrl or DOX group; **p < 0.01, compared with GD5 i.t. or i.p. groups; #p < 0.05, compared with Co i.p., log-rank test.
Combination Therapy Initiates Systemic Antitumor Effect
Implanted EL4 cells on the both side of mice back to prepare both-side-tumor-bearing mice model. Right-side-tumors were treated with drugs, whereas left-side-tumors were kept untreated to evaluate the systemic effect. For the right-side-tumor, the all drug treatments showed a significant antitumor effect compared with PBS controls. Either Co i.t. or Co i.p. group exhibited improved tumoricidal effectiveness compared with single-drug treatment groups after which the tumors began to regrow on D10. No significant difference was observed between the Co i.t. and Co i.p. groups (Fig. 4A).
Therapeutic efficacy of combination therapies. Tumor-bearing mice were treated as described in Figure 1. Tumor-bearing mice were randomly assigned to 6 groups, PBS, GD5 i.t., GD5 i.p., DOX i.t., Co i.t., or Co i.p treatment group. After 2 weeks, the volume of the directly treated right tumors (A) and the untreated left tumors (B) were measured. Experimental groups contained at least 5 mice and are representative of at least 2 independent experiments. *p < 0.007, one-way ANOVA, NS, non-significant.
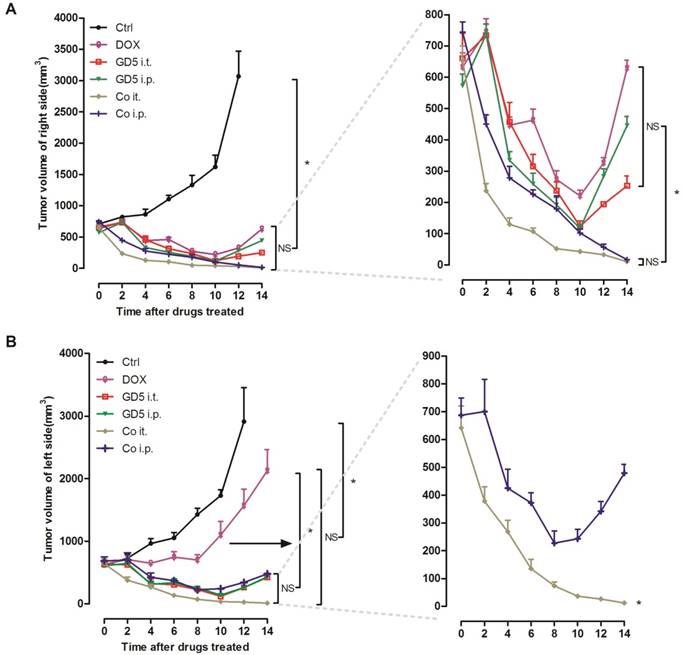
For the left-side tumor, all drug treatments showed a significant antitumor effect compared with controls, while the tumors volume in the DOX group grew significantly larger than in the other 4 groups, especially after D8. Compared with the Co i.p., Co i.t. treatment significantly suppressed the tumors volume, the tumors were undetectable to the naked eye on D14, but in contrast the tumors began to regrow after D8 in the Co i.p. group (Fig. 4B).
Long-term Surviving Mice Have Robust Anti-tumor Immunity
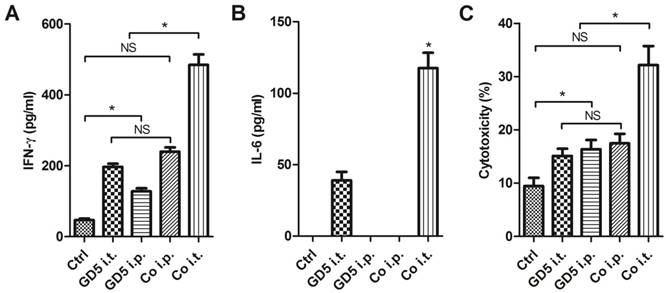
Long-term survival observation later, the surviving mice were sacrificed, and lymphocytes were collected as effector cells, subsequently effector cells were co-cultured with EL4 mouse T cell lymphoma cells at an effector: target cells ratio of 10:1, lymphocytes collected from untreated mice used as control. Co-culture 24 h later, the medium was collected and centrifuged to remove the insoluble components. The cytokine in supernatant was subsequently assayed by ELISA. The results indicated that the lymphocytes collected from Co i.t. group generated a higher level of IFN-γ, which was approximately 10-fold compared with control and 2- to 3-fold compare with that from the GD5 i.t., GD5 i.p. or Co i.p. groups (Fig. 5A). For IL-6 production, it was not observed in Ctrl, GD i.p. and Co i.p. groups, but was observed in GD i.t. and Co i.t. groups with a 3-fold increase in Co i.t. group (Fig. 5B). For the cytotoxicity, EL4 tumor cells were significantly more sensitive to the effector cells from the drugs administration groups than Ctrl group; but no significant difference among GD5 i.t., GD5 i.p. and Co i.p. groups were observed. Only Co i.t. group and supra three groups have significant difference (Fig. 5C).
Surviving mice were provided with a robust anti-tumor immune ability. Tumor-bearing mice were treated as described in Figure 1. Tumor-bearing mice were randomly assigned to 5 groups, PBS, GD5 i.t., GD5 i.p., Co i.t., or Co i.p treatment group. Lymphocytes were harvested from the spleen of long-term surviving mice, and were co-cultured with EL4 cells at an effector: target ratio of 10:1. The cytokine induction of IFN-γ (A) or IL-6 (B) and cytotoxicity (C) were tested. *p < 0.01, n ≥ 7, one-way ANOVA, NS, non-significant.
Discussion
Immunotherapy in combination with conventional drugs showed great promise for lymphoma. For B cell lymphoma, Pam3CSK4, a TLR1/2 agonist was applied with Ara-C, a conventional chemotherapeutic agent, resulting in a synergistic anticancer effect by up-regulated immunomodulatory molecules [26]. In murine lymphoma model, the cyclophosphamide or DOX therapy in combination with TNF-α enhanced the antitumor effects against large established tumors [27,28]. The standard therapies, DOX, cyclophosphamide, vincristine and prednisone (CHOP) were also used in combination with bryostatin 1 for human diffuse large cell lymphoma, or with a monoclonal antibody, rituximab (R-CHOP), for CD20+ B cell lymphoma [29,30].
Targeting the TLRs is a rationale for producing more efficient immune response. To date, few remarkable investigations have been managed with regard to employing TLRs in lymphoid malignancies. Combinational regimens including both TLR9 agonist CpG ODNs, such as 1018 ISS [21] and 1826 [23], and TLR7 agonist imidazoquinolines, such as R848 [22], accompanying with radiotherapies, and conventional chemotherapeutic designs such as denileukindiftitox (ONTAK) [31], rituximab and alemtuzumab seem to surpass single treatments alone. Here, we attempt to establish improved dosage regimen for lymphoma using purine-scaffold TLR7 agonist combined with chemotherapeutic drugs in murine model. In this model, a therapeutic regimen using DOX in combination with TLR7 agonist GD5 significantly improved survival compared with DOX or GD5 administration alone.
GD5 directly activated TLR7 signal pathway, inducing Th1 immune responses, activating B/T cells and DCs as early as 2 h after administration. Moreover, the lymphocytes collecting from long-term survival mice treated by GD5 generated a higher level of IFN-γ and cytotoxicity than control. The tumors appear to regrow 10 days after i.t. and i.p. treatment with GD5, and deaths occur 25 days after treatment, but the tumors develop slowly in long-term survival mice, and the 70% mice survive after 70 days. Moreover, significant difference was observed between Co i.t. group and Co i.p. group in survival time. Thus, no matter alone and combination with DOX, GD5 is responsible for EL4 shrinkage.
TLR7 was widely expressed in lymphoma, such as diffuse large B cell lymphoma (DBCL), peripheral T cell lymphoma (PTCL), follicular lymphoma (FL) and chronic lymphocytic leukaemia (CLL) [32-34]. Previous study found that TLR7/8 agonist R848 may lead to terminal differentiation and suppressed cell proliferation in AML [35], but another TLR7 agonist, imiquimod had no direct effect on acute myeloid leukaemia (AML) cells.
The dosage regimen of TLR agonists must be considered for the anticancer immunotherapy because they could induce quick and systemic proinflammatory cytokine expression [36]. In our dosage regimen, low dose of DOX and every 2 d injection that was approximately 50% less than previous therapy [30]. Previous study reported that local administration of imiquimod or systemic administration of R848 with anti-CD40 immunotherapy or radiotherapy exhibited a strong antitumor effect in a murine model [22,37]. Our results suggested that local administration with DOX and GD5 exhibited the best therapeutic performance by generating a strong local and systemic immune response in the murine among our experimental group setting. Although the underlying mechanisms of the synergy of purine-scaffold TLR7 agonist with doxorubicin on systemic inhibition of EL4 are unclear, from what is known at present, we speculate some factors that including (1) the accumulation of ATP-driven and purinergic receptor-dependent CD11c+CD11b+Ly6Chi tumor-infiltrating leukocytes (TILs) after DOX administration, which efficiently present tumor antigens to CD8+ T cells [38], (2) the activation and maturation of immune cell (B, T cells and DCs), and the expression of cytokine (IFN-γ, TNF-α, IL-12 and IL-6) after GD5 treatment, (3) the enhancement of antigen presentation locally, after GD5 intratumorally administration, where the tumor-specific antigens were concentrated by DOX-induced tumor cell death. Furthermore, CD11b+Ly6G+ cells respond to TLR3 agonist and exhibit tumoricidal activity to EL4 that occurred independently of CD8α+/CD103+ DCs and CTLs [39]. Therefore, induction of CD11b+Ly6G+ cell-mediated cytolysis by TLR3 agonist in combination with CD8+ T cell-based therapy may be beneficial for EL4 shrinkage.
Conclusions
In summary, our results demonstrated that combination therapy with TLR7 agonist GD5 and conventional drugs, like DOX can enhance the antitumor effectiveness, such as generating strong cytokines and enhancing immune response, leading to local and systemic immune response to eradicate both local and distant tumors. Our research suggested that purine-scaffold TLR7 agonist can improve the effectiveness of the current standard CHOP therapy, and develop as a promising dosage regimen for T cell lymphoma.
Abbreviations
AML: acute myeloid leukaemia; CHOP: cyclophosphamide; CLL: chronic lymphocytic leukaemia; Co: combined administration; CTL: cytotoxic T lymphocyte; CTLA4: cytotoxic T lymphocyte antigen 4; DBCL: diffuse large B cell lymphoma; DC: dendritic cell; DOX: doxorubicin; FL: follicular lymphoma; GD5: gaodong 5; IFN-γ: interferon gamma; IL: interleukin; i.t.: intratumorally; i.p.: intraperitoneally; MDSCs: myeloid-derived suppressor cells; NK: natural killer cell; PAMPs: pathogen associated molecular patterns; PBS: phosphate buffered saline; PTCL: peripheral T cell lymphoma; TFN-α: tumor necrosis factor-α; Th1: T helper 1; TILs: tumor-infiltrating leukocytes; TLRs: toll-like receptors; Tregs: regulatory T cells.
Acknowledgements
This work was supported by grants from the Basic Research Program of Shenzhen (JCYJ20130326110139687, JSGG20160226161357949); the China Postdoctoral Science Foundation (2014M552234).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235-42
2. Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24:203-16
3. van Meerten T, Hagenbeek A. CD20-targeted therapy: the next generation of antibodies. Semin Hematol. 2010;47:199-210
4. Harsini S, Beigy M, Akhavan-Sabbagh M, Rezaei N. Toll-like receptors in lymphoid malignancies: double-edged sword. Crit Rev Oncol Hematol. 2014;89:262-83
5. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC. et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379-88
6. Ansell SM, Hurvitz SA, Koenig PA, LaPlant BR, Kabat BF, Fernando D. et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2009;15:6446-53
7. Honeychurch J, Glennie MJ, Johnson PW, Illidge TM. Anti-CD40 monoclonal antibody therapy in combination with irradiation results in a CD8 T-cell-dependent immunity to B-cell lymphoma. Blood. 2003;102:1449-57
8. Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H. et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin's lymphoma. J Clin Oncol. 2009;27:4371-7
9. Bekeredjian-Ding I, Jego G. Toll-like receptors: sentries in the B-cell response. Immunology. 2009;128:311-23
10. Hamm S, Rath S, Michel S, Baumgartner R. Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol. 2009;6:257-65
11. Girart MV, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D regulates IFN-gamma secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-12. J Immunol. 2007;179:3472-9
12. Qi H, Denning TL, Soong L. Differential induction of interleukin-10 and interleukin-12 in dendritic cells by microbial Toll-like receptor activators and skewing of T-cell cytokine profiles. Infect Immun. 2003;71:3337-42
13. Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ. et al. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101-9
14. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033-6
15. Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA. et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci USA. 2003;100:6646-51
16. Bishop GA, Ramirez LM, Baccam M, Busch LK, Pederson LK, Tomai MA. The immune response modifier resiquimod mimics CD40-induced B cell activation. Cell Immunol. 2001;208:9-17
17. Kurimoto A, Ogino T, Ichii S, Isobe Y, Tobe M, Ogita H. et al. Synthesis and evaluation of 2-substituted 8-hydroxyadenines as potent interferon inducers with improved oral bioavailabilities. Bioorg Med Chem. 2004;12:1091-9
18. Miller RL, Gerster JF, Owens ML, Slade HB, Tomai MA. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol. 1999;21:1-14
19. Nishiya T, DeFranco AL. Ligand-regulated chimeric receptor approach reveals distinctive subcellular localization and signaling properties of the Toll-like receptors. J Biol Chem. 2004;279:19008-17
20. Zhao BG, Vasilakos JP, Tross D, Smirnov D, Klinman DM. Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer. 2014;2:12
21. Friedberg JW, Kelly JL, Neuberg D, Peterson DR, Kutok JL, Salloum R. et al. Phase II study of a TLR-9 agonist (1018 ISS) with rituximab in patients with relapsed or refractory follicular lymphoma. Br J Haematol. 2009;146:282-91
22. Dovedi SJ, Melis MH, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J. et al. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood. 2013;121:251-9
23. Mason KA, Neal R, Hunter N, Ariga H, Ang K, Milas L. CpG oligodeoxynucleotides are potent enhancers of radio- and chemoresponses of murine tumors. Radiother Oncol. 2006;80:192-8
24. Gao D, Diao YW, Li W, Gao NN, Liu Y, Wang ZL. et al. Toll-like receptor 7 inactive ligands enhanced cytokine induction by conjugation to weak antigens. ChemMedChem. 2015;10:977-80
25. Carlsson G, Gullberg B, Hafstrom L. Estimation of liver tumor volume using different formulas-an experimental study in rats. J Cancer Res Clin Oncol. 1983;105:20-3
26. Lee SK, Chwee JY, Ma CA, Le Bert N, Huang CW, Gasser S. Synergistic anticancer effects of Pam3CSK4 and Ara-C on B-cell lymphoma cells. Clin Cancer Res. 2014;20:3485-95
27. Ehrke MJ, Verstovsek S, Krawczyk CM, Ujházy P, Zaleskis G, Maccubbin DL. et al. Cyclophosphamide plus tumor necrosis factor-alpha chemoimmunotherapy cured mice: life-long immunity and rejection of re-implanted primary lymphoma. Int J Cancer. 1995;63:463-71
28. Ehrke MJ, Verstovsek S, Ujhazy P, Meer JM, Eppolito C, Maccubbin DL. et al. Doxorubicin plus tumor necrosis factor alpha combination treatments in EL4-lymphoma-bearing C57BL/6 mice. Cancer Immunol Immunother. 1998;45:287-98
29. Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803-43
30. Mohammad RM, Wall NR, Dutcher JA, Al-Katib AM. The addition of bryostatin 1 to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy improves response in a CHOP-resistant human diffuse large cell lymphoma xenograft model. Clin Cancer Res. 2000;6:4950-6
31. Kreitman RJ. Recombinant toxins for the treatment of cancer. Curr Opin Mol Ther. 2003;5:44
32. Akhter A, Masir N, Elyamany G, Phang KC, Mahe E, Al-Zahrani AM. et al. Differential expression of Toll-like receptor (TLR) and B cell receptor (BCR) signaling molecules in primary diffuse large B-cell lymphoma of the central nervous system. J Neurooncol. 2015;121:289-96
33. Spaner DE, Shi Y, White D, Mena J, Hammond C, Tomic J. et al. Immunomodulatory effects of Toll-like receptor-7 activation on chronic lymphocytic leukemia cells. Leukemia. 2006;20:286-95
34. Smith TJ, Yamamoto K, Kurata M, Yukimori A, Suzuki S, Umeda S. et al. Differential expression of Toll-like receptors in follicular lymphoma, diffuse large B-cell lymphoma and peripheral T-cell lymphoma. Exp Mol Pathol. 2010;89:284-90
35. Ignatz-Hoover JJ, Wang H, Moreton SA, Chakrabarti A, Agarwal MK, Sun K. et al. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia. 2015;29:918-26
36. Wu CC, Hayashi T, Takabayashi K, Sabet M, Smee DF, Guiney DD. et al. Immunotherapeutic activity of a conjugate of a Toll-like receptor 7 ligand. Proc Natl Acad Sci USA. 2007;104:3990-5
37. Broomfield SA, van der Most RG, Prosser AC, Mahendran S, Tovey MG, Smyth MJ. et al. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J Immunol. 2009;182:5217-24
38. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H. et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729-41
39. Shime H, Matsumoto M, Seya T. Double-stranded RNA promotes CTL-independent tumor cytolysis mediated by CD11b+Ly6G+ intratumor myeloid cells through the TICAM-1 signaling pathway. Cell Death Differ. 2017;24:385-96
Author contact
![]() Corresponding author: Dong Gao, Shenzhen Hornetcorn Biotechnology Co., Ltd., Shihua Road 14, Shenzhen 518045, Guangdong, China, Tel.: [+86 755 21676129], Fax: [+86 755 21672020], E-mail: gaodong5211com; Chih-Chang Wei, Shenzhen Hornetcorn Biotechnology Co., Ltd., Shihua Road 14, Shenzhen 518045, Guangdong, China, Tel.: [+86 755 21676129], Fax: [+86 755 21672020], E-mail: richardweicccom.
Corresponding author: Dong Gao, Shenzhen Hornetcorn Biotechnology Co., Ltd., Shihua Road 14, Shenzhen 518045, Guangdong, China, Tel.: [+86 755 21676129], Fax: [+86 755 21672020], E-mail: gaodong5211com; Chih-Chang Wei, Shenzhen Hornetcorn Biotechnology Co., Ltd., Shihua Road 14, Shenzhen 518045, Guangdong, China, Tel.: [+86 755 21676129], Fax: [+86 755 21672020], E-mail: richardweicccom.