Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(17):3405-3409. doi:10.7150/jca.21145 This issue Cite
Research Paper
p300 Knockout Promotes Butyrate Resistance
Darina L. Lazarova, Michael Bordonaro ![]()
Department of Basic Sciences, Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509, USA.
Received 2017-5-23; Accepted 2017-8-31; Published 2017-9-20
Abstract
Dietary fiber is linked to a reduced risk of colorectal cancer (CRC), and this protective activity is likely due to its fermentation product, butyrate. Dependent upon the hyperactivation of Wnt signaling, butyrate represses CRC cell growth and induces apoptosis. However, resistance to butyrate activity may allow for CRC development even in the context of relatively high fiber intake. We have previously determined that CRC cells resistant to butyrate are deficient in p300 expression. The histone acetylase p300 influences colonic cell signaling and physiology through effects on Wnt signaling. In this short research communication, we report that p300 knockout CRC cells exhibit butyrate resistance, and the re-introduction of p300 expression in p300 knockout CRC cells restores butyrate sensitivity. Microarray data on gene expression associated with butyrate sensitivity are presented and discussed.
Keywords: butyrate, resistance, colorectal cancer, butyrate, CBP, p300.
Introduction
Dietary fiber intake is linked to a reduced risk of colorectal cancer (CRC) [1], and this protective effect is likely mediated by the fermentation product of fiber in the colon, butyrate [2 and refs. therein]. Butyrate is a short-chain fatty acid and histone deacetylase inhibitor (HDACi). The agent induces apoptosis and represses clonal growth of CRC cells, dependent upon its ability to hyperactivate Wnt signaling [3]. However, resistance to the chemopreventive action of butyrate derived from a high-fiber diet may contribute to CRC [1]. To understand the mechanisms of resistance, we have derived a butyrate resistant cell line (HCT-R) from butyrate-sensitive HCT-116 CRC cells. HCT-R cells are not only resistant to physiological levels of butyrate, but also exhibit cross-resistance to clinically relevant HDACis [3, 4].
The hyperactivation of Wnt signaling by butyrate is influenced by CREB binding protein (CBP) and p300, histone acetylases that associate with beta-catenin, a major component of Wnt signaling transcriptional complexes [8]. Modulation of CBP- or p300-mediated Wnt signaling affects the ability of butyrate to induce Wnt activity and apoptosis [5-7]. Since we have observed that HCT-R cells are deficient in p300 expression, we hypothesized that one mechanism by which HCT-R cells develop resistance to butyrate is through downregulation of p300 expression.
To ascertain the role of p300 expression in butyrate resistance, we analyzed two p300 knockout lines derived from HCT-116 cells. In this short research communication, we demonstrate that p300 knockout lines exhibit significant butyrate resistance. Microarray analysis revealed a number of gene targets that may mediate the effect of p300 deficiency on butyrate resistance. Our hypothesis was further corroborated by findings that the re-introduction of p300 expression in knockout cells restores butyrate sensitivity.
Materials and Methods
Cells, plasmids, transfection, luciferase assay
Cell lines [9] were kindly provided by Dr. C. Caldas via Cancer Research UK. Wnt reporters pTOPFLASH and pFOPFLASH were obtained from Dr. H. Clevers; pRLTK from Promega. Transfections and luciferase assays were performed as previously described [3-7]. Butyrate treatment (5 mM) was 17.5 hr for Wnt activity reporter analyses, and 24 hr for other experiments.
Reagents
Crystal violet was from Fisher. Butyrate was obtained from Sigma, dissolved in purified water, filter sterilized, and used at 5 mM concentration.
Apoptosis and proliferation
Apoptotic analyses were performed using the caspase 3/7 GLO kit (Promega), as previously described [5-7]. Cell proliferation assays were performed with the QuickCell Proliferation Kit (Biovision) as previously described [5-7].
Clonogenics
100 cells/well were plated in 6 well dishes, mock or butyrate (5 mM) treated for 24 hr, and then incubated in media without butyrate for 14 days until colonies form. Cell colonies were stained with crystal violent and counted. Percent suppressed clonal growth was calculated.
Western blotting
Western blotting was performed as previously described [3]; band images were adjusted for brightness and contrast to enhance visualization.
Microarray
Cell treatment, RNA isolation, and full human genome microarray analysis (Genus Biosystems) was performed as previously described [4,7,10].
Statistics
Student's T-test was utilized, with statistical significance set at P < 0.05.
Results
HCT-116, D10, and F5 data and microarray
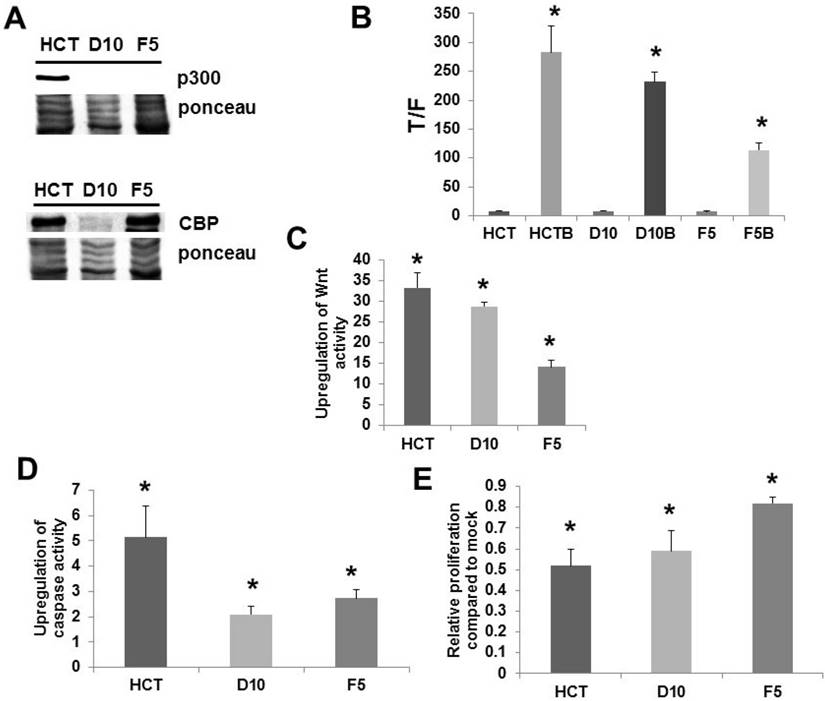
We confirmed (Fig. 1A) the lack of p300 expression in the knockout lines D10 and F5 [9] at the protein level; in addition, we ascertained that the cells expressed CBP (Fig. 1A). All cell lines exhibited significant upregulation of Wnt activity by butyrate (Fig. 1B). However, we have previously reported that it is the fold-induction of Wnt activity, and not the final levels of induced Wnt signaling, that determines the effects of butyrate in CRC cells [2-7]. F5 cells exhibited decreased fold-upregulation of Wnt activity compared to HCT-116 cells, and the greater than two-fold difference in Wnt signaling upregulation between these lines was highly significant (P < 0.001) (Fig. 1C). D10 cells exhibited fold-upregulation of Wnt activity that was statistically equivalent to HCT-116 cells (P < 0.001) (Fig. 1C). All cell lines exhibited increased caspase activity upon exposure to butyrate (Fig. 1D). However, induction of apoptosis by butyrate was markedly inhibited in the two knockout lines; the fold-increase was more than two-fold lower in D10 knockout cells compared to wild-type HCT-116 cells (P < 0.005), while the fold-increase in F5 cells was close to two-fold lower (P < 0.02). Whereas all lines exhibited reduced proliferation upon butyrate treatment (Fig. 1E), F5 cells exhibited a significantly greater degree of proliferation when exposed to butyrate compared to HCT-116 cells (P < 0.002). In contrast, HCT-116 and D10 cells exhibited statistically equivalent proliferation with butyrate treatment. A pilot microarray analysis was conducted on the cell lines (Fig. S1); discussion of these data are included in Supplementary Information, Part I.
Analysis of p300 knockout cell lines. (A) Western blot of total (100 μg) protein from wild-type p300 HCT-116 (HCT) and p300 knockout (D10 and F5) CRC cells probed with an anti-p300 polyclonal antibody (Santa Cruz) (top) and anti-CBP polyclonal antibody (Santa Cruz). A representative experiment is shown. (B) Wnt transcriptional activity in the cell lines. Statistical analysis was performed on normalized TOPFLASH (T)/FOPFLASH (F) ratios; data are from six separate experiments for HCT-116 and three separate experiments for D10 and F5 cells. (C) Fold-induction of Wnt activity by butyrate based on the data from (B). (D) Knockout of p300 inhibits butyrate-induced apoptosis. The fold-induction of apoptosis after treatment with butyrate is shown. Data are from six separate experiments for HCT-116 and three separate experiments for D10 and F5. (E) Knockout of p300 inhibits the ability of butyrate to repress cell proliferation in F5 knockout cells. The relative change in proliferation (growth of butyrate-treated cells divided by that of mock-treated cells) is shown. Data are from six separate experiments for HCT-116 and three separate experiments for D10 and F5. Bars, SDs. * = statistical significance.
p300 rescue cells
From the two p300 knockout cell lines, F5 cells exhibited the greater degree of butyrate resistance (Fig. 1). Therefore, to further ascertain the role played by p300 in butyrate resistance, we utilized F5 cells that were rescued through stable transfection with a p300 expression vector [9] (Fig. 2a).
Analysis of p300 rescue vs. p300 knockout CRC cells. For A-E, experiments were performed as described for Fig. 1. (A) A representative Western blot is shown. (B) Wnt activity levels, data are from three separate experiments. (C) Fold-induction of Wnt activity by butyrate based on the data from (B). (D) p300 rescue cells do not exhibit increased fold-upregulation of apoptosis after butyrate treatment (top); however, these cells exhibit increased absolute levels of apoptosis after butyrate treatment (bottom). Data are from three separate experiments. (E) p300 rescue restores sensitivity to butyrate-induced repression of cell proliferation. Data are from four separate experiments. (F) Clonogenic analyses, performed as described in Methods. Data are from three separate experiments. Bars, SDs. * = statistical significance
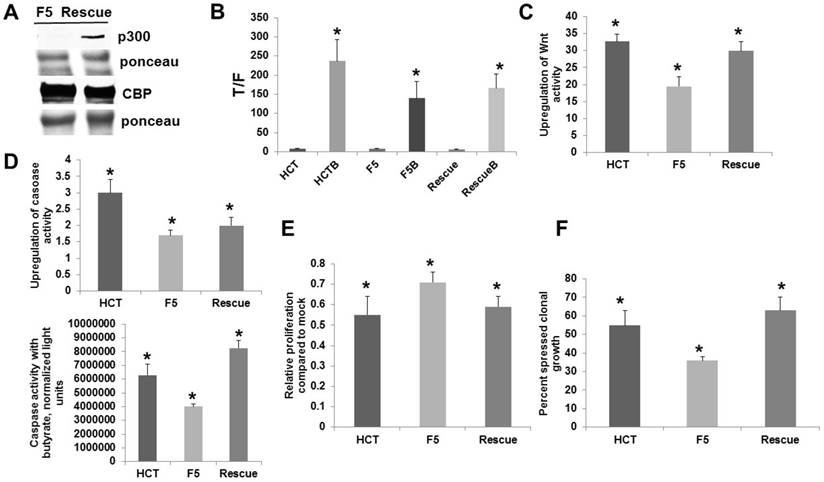
The p300 rescue cells were compared to the parental p300 knockout F5 and the wild-type HCT-116 cells with respect to Wnt signaling, apoptosis, and growth. All cells demonstrated significant enhancement of Wnt activity by butyrate (Fig. 2B). Importantly, p300 rescue cells exhibited significantly increased fold-upregulation of Wnt activity by butyrate compared to F5 cells (P < 0.01), and the fold-upregulation of Wnt signaling was statistically equivalent between the rescue cells and HCT-116 cells (Fig. 2C). All cells exhibited statistically significant enhancement of apoptosis after exposure to 5 mM butyrate (Fig. 2D, top). As expected, F5 cells underwent a lower fold-induction of apoptosis compared to HCT-116 cells (P < 0.01). The p300 rescue cells also exhibited lower fold-induction of apoptosis compared to wild-type HCT-116 cells (P < 0.03). However, rescue cells had high levels of basal caspase activity, and the increased apoptosis in the mock-treated state depressed the fold-induction of apoptosis by butyrate. In terms of final levels of caspase activity in the presence of butyrate (Fig. D, bottom), rescue cells exhibited the highest levels of butyrate-induced apoptosis, significantly higher than both HCT-116 cells (P < 0.03) and p300 knockout F5 cells (P < 0.001). Therefore, whereas rescue expression of p300 did not restore wild-type fold-induction of apoptosis, it resulted in a marked increase of apoptosis in butyrate-treated cells.
The ability of butyrate to repress cell proliferation was enhanced by the restored p300 expression (Fig. 2E). Butyrate repressed proliferation in all cell lines (P < 0.001). Importantly, proliferation in the presence of butyrate was statistically similar between HCT-116 and p300 rescue cells; whereas, butyrate-resistant p300 knockout F5 cells exhibited the greatest proliferation in presence of butyrate compared to HCT-116 (P < 0.03) and rescue (P < 0.05) cells.
To further evaluate the significance of these findings, we analyzed clonogenic growth, which is influenced by both proliferation and apoptosis. Treatment with butyrate resulted in a 55% reduction in colony formation in HCT-116 cells; whereas, F5 cells exhibited a 36% reduction in clonal growth. In contrast to parental F5 cells, p300 rescue cells were more sensitive to butyrate, exhibiting a 63% reduction in clonal growth (Fig. 2F). The differences in clonal growth for HCT-116 vs. F5 cells were statistically significant (P < 0.02), as were the differences in clonal growth between the F5 cells and the rescue cells (P < 0.005). The clonal growth of wild-type HCT-116 and p300 rescue cells did not differ in a statistically significant manner.
Discussion
p300 knockout D10 and, especially, F5 cells exhibit butyrate resistance without previous exposure to the short-chain fatty acid. Our findings indicate that the re-introduction of p300 expression into p300 knockout F5 cells restores butyrate sensitivity. However, additional factors must contribute to butyrate resistance [4], since D10 cells exhibit less butyrate resistance than F5 cells, and F5 cells exhibit less resistance than HCT-R cells [3, 4]; however, all three lines are deficient in p300 expression. Despite these differences in resistance between the cell lines, our findings indicate that the loss of p300 expression alone contributes to a significant degree of butyrate resistance, likely through decreased p300-mediated Wnt activity [6].
Butyrate resistance may contribute to CRC development, particularly in the proximal colon, where the levels of diet-derived butyrate are at their highest [11]. Furthermore, microsatellite instability (MSI) is more common in right-sided colon tumors, and MSI CRCs have a higher frequency of p300 mutation [12]; whereas, 5.4% of CRC patients have alterations in EP300 (p300) [13]. A fraction of CRCs also express p300 at relatively low levels [14], suggesting that epigenetic suppression of promoter activity [15] and/or loss of heterozygosity may contribute to downregulation of p300. Loss of heterozygosity across the EP300 locus has been reported for 38% of primary colon tumors [15].
Overexpression of nuclear p300 is associated with a more favorable prognosis (disease-free survival rate) specifically in colon, but not rectal, cancer [16]. This distinction is important since butyrate resistance is expected to occur in the proximal portions of the colon, where levels of dietary butyrate are at their highest [11]. If resistance occurs where colonic neoplasms are exposed to higher levels of dietary butyrate, and if resistance is associated with repressed expression of p300, it is expected that the colon, not the rectum, would exhibit reduced expression of p300 associated with worse prognosis [16].
In Supplementary Information (Part II), we discuss how these findings fit into our work on butyrate and Wnt signaling. Understanding the role of p300 in butyrate resistance may lead to better preventive and/or therapeutic approaches against CRC.
Abbreviations
EMT: epithelial-mesenchymal transition; CBP: CREB binding protein; CRC: colorectal cancer; HDACI: histone deacetylase inhibitor; MSI: microsatellite instability.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by funding from Geisinger Commonwealth School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bingham SA, Day NE, Luben R. et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition [EPIC]: an observational study. Lancet. 2003;361:1496-501
2. Bordonaro M, Lazarova D, Sartorelli AC. Hyperinduction of WNT signaling: a new paradigm for the treatment of colorectal cancer? Oncol Res. 2008;17:1-9
3. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by WNT signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66
4. Chiaro C, Lazarova DL, Bordonaro M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem Biophys Res Commun. 2012;428:121-6
5. Lazarova DL, Chiaro C, Wong T. et al. CBP activity mediates effects of the histone deacetylase inhibitor butyrate on Wnt activity and apoptosis in colon cancer cells. J. Cancer. 2013;4:481-90
6. Lazarova DL, Wong T, Chiaro C. et al. p300 influences butyrate-mediated WNT hyperactivation in colorectal cancer cells. J Cancer. 2013;4:491-501
7. Lazarova D, Lee A, Wong T. et al. Modulation of Wnt activity and cell physiology by butyrate in LT97 microadenoma cells. J. Cancer. 2014;5:203-13
8. Emami KH, Nguyen C, Ma H. et al. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc Natl Acad Sci USA. 2004;101:12682-7
9. Krubasik D, Iyer NG, English WR. et al. Absence of p300 induces cellular phenotype changes characteristic of epithelial to mesenchyme transition. Br J Cancer. 2006;94:1326-1332
10. Lazarova DL, Chiaro C, Bordonaro M. Butyrate induced changes in Wnt-signaling specific gene expression in colorectal cancer cells. BMC Research Notes. 2014;7:226
11. Lazarova DL, Bordonaro M. Extreme fluctuations in Wnt/beta-catenin signaling as an approach for colon cancer prevention and therapy. Advanced Studies in Biology. 2012;4:351-62
12. Ionov Y, Matsui S-I, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. PNAS. 2004;101:1273-1278
13. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483-8
14. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225-31
15. Bryan EJ, Jokubaitis VJ, Chamberlain NL. et al. Mutation analysis of EP300 in colon, breast and ovarian carcinomas. Int J Cancer. 2002;102:137-141
16. Huh JW, Kim HC, Kim SH. et al. Prognostic impact of p300 expression in patients with colorectal cancer. J Surg Oncol. 2013;108:374-7
Author contact
![]() Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509, Tel: 570-504-9646 Fax: 570-504-9636 Email: mbordonaroedu
Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509, Tel: 570-504-9646 Fax: 570-504-9636 Email: mbordonaroedu