Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3261-3267. doi:10.7150/jca.21261 This issue Cite
Research Paper
Improvement in prediction of prostate cancer prognosis with somatic mutational signatures
Shengping Zhang1*, Yafei Xu2*, Xinjie Hui2, Fei Yang3, Yueming Hu2, Jianlin Shao4, Hui Liang1 ![]() , Yejun Wang2
, Yejun Wang2 ![]()
1. Dept. Surgical Urology, The Affiliated Longhua District People's Hospital of Southern Medical University, Shenzhen 518109, China;
2. Dept. Cell Biology and Genetics, Shenzhen University Health Science Center, Shenzhen 518060, China;
3. Dept. Surgical Urology, The third affiliated hospital, Sun Yat-Sen University, Guangzhou 510630, China;
4. First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China.
*Shengping Zhang and Yafei Xu contributed equally to this article.
Received 2017-5-31; Accepted 2017-8-29; Published 2017-9-15
Abstract
Prostate cancer is a leading male malignancy worldwide, while the prognosis prediction remains quite inaccurate. The study aimed to observe whether there was an association between the prognosis of prostate cancer and genetic mutation profile, and to build an accurate prognostic predictor based on the genetic signatures. The patients diagnosed of prostate cancer from The Cancer Genomic Atlas were used for prognostic stratification, while the somatic gene mutation profiles were compared between different prognostic groups. The genetic features were further used for training machine-learning models to predict prostate cancer prognosis. No significant gene with somatic mutation rate difference was found between prognostic groups of prostate cancer. Total 43 atypical genes were screened for building a support vector machine model to predict prostate cancer prognosis, with an average accuracy of 66% and 64% for 5-fold cross-validation or training-testing evaluation respectively. When combined with the National Institute for Health and Care Excellence (NICE) features, the model could be further improved, with the 5-fold cross-validation accuracy of ~71%, much better than NICE itself (62%). To our knowledge, for the first time, the research studied the relationship of genome-wide somatic mutations with prostate prognosis, and developed an effective prognostic prediction model with the atypical genetic signatures.
Keywords: prostate cancer, somatic mutation, prognosis prediction, atypical features, support vector machine
Introduction
Prostate cancer (PCa) is one of the most common malignancy in male worldwide, with ~ 1,000,000 cases diagnosed annually [1]. In developed countries, PCa is the second leading cause of cancer-related deaths among men [2]. Both genetics and demographic factors such as age, family history and race, are closely related with the incidence and progress of PCa [3-4]. As our understanding has been broadened gradually on the underlying biology of PCa, various treatment strategies have also been developed, such as radical prostatectomy, hormone deprivation therapy, radiation therapy and chemotherapy. However, the prognosis of PCa is still far away from being satisfying, and most tumors relapse in 2 years to the castration-resistant state [5].
Currently, over 80% of PCa are localised or locally advanced non-metastatic diseases and the patients face the selection of the best treatment regimen from a wide array [6]. Risk stratification plays an important role in the clinical decision making and treatment options, which is mainly determined by a general impression currently, with the combination of a couple of clinical parameters, such as PSA concentration, clinical stage, biopsy Gleason score, patient age, number of positive prostate biopsies and so on [7-10]. The most widely used stratification system for primary non-metastatic PCa is endorsed by the National Institute for Health and Care Excellence (NICE) guidelines, which use presenting PSA concentration, Gleason grade, and clinical T stage to classify PCa patients as low, intermediate, or high risk [11]. Some new methods were proposed on the basis of the NICE stratification system, which displayed improved prognostic power [6]. Despite the success of these risk stratification systems in prognosis prediction, the tumors within the same risk groups still showed remarkably different clinical courses [12-14]. Therefore, new prognostic prediction tools are still urgently needed to further improve the accuracy and sensitivity of classification of PCa.
Classically, the progression of various cancer types is ascribed to the sequential accumulation of genetic alterations. Somatic mutation signatures have been successfully applied in the development of prognostic prediction tools for various cancers, such as breast cancer, lung cancer, nasopharyngeal carcinoma, etc [15-17]. Gene signatures have also been attempted in PCa risk stratification [18-21]. For example, Irshad et al identified a three-gene panel, including FGFR1, PMP22 and CDKN1A, which could accurately predict the outcome PCa with low Gleason scores [20]. Berg et al found that over-expression of ERG was associated with an increased risk of disease progression during active surveillance for PCa patients [22]. In another study, a model with 100-gene signature, which classified PCa patients into five separate subgroups with distinct genomic alterations and expression profiles, showed better performance in prediction of diseases with poor prognosis than traditional predictors based on PSA and Gleason scores [23]. The above studies demonstrate that genetic variation plays an important role in the classification of PCa and may display immense potential of clinical prediction. However, the current widely-used risk stratification systems in PCa were almost exclusively based on routine clinic-pathological parameters, without attention to the genetic variation.
In this research, an extensive comparison was performed on the somatic mutation profiles in PCa with different prognosis, with the prostate adenocarcinoma (PRAD) data from The Cancer Genome Atlas (TCGA). No gene was found with significant somatic mutation rates between groups (False Positive Rate, FDR < 0.05). However, a combined filtering strategy generated 43 genes, which were further used as features for prognostic model development and reached good classification performance. With a 5-fold cross validation, the genetic model based on the 43 features showed an average AUC (Area Under Curve) of ROC (Receiver Operating Characteristic) curves and accuracy of ~0.70 and ~0.66 receptively, better than NICE (accuracy: ~0.62). A combined model with both the genetic signatures and NICE could reach better average performance (AUC: ~0.75; accuracy: ~0.71). Taking together, the study suggested that the somatic mutation signatures could largely facilitate the prognostic prediction of PCa, independently or combined with other clinical features.
Materials and Methods
Datasets, stratification and somatic mutation rate comparison
The clinical data for the patients with PCa were downloaded from TCGA (The Cancer Genome Atlas) website. The somatic mutation data between tumor-normal pairs of each PCa case were also downloaded. The mutations causing codon changes, frame-shifts, and premature translational terminations were retrieved for further analysis. Cases were stratified based on either 'tumor status' or 'biochemical recurrence'. For 'tumor status', 'with tumor' group included the patients detected with residual or recurrent tumors before death or at last follow-up; the rest were classified into 'tumor free' group. For 'biochemical recurrence' stratification, two groups were designated with 'recurrence' and 'non-recurrence' representing the cases with or without recurrence respectively. The clinical examination results were also used for NICE risk stratification ('low', 'medium' and 'high' risks) [11]. To compare the somatic gene mutation frequency between prognostic groups, a matrix was prepared to record the mutations of all the genes for each case, followed by counting the number of cases with mutations for each gene in each group. Both Chi-square test with Benjamini & Hochberg correction and EBT were used for rate comparisons, and a False Discovery Rate (FDR) or p value < 0.05 was set as the significance level for Chi-square or EBT test respectively [24, 25].
Feature selection
A multi-factor filtering strategy was proposed to select the genetic features for prognosis-prediction model training. The genes were filtered when any of the following criteria was met: (1) the mutation rates in both groups were lower than 5%; (2) the absolute difference between the mutation rates of two groups was lower than 5%; (3) the significance of Chi-square test without FDR correction was higher than 0.1. Both TopN and mRMR strategy were also used for feature selection and model comparison [25, 26]. For TopN strategy, the top N genes with smallest p values (EBT) for mutation rate comparison were selected as the features [25]. The mRMR software package was downloaded, installed and used for mRMR feature selection [26].
Training of Support Vector Machine models
The n genes were selected as genetic features for model training. For each case Pj (j = 1, 2, …,mi) belonging to a certain category Ci, where i equaled to 1 or 0, and mi represented the total number of cases of the category Ci, the genetic features were represented as a binary vector Fj (g1,g2,…,gn) in which gk (k = 1, 2, …, n) represented the kth genetic feature, taking the value of 1 if the corresponding gene was mutated and 0 otherwise. There was an mi*n matrix for category Ci. When NICE was used as an additional feature, the size of matrix was enlarged to mi*(n+1), and the NICE feature was also represented in a binary form in the additional column, for which 1 and 0 represented 'high' and 'low'/'medium', respectively.
An R package, 'e1071', was used for training Support Vector Machine (SVM) models using each training dataset (http://cran.r-project.org). For each training-testing experiment, the training dataset was used for both kernel selection and parameter optimization as described previously [25]. Four kernels, including 'radial' (Radial Base Function, RBF), 'linear', 'polynomial' and 'sigmoid', were individually tested for the best-optimized parameters with a 10-fold cross-validation grid search strategy. The performance of different kernels with best-optimized parameters was then compared and the best kernel (with optimal parameters) was selected for further model training and prediction on the testing dataset.
Model performance assessment
A 5-fold cross validation and training-testing strategy were used for model performance evaluation. For 5-fold cross validation, the original feature-represented matrix for each category were randomly split into five parts with identical size. Every four parts of each category were combined and served as a training dataset while the rest one of each category was used for testing and performance evaluation. For the training-testing strategy, 2/3 of the original cases belonging to each category were randomly selected for mutation frequency comparison or feature selection and consequential representation, and served as the training datasets. Matrices were prepared for the rest 1/3 of the cases with the features newly identified with corresponding training datasets, and used for testing.
The relatively balanced items, Receiver Operating Characteristic (ROC) curve, the area under ROC curve (AUC) and Accuracy, were utilized to assess the predictive performance. An ROC curve is a plot of Sensitivity versus (1 - Specificity) and is generated by shifting the decision threshold. AUC gives a measure of classifier performance. Accuracy was defined as (TP + TN)/(TP+FP+TN+FN), where TP, TN, FP and FN represented true positives, true negatives, false positives and false negatives respectively. The performance of genetic or combined models was recorded as the average 5-fold cross-validation or training-testing results, while that of pure NICE model was represented as the average 10-fold bootstrapping results. Students' t-tests were performed for the performance comparison with a preset significance level of 0.05.
Results
Prognostic stratification of PCa
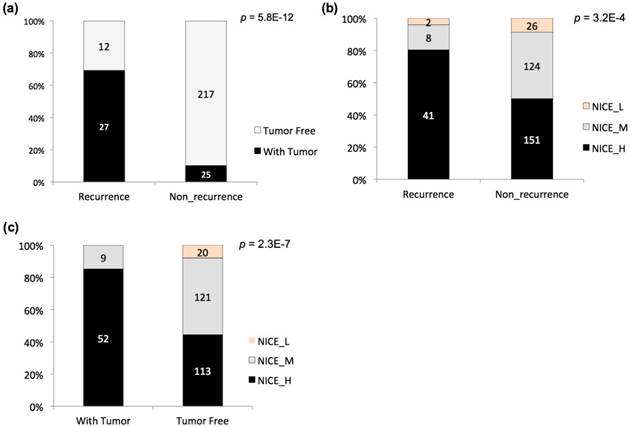
The post-operation survival rate is high for PCa patients, so the biochemical recurrence or non-recurrence and other indicators have been used as more effective prognostic statistics [23]. Two indicators, biochemical recurrence / non-recurrence, and tumor status of the last follow-up (with tumor / tumor free), were also adopted to stratify the TCGA PCa patients (Fig 1a). A significant dependence was observed between the two stratification criteria, with apparent enrichment of 'with tumor' patients in the 'recurrence' group (i.e., 'tumor free' patients in the 'non-recurrence' group) (Fig 1a; p = 5.8E-12, Chi-square test). NICE has been widely used for guiding the prognostic assessment for PCa in clinical practice. The TCGA cases were also evaluated with NICE, followed by comparison with the stratification results based on recurrence or tumor status (Fig 1b-c). With each type of stratification, NICE levels showed significant association with the prognostic groups (Fig 1b-c; p = 3.2E-4 and 2.3E-7 for NICE vs. recurrence status, and NICE vs. tumor status, respectively). Taking together, the results suggested that both recurrence status and tumor status could be considered as indicators used for prognostic stratification of PCa.
Prognostic stratification of TCGA PCa cases. (a) Stratification of PCa cases based on biochemical recurrence status and tumor status at last follow-up and the relationship. (b) Stratification of PCa cases based on biochemical recurrence status and NICE criteria and the relationship. (c) Stratification of PCa cases tumor status at last follow-up and NICE criteria and the relationship. The accumulative bar diagrams were shown with the sum percentage of 100%. The number of cases for each subgroup was indicated. Chi-square tests were performed, with the p values indicated at the right upper corner.
Classification of PCa prognosis with atypical somatic mutation signatures
A majority of the TCGA PCa cases were also profiled for the tumor somatic mutations. To observe whether there is an association between PCa prognosis and somatic mutation profiles, the cases with somatic mutation data were stratified according to the prognostic indicators (Table 1). Statistical comparisons were further performed between prognostic groups per gene for the mutation rates. However, with either strategy of stratification, no any gene with significant mutation rate difference was called between prognostic groups (Table 1; Supplemental file 1-4).
Sample size summary and comparison of somatic mutation profiles between PCa prognostic groups
| Recurrence Status | Tumor Status | ||
|---|---|---|---|
| Recurrence # | 58 | With Tumor # | 80 |
| Non_recurrence # | 366 | Tumor Free # | 308 |
| Sign. Genes # | 0 | Sign. Genes # | 0 |
Note: Rate comparisons were performed with both Chi-square tests with FDR correction and EBT.
To further observe whether the atypical somatic mutation profiles were associated with PCa prognosis and therefore useful for classification with machine learning strategies, an integrated feature-filtering pipeline was adopted to screen the possibly more meaningful signatures. The prognosis data stratified by tumor status was used for further analysis since the 'poor prognosis' group ('With Tumor') contained more samples than the corresponding group stratified by recurrence status ('Recurrence') (Table 1). In total, 43 genes with subtle mutation difference between prognostic groups were identified with the filtering pipeline (Table 2).
The list of 43 genes used for PCa prognosis classification
| Signature genes | |||
|---|---|---|---|
| AHNAK2 | FAM47C | MUC2 | SACS |
| ANKRD30A | FAT2 | MUC4 | SALL1 |
| ANKRD36C | FAT4 | MYH11 | SCN5A |
| APOB | FBN3 | MYT1L | SPOP |
| ATP13A5 | FLG2 | NOD1 | SRCAP |
| BAI3 | FRG1B | PCDHA12 | TP53 |
| CACNA1A | HSPG2 | PIK3CA | TRPM6 |
| CACNA1E | KMT2D | PTEN | USH2A |
| CDH23 | KRTAP4-9 | PTH2 | ZNF208 |
| CNTNAP5 | LPHN3 | PTPRC | ZNF91 |
| EPB41L3 | MUC16 | RYR1 | |
SVM models were trained with the 43 features represented by their somatic mutation profile. The SVM models based on the 43 atypical features could well discriminate prognosis of PCa, achieving an average AUC of 0.696 and accuracy of 0.662 with a 5-fold cross-validation strategy (Fig 2a-b). When the feature size was reduced, the model performance also declined strikingly (Fig 2a-b). Interestingly, the models also performed much better than the ones based on the same size of genes with smallest p value for mutation rate comparison between prognostic groups (topN), or those based on 43 genes with smallest redundancy filtered with mRMR (Fig 2c).
Prediction of PCa prognosis with models based on genetic features. (a) ROC curves of 5-, 10, 20, 30 and 43-gene genetic models (f5, f10, f20, f30 and f43, respectively). The average results of 5-fold cross validations were shown. (b) AUC and general accuracy of prognosis prediction models with varied feature size. (c) Comparison of AUC and general accuracy of the f43 model and those based on topN and mRMR feature selection strategies. (d) Performance of models based on 5-fold cross validation (CV) and 5-fold training-testing (TT).
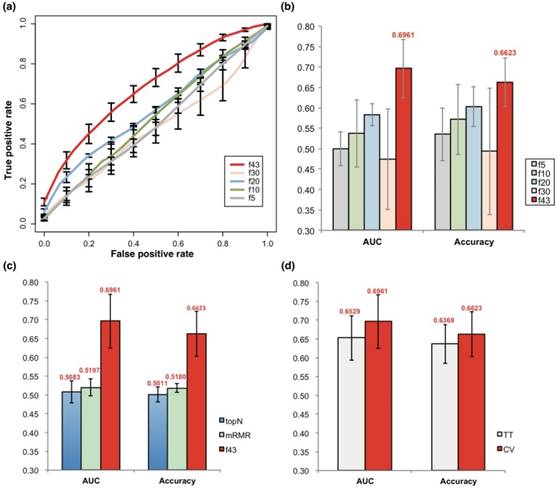
Training-testing evaluations were also performed to test the effectiveness of the models and the feature selection strategy. With the size of features varying from 29 to 47, the models averagely reached an AUC of 0.653, only slightly lower than the 5-fold cross-validation results, but better than the neutral, topN or mRMR models (Fig 2d).
Improvement of prognostic prediction of PCa with a combination of NICE and somatic mutation signatures
Currently, NICE was most often used for PCa prognosis prediction. NICE was also used to predict the prognosis of TCGA PCa cases, but only with accuracy of 55.3% and 61.5% for recurrence and tumor status stratification respectively (Fig 3a). The performance was even worse than the 5-fold cross-validation or training-testing models.
Prediction of PCa prognosis with models based on the combined NICE and genetic features. (a) The classification performance of NICE on PCa prognosis stratified by tumor status or recurrence. Bootstrapping analysis was performed and the results were represented as mean ± sd. (b) ROC curves of 43-gene genetic models (f43) and models based on the combined NICE and genetic features (f43+NICE). The average results of 5-fold cross validations were shown. (c) Comparison of the general accuracy of different prognosis prediction models. Students' t-tests were performed, and asterisk represented p < 0.05.
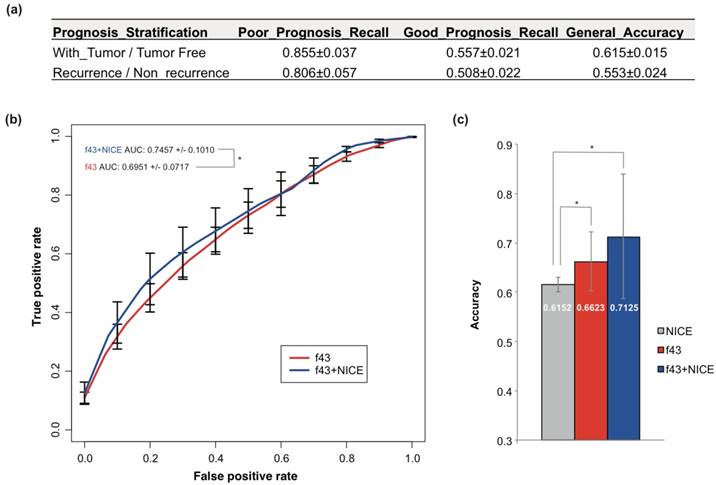
The NICE stratification results were also considered as an independent feature and combined with the 43 genetic signatures. A new SVM model was built, which showed apparent improvement for the performance compared with NICE or models based on genetic signatures solely (Fig 3b-c). The 5-fold cross-validation AUC and accuracy achieved 0.746 and 0.713 respectively (Fig 3b-c).
Discussion
PCa is an important cancer type, with a high worldwide morbidity. The 5-year survival has been improved significantly recently. However, there is still a big challenge to reduce the long-term mortality and recurrence, and to increase the percentages of tumor-free survival. NICE has been for a long time used as risk stratification of PCa patients on prognosis. However, the accuracy needs to be improved. With TCGA PCa data, we also found that NICE stratification could only correctly predict the prognosis for ~ 60% of the patients (Fig 3a).
The somatic mutations have been well characterized for PCa patients [4,21]. However, it remained largely unknown whether the prognosis of PCa was also related with genetic background. One major objective of the current research was to answer the question. Prognostic groups were stratified by either 'tumor status' or 'recurrence'; however, no gene was discovered that showed significant somatic mutation rate difference between groups with different prognosis. There could be no association between prognoses of PCa with somatic mutation profiles, but alternatively, other factors could also explain the observations. For example, the prognosis of PCa could be further improved with new stratification criteria. The small number of PCa cases and the general low somatic gene mutation rates in PCa could also have led to the dramatic low power [25]. Therefore, no solid conclusions could be drawn before more objective-targeted studies are performed with enlarged size of cases and observation of elongated period of survival. In fact, with the atypical mutation rate difference between prognostic groups, the models trained in this research could still well distinguish the prognosis, with accuracy even higher than NICE (Fig 3c). The results indirectly suggested the dependency of prognosis with genetic mutation profile.
In total, 43 atypical features were used for the model predicting PCa prognosis. Although many of the genes have been reported to function in different tumor types and progresses, a functional clustering analysis showed a significant enrichment of genes participating in calcium ion binding and transporting (GO:0015085, p = 2.59e-02; GO:0005509, p = 3.15e-02; PANTHER Overrepresentation Test, http://pantherdb.org/; data not shown). The combination of these genetic features with NICE factors appeared to improve the prognosis prediction significantly when compared with models based only on genetic features or NICE (Fig 3c). A tool was also developed to facilitate the testing of the new method in PCa prognosis prediction (http://www.szu-bioinf.org/PCpp). There are several drawbacks with the current model that need to be improved in the future. First of all, the current model was only evaluated with a single dataset from TCGA since it is difficult to find another dataset with both full genomic information and clinical data. 5-fold cross validation and training-testing were performed to correct the overfitting problem; however, new independent datasets are still in need to make more accurate evaluation. The size of genetic features was also a little large, and new experiments with enlarged size of cases could assist the finding of fewer more effective features.
Abbreviations
PCa: Prostate Cancer; AUC: Area Under the Curve; ROC: Receiver Operating Characteristic; TCGA: The Cancer Genome Atlas; SVM: Support Vector Machine; RBF: Radial Base Function; TF: Tumor Free; WT: With Tumor.
Author Contributions
YW, SZ and HL conceived the project. SZ, YX, YH, FY and YW collected the data and performed the data analysis. XH and YW performed statistical analysis. HL provided the clinical support. SZ, YX, JS and YW developed the models. SZ, JS and YW developed the software tools. SZ, YX and YW wrote the manuscript and all the authors revised it. All the authors approved the final version of manuscript.
Supplementary Material
Supplementary tables.
Acknowledgements
We are greatly indebted to the TCGA (The Cancer Genome Atlas) team for their maintaining and sharing valuable cancer genomics data.
Fundings
The project was supported by a Natural Science Funding of Shenzhen (grant no. JCYJ20160422091914681), a Natural Science Funding of Guangdong Province (grant no. 2017A030310468), and a Natural Science Foundation of SZU (grant no. 2016082) to YX, and a Natural Science Foundation of SZU (grant no. 2015059) to YW. HL was supported by a Shenzhen Knowledge Innovation Program (grant no. JCYJ20150402144905865).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mistry M, Parkin D M, Ahmad AS. et al. Cancer incidence in the United Kingdom: projections to the year 2030. Br J Cancer. 2011;105:1795-1803
2. Siegel RL, Miller K D, Jemal A. Cancer statistics. CA Cancer J Clin. 2015;65:5-29
3. Hoffman RM. Clinical practice. Screening for prostate cancer. N Engl J Med. 2011;365:2013-9
4. Al Olama AA, Kote-Jarai Z, Berndt SI. et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46:1103-9
5. Damber JE, Aus G. Prostate cancer. Lancet. 2008;371:1710-21
6. Gnanapragasam VJ, Lophatananon A, Wright KA. et al. Improving Clinical Risk Stratification at Diagnosis in Primary Prostate Cancer: A Prognostic Modelling Study. PLoS Med. 2016;13:e1002063
7. Attard G, Parker C, Eeles RA. et al. Prostate cancer. Lancet. 2016;387:70-82
8. Gleason DF. Classification of prostatic carcinomas. Cancer Chemother Rep. 1966;50:125-8
9. Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111:58-64
10. Catalona WJ, Richie JP, Ahmann FR. et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J Urol. 1994;151:1283-90
11. National Institute for Health and Care Excellence. Prostate cancer: diagnosis and treatment. NICE guidelines [CG175]. London: National Institute for Health and Care Excellence. 2014
12. Kattan MW, Eastham JA, Stapleton AM. et al. A preoperative nomogram for disease recurrence following radical prostatectomy for prostate cancer. J Natl Cancer Inst. 1998;90:766-71
13. Reese AC, Pierorazio PM, Han M. et al. Contemporary evaluation of the National Comprehensive Cancer Network prostate cancer risk classification system. Urology. 2012;80:1075-9
14. Hernandez DJ, Nielsen ME, Han M. et al. Contemporary evaluation of the D'amico risk classification of prostate cancer. Urology. 2007;70:931-5
15. Zhao SG, Chang SL, Spratt DE. et al. Development and validation of a 24-gene predictor of response to postoperative radiotherapy in prostate cancer: a matched, retrospective analysis. Lancet Oncol. 2016;17:1612-20
16. Curtis C, Shah SP, Chin SF. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346-52
17. Liu N, Chen NY, Cui RX. et al. Prognostic value of a microRNA signature in nasopharyngeal carcinoma: a microRNA expression analysis. Lancet Oncol. 2012;13:633-41
18. Glinsky GV, Glinskii AB, Stephenson AJ. et al. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest. 2004;113:913-23
19. Tomlins SA, Mehra R, Rhodes DR. et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41-51
20. Irshad S, Bansal M, Castillo-Martin M. et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5:202ra122
21. Taylor BS, Schultz N, Hieronymus H. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22
22. Berg KD, Vainer B, Thomsen FB. et al. ERG protein expression in diagnostic specimens is associated with increased risk of progression during active surveillance for prostate cancer. Eur Urol. 2014;66:851-60
23. Ross-Adams H, Lamb AD, Dunning MJ. et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine. 2015;2:1133-44
24. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289-300
25. Hui X, Hu Y, Sun MA. et al. EBT: A Statistic Test Identifying Moderate Size of Significant Features with Balanced Power and Precision for Genome-wide Rate Comparisons. Bioinformatics. 2017 doi: 10.1093/bioinformatics/btx294
26. Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol. 2005;3:185-205
Author contact
![]() Corresponding authors: Yejun Wang, Dept. Cell Biology and Genetics, Shenzhen University Health Science Center, Shenzhen 518060, China. Email: wangyjedu.cn; Hui Liang, Dept. of Surgical Urology, The Affiliated Longhua District People's Hospital of Southern Medical University, Shenzhen 518109, China. Email: lianghui1976com
Corresponding authors: Yejun Wang, Dept. Cell Biology and Genetics, Shenzhen University Health Science Center, Shenzhen 518060, China. Email: wangyjedu.cn; Hui Liang, Dept. of Surgical Urology, The Affiliated Longhua District People's Hospital of Southern Medical University, Shenzhen 518109, China. Email: lianghui1976com