Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(17):3430-3440. doi:10.7150/jca.21125 This issue Cite
Review
The Glycolytic Switch in Tumors: How Many Players Are Involved?
Li Yu1 ![]() *, Xun Chen2*, Xueqi Sun1, Liantang Wang 1, Shangwu Chen3
*, Xun Chen2*, Xueqi Sun1, Liantang Wang 1, Shangwu Chen3 ![]()
1. Department of Pathology, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People's Republic of China;
2. Guanghua School and Hospital of Stomatology, Sun Yat-sen University, Guangzhou 510055, People's Republic of China;
3. State Key Laboratory for Biocontrol, Guangdong Key Laboratory of Pharmaceutical Functional Genes, Key Laboratory of Gene Engineering of the Ministry of Education, Department of Biochemistry, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, People's Republic of China.
* The two authors contributed equally to this work.
Received 2017-5-22; Accepted 2017-8-31; Published 2017-9-20
Abstract
Reprogramming of cellular metabolism is a hallmark of cancers. Cancer cells more readily use glycolysis, an inefficient metabolic pathway for energy metabolism, even when sufficient oxygen is available. This reliance on aerobic glycolysis is called the Warburg effect, and promotes tumorigenesis and malignancy progression. The mechanisms of the glycolytic shift in tumors are not fully understood. Growing evidence demonstrates that many signal molecules, including oncogenes and tumor suppressors, are involved in the process, but how oncogenic signals attenuate mitochondrial function and promote the switch to glycolysis remains unclear. Here, we summarize the current information on several main mediators and discuss their possible mechanisms for triggering the Warburg effect.
Keywords: the Warburg effect, reprogramming of glucose metabolism, aerobic glycolysis, tumor metabolism, glycolytic switch.
Introduction
Reprogramming of glucose metabolism is a key event in tumorigenesis. Cancer cells undergo a metabolic switch from oxidative phosphorylation (OXPHOS) to glycolysis in which a molecule of glucose is degraded to two molecules of pyruvate (Fig 1). Depending on the supply of oxygen for the cells, pyruvate is either reduced to lactate in the absence of oxygen via an anaerobic glycolysis pathway, or oxidized to yield acetyl-coenzyme A in the presence of oxygen and then oxidized completely to CO2 and H2O via citric acid cycle. The majority of cancer cells depend on high rates of glycolysis for growth and survival, even when there is sufficient oxygen [1, 2]. This type of aerobic glycolysis is called the Warburg effect, and the mechanisms underlying this reprogramming are not fully understood. The Warburg effect has long been linked to hypoxia, but it is not solely adaptive to hypoxia, as it also occurs under normoxic conditions [1, 2]. Although mitochondrial dysfunction in cancer cells can cause a shift in energy metabolism, a majority of tumor cells demonstrate normal mitochondrial function and OXPHOS [3-5], and the high glycolytic flux in cancer cells does not mean impairment of OXPHOS [6]. The high rates of glycolysis provide advantages for the survival and growth of cancer cells [7]. Three possible explanations for tumor cell use of the glycolysis pathway, an inefficient metabolic pathway, have been proposed [8, 9]. First, compared to OXPHOS, the rate of ATP production through glycolysis is much more rapid [10]. Secondly, high glycolytic flux provides sufficient glycolytic intermediates to meet the biosynthesis needs of the rapidly proliferating cells [11-13]. Finally, NADPH, derived from the enhanced pentose phosphate pathway (PPP) due to the accumulation of glycolytic intermediates, enables cancer cells to maintain adequate levels of reduced forms of glutathione for resistance to chemotherapeutic agents.
Main steps in glycolysis and possible key enzymes regulated in the Warburg effect. The three reactions catalyzed by hexokinase (HK), phosphofructokinase-1 (PFK1), and pyruvate kinase (PK) in this process are rate-limiting steps. During glycolysis, four molecules of ATP are produced per molecule of oxidized glucose via substrate-level phosphorylation, and the net yield is two molecules of ATP after deduction of two ATPs consumed in phosphorylation. The fate of pyruvate depends largely on the availability of oxygen for the cells. Pyruvate is reduced to lactate under hypoxia via an anaerobic glycolysis pathway or, under aerobic conditions, oxidized to yield acetyl-coenzyme A, which is then oxidized completely to CO2 via the citric acid cycle, resulting in the production of large amounts of ATP. G-6-P, glucose-6-phosphate; G6DP, glucose-6-phosphate dehydrogenase; GLUT, glucose transporter; Fru-2,6-P2, fructose-2,6-bisphosphate; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; TPI, triose phosphate isomerase.
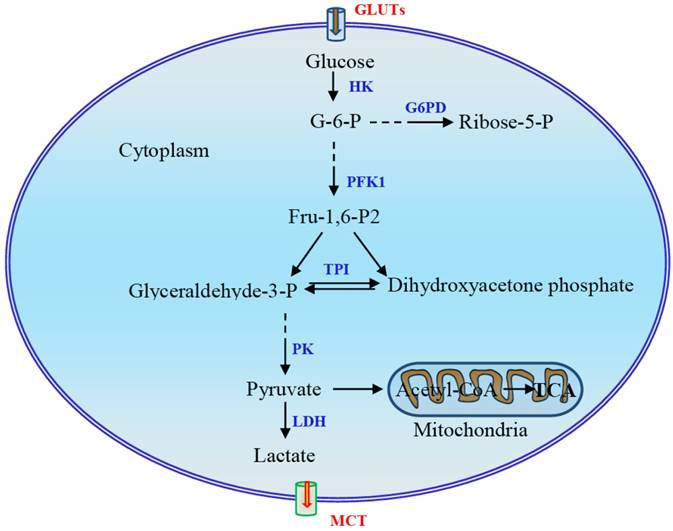
Several mechanisms have been proposed to enable cancer cells to maintain high glycolytic flux [11]. First, phosphofructokinase-1 (PFK1) serves as a critical driver of glycolytic flux. The expression of PFK2 is upregulated in cancer cells and promotes the production of fructose-2,6-bisphospate, which acts as a potent allosteric activator of PFK1 to overcome negative allosteric feedback inhibition of PFK1 by high ATP levels. Second, re-generation of NAD+ and lactate production mediated by lactate dehydrogenase (LDH) is instrumental in maintaining glycolysis. In addition, the expression of pyruvate kinase M2 (PKM2) is upregulated in cancer cells. Allosteric and covalent inhibition of PKM2 channels glycolytic intermediates upstream of pyruvate into biosynthetic pathways [11]. Although metabolic reprogramming has long been observed as a feature of neoplasia and tumor growth, the mechanism triggering and modulating this process remains largely unclear. In this review, we mainly focus on the mechanism underlying the regulation of glycolytic switch in tumors. In addition to signal molecules and transcription factors HIF-1α, c-Myc, Akt, and mTOR, the main regulators which have been well documented, several other regulators including oncogene K-Ras, tumor suppressor p53, energy sensor adenosine monophosphate activated protein kinase (AMPK), non-coding RNAs, and sirtuin family proteins and deacetylation will also be discussed.
Master regulator HIF-1α
Hypoxia-inducible factor-1 (HIF1) consists of two subunits, HIF-1α and HIF-1β, also known as ARNT. Under physiological oxygen levels, the HIF-1α subunit is sensitive to oxygen concentration and is hydroxylated by prolyl-hydroxylases (PHD) and targeted for proteasomal degradation. Increase of reactive oxygen species (ROS) under hypoxia inhibits PHD and stabilizes the HIF-1α subunit. HIF-1α is a master regulator of glycolysis and plays an important role as an activator of aerobic glycolysis and lactate production. It potentiates the transcription of glucose transporters (GLUT) and glycolytic enzymes including GLUT1, hexokinase II (HKII), pyruvate dehydrogenase kinase (PDK), and PKM2 [14-16]. The upregulation of HIF-1-mediated HKII results in a high glycolytic rate in hypoxic solid tumor [17]. Phosphorylation of pyruvate dehydrogenase leads to its inactivation and inhibits conversion of pyruvate into acetyl-CoA for the tricarboxylic acid (TCA) cycle [18, 19]. PKM2 is less catalytically active than is PKM1. A higher level of PKM2 in tumor cells, leading to accumulation of carbohydrate intermediates, facilitates the biosynthesis of macromolecules and tumor cell proliferation. HIF-1α drives expression of many glycolytic enzymes, and hypoxic glycolysis is, in turn, necessary for maintaining HIF-1α activity. This constitutes a novel feed-forward mechanism of glycolysis-HIF-1α signaling (Fig 2) [20].
HIF-1α is a master regulator of the Warburg effect and plays a critical role as an activator of aerobic glycolysis. Hypoxia increases the production of ROS, which stabilizes HIF-1α. HIF-1α induces expression of glucose transporters and glycolytic enzymes, facilitating glycolysis that is, in turn, essential for maintaining HIF-1α activity. Many oncogenes and tumor suppressors are involved in the regulation of HIF-1α. EZH2, enhancer of zeste 2 polycomb repressive complex 2; GRIM-19, gene associated with retinoid-interferon-induced mortality-19; LSD1, lysine specific demethylase 1; ROS, reactive oxygen species; RPS7, ribosomal protein S7; TKTL1, transkelolase-like 1; VEGF, vascular endothelial growth factor; VHL, von Hippel-Lindau; WWOX, WW domain-containing oxidoreductase.
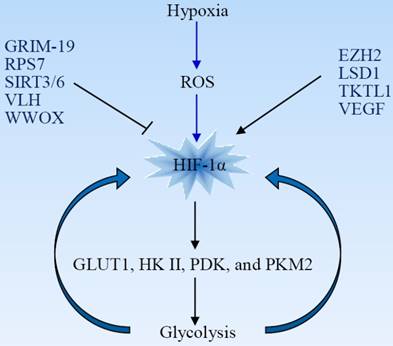
HIF-1α activity is tightly regulated by oncogenes and other factors. For example, the WW domain-containing oxidoreductase (WWOX), lacking in many cancer types, interacts with HIF-1α and modulates its levels and transactivation function. WWOX absence is associated with enhanced glycolysis, and WWOX-deficient cells are more tumorigenic [21]. Enhancer of zeste 2 polycomb repressive complex 2 (EZH2), a multifaceted oncogenic protein, promotes glioblastoma tumorigenesis and malignant progression through activation of HIF-1α and the Warburg effect. HIF-1α activation is necessary for EZH2-mediated metabolic adaption [22]. Ribosomal protein S7 (RPS7) inhibits glycolysis in colorectal cancer by suppressing the expression of HIF-1α as well as of GLUT4 and lactate dehydrogenase B (LDHB) [23]. Vascular endothelial growth factor (VEGF) enhances glycolysis in pancreatic cancer via upregulation of HIF-1α [24]. Histone demethylase JMJD1A facilitates glycolysis via coactivation of HIF-1α and promotes cancer progression [25].
HIF-1α activity can be regulated by modifying its stabilization. Transkelolase-like 1 (TKTL1) contributes to carcinogenesis through increased HIF-1α stabilization and the upregulation of downstream glycolytic enzymes and aerobic glycolysis [26]. The von Hippel-Lindau (VHL) gene is a tumor suppressor involved in the regulation of HIF-1α stability. VHL protein serves as an E3 ligase that ubiquitinates HIF-1α and results in its degradation by the proteasome. HIF-1α becomes constitutively activated in the absence of VHL [27]. The gene associated with retinoid-interferon-induced mortality-19 (GRIM-19), a potential tumor suppressor, promotes VHL-mediated HIF-1α ubiquitination and degradation in glioblastoma cells [28]. Lysine specific demethylase 1 (LSD1), a histone demethylase, prevents HIF-1α from subsequent acetylation-dependent degradation and maintains the HIF1α-dependent glycolytic process [29].
Akt and mTOR signaling
Akt is a serine/threonine kinase that promotes cancer growth and has been called 'Warburg kinase,' because it facilitates a glycolytic switch in tumor cells under normoxic conditions [30, 31]. Akt activation promotes the expression and activity of glucose transporters and glycolytic enzymes. The transcription of GLUT1, a widely expressed glucose transporter, is enhanced upon the activation of Akt [32, 33]. Lack of S-phase kinase-associated protein 2 (Skp2), an E3 ligase, impairs Akt activation, GLUT1 expression, glycolysis, and cancer progression [34]. Akt signaling induces the expression of HKII, a rate-controlling enzyme of glycolysis [33, 35]. Akt phosphorylates and activates PFK2 to produce fructose-2,6-bisphosphate, an allosteric activator of PFK1 [36]. Active Akt accumulates in the mitochondria during hypoxia and phosphorylates pyruvate dehydrogenase kinase 1 (PDK1) to inactivate the pyruvate dehydrogenase complex, switching tumor metabolism toward glycolysis [37]. Importantly, Akt-mediated aerobic glycolysis does not affect the rate of OXPHOS. Increased glycolytic flux is required for rapidly proliferating tumor cells to obtain essential metabolic intermediates. Akt-mediated enhanced aerobic glycolysis results in acquired radioresistance of tumor cells [38]. Constitutively active Akt leads to cell death in low-glucose conditions [31].
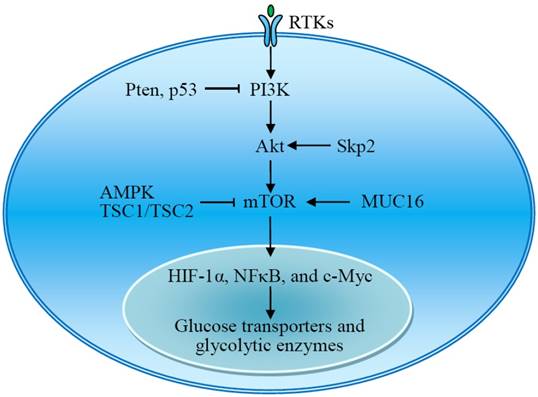
The mammalian target of rapamycin (mTOR) is also a serine/threonine kinase downstream of Akt and consists of two complexes, mTORC1 and mTORC2. mTOR acts as a central activator of the Warburg effect by inducing expression of glycolytic enzymes under normoxic conditions. mTOR-mediated upregulation of PKM2, a rate-limiting glycolytic enzyme expressed exclusively in tumor cells, is critical to aerobic glycolysis and tumor growth [39]. Tuberous sclerosis protein 1 and 2 complex (TSC1/TSC2) negatively regulates the expression of GLUT3 through the inactivation of mTORC1 signaling [40]. The transmembrane mucin MUC16 increases glycolysis through activation of mTOR. The mTOR-mediated expression of glycolytic proteins involves activation of HIF-1α, NFκB, and c-Myc [39-42]. Upon stimulation, the receptor tyrosine kinases (RTKs) activate membrane PI3K, which recruits and activates Akt. Thus, RTKs-PI3K-Akt-mTOR signaling plays a critical role in the regulation of aerobic glycolysis and tumor growth (Fig 3) [43-45].
The RTKs-PI3K-Akt-mTOR signal pathway plays an important role in the regulation of aerobic glycolysis. Akt and mTOR are central activators of the Warburg effect, promoting the expression of glucose transporters and glycolytic enzymes, which is regulated by many signal molecules. AMPK, adenosine monophosphate activated protein kinase; MUC16, mucin 16; mTOR, mammalian target of rapamycin; RTKs, receptor tyrosine kinases; Skp2, S-phase kinase-associated protein 2; TSC1/TSC2, tuberous sclerosis protein 1 and 2 complex.
Oncogenes and tumor suppressors
Oncogenic K-Ras promotes metabolic reprogramming in tumors [46, 47]. Mutated K-Ras has been found to upregulate the expression of the GLUT1 and facilitate cell survival in low-glucose culture conditions via increased glucose uptake and glycolysis [48]. Thus, K-Ras mutated tumor cells are highly vulnerable to the glycolytic inhibitor [48]. The small guanosine triphosphatase (GTPase) ADP-ribosylation factor 6 (ARF6) is a target of mutanted K-Ras and promotes the Warburg effect and pancreatic cancer growth [49]. The K-Ras G12D mutation stimulates glucose uptake and drives glycolytic intermediates into the nonoxidative PPP [50]. K-Ras (G12V) activation leads to mitochondrial dysfunction, promoting a metabolic switch from OXPHOS to glycolysis and enhancing the tumorigenicity of the transformed cells [51]. The K-Ras G13D mutation is associated with increased expression of glycolytic proteins in colorectal cancer [52].
Tumor suppressor p53 negatively regulates cellular glycolysis, contributing to tumor metabolic reprogramming via promotion of mitochondrial OXPHOS and suppression of glycolysis via several routes [53, 54]. It downregulates the expression of glucose transporters GLUT1, GLUT3, and GLUT4 [55, 56] and promotes the ubiquitination-mediated degradation of phosphoglycerate mutase (PGM) [57]. p53 also directly inhibits glucose-6-phosphate dehydrogenase (G6PD), the first and rate-limiting enzyme in the PPP [58]. Inactivation of p53 and the resultant enhanced PPP glucose flux may increase glucose consumption and channel glucose to biosynthesis in tumor cells. In addition, p53 may inhibit glycolysis through its target genes [59-61]. For example, p53 induces Ras-related associated with diabetes (RRAD), which in turn inhibits the translocation of GLUT1 and glycolysis in lung cancer cells [59]. p53 downregulates glycolysis by transcribing TP53-induced glycolysis and apoptosis regulator (TIGAR) [60, 62]. TIGAR degrades fructose-2,6-bisphosphate (Fru-2,6-P2) to fructose-6-phosphate and causes a significant reduction in cellular Fru-2,6-P2 levels. Fru-2,6-P2 serves as an allosteric activator of PFK1 and promotes the production of fructose-1,6-bisphosphate in glycolysis. p53 also negatively regulates the PI3K-Akt-mTOR pathway through its target genes. p53 activates adenosine monophosphate activated protein kinase (AMPK), a major upstream negative regulator of mTOR, and induces Pten and TSC2 to negatively regulate PI3K-Akt signaling and mTOR activity [63, 64]. HKII-mediated aerobic glycolysis is required for Pten-/p53-deficiency-driven tumor growth in xenograft mouse models of prostate cancer [65]. Pten deletion promotes HKII mRNA translation via the activation of the Akt-mTORC1-4EBP1 axis [65]. Absence of p53 enhances HKII mRNA stability through the inhibition of miR143 biogenesis [65].
Given the high mutation rate of p53 in human tumors, the loss of p53 function could be an important factor contributing to the Warburg effect. It has been determined that tumor-associated mutant p53 (mutp53) drives the Warburg effect under normoxia, and inhibition of glycolysis impairs mutp53-promoting tumorigenesis [66]. Mutant R175H and R273H p53 proteins trigger PKM2 phosphorylation via mTOR signaling [67]. CD147 promotes reprogramming of glucose metabolism by inhibiting the p53-dependent signaling pathway [68].
Upregulation of glucokinase, PK, and PFK2 levels was observed in the liver of c-Myc transgenic mice about two decades ago, suggesting that transcription factor c-Myc is a regulator of glycolytic enzymes [69]. c-Myc promotes glucose uptake via the upregulation of GLUT1 [14, 70] and potentiates transcription of glycolytic enzymes HKII, PFK [14], and lactate dehydrogenase A (LDHA) [14, 71, 72]. c-Myc upregulates the expression of monocarboxylate transporter (MCT) through direct transcriptional activation or by suppressing transcription of miR-29a and miR-29c [73]. c-Myc promotes transcription of polypyrimidine tract binding protein (PTB), which binds to PKM pre-mRNA and switches PKM splicing to favor the PKM2 variant, ensuring a high PKM2/PKM1 ratio [74, 75]. Inhibition of c-Myc in tumor cells blunts hypoxia-dependent glycolytic reprogramming and is a potential strategy for tumor therapy [76, 77].
Some molecules promote glycolysis via regulation of c-Myc activity. Proto-oncogene human pituitary tumor-transforming gene (PTTG) regulates GLUT1 and several glycolytic enzymes via the c-Myc pathway [78]. N-Myc downstream-regulated gene (NDRG) family members can manipulate Myc-mediated tumor metabolic pathways and ultimately modify the Warburg effect [79]. NDRG2, a tumor suppressor, acts as a critical regulator of glycolysis via repression of c-Myc through downregulation of c-Myc transcriptional activator β-catenin, consequently suppressing the expression of GLUT1, HKII, PKM2, and LDHA in colorectal cancer cells [80]. Inhibitor of differentiation 1 (Id1), a transcription factor, promotes a metabolic shift to aerobic glycolysis in hepatocellular carcinoma cells by regulating the expression levels of c-Myc [81]. lncRNA-MIF, a c-Myc-activated long non-coding RNA, inhibits aerobic glycolysis by promoting c-Myc degradation. lncRNA-MIF acts as a molecular sponge for miR-586, competing with Fbxw7 mRNA for miR‐586. Fbxw7 serves as an E3 ligase for c-Myc that promotes c-Myc degradation [82].
Energy sensor
Cellular energy metabolism is strictly regulated. Adenosine monophosphate activated protein kinase (AMPK) is a metabolic sensor that helps maintain cellular energy homeostasis [83]. Increases in AMP:ATP and ADP:ATP ratios activate AMPK, potentiating the metabolic process from an anabolic condition to a catabolic state by switching off the synthesis of lipids, carbohydrates, ribosomal RNA, and proteins [84, 85]. This leads to downregulation of glycolytic enzymes and glucose transporters. Thus, AMPK negatively regulates aerobic glycolysis in tumor cells and suppresses tumor growth in vivo [86]. Inactivation of AMPK promotes a metabolic shift to aerobic glycolysis, which requires normoxic stabilization of HIF-1α [86].
AMPK is involved in the regulation of glycolysis in many tumors, but its underlying mechanism remains unclear. The role of AMPK in glycolytic shift is also controversial. Several studies report a glycolysis-promoting effect of AMPK. For example, AMPK supports the growth of aggressive experimental tumors in part through positive regulation of glycolysis [87]. Manganese superoxide dismutase (MnSOD/SOD2) upregulation in cancer cells increases mitochondrial ROS that sustains AMPK activation and the metabolic shift to glycolysis [88]. Astrocyte elevated gene-1 (AEG-1) mediates glycolysis and tumorigenesis in colorectal carcinoma cells via AMPK signaling [89]. Prostate cancer cell growth mediated by androgen receptor signaling is involved in an AMPK-mediated metabolic switch to glycolysis [90]. miR-101-3p targets AMPK in triple negative breast cancer to regulate glycolysis [91]. AMPK is also essential to balance glycolysis and mitochondrial metabolism in acute T cell lymphoblastic leukemia [92].
Non-coding RNAs
microRNAs (miRNAs) are involved in the genesis of various cancers and may inhibit aerobic glycolysis via regulation of glucose uptake and glycolytic enzymes (Fig 4) [93]. miR-22, miR-144, and miR-1291 directly target glucose transporter GLUT1 in breast [94], ovarian [95], and renal cancer cells [96], respectively, while miR-195-5p targets GLUT3 in bladder cancer cells [97]. Thus, downregulation of these miRNAs in tumors stimulates aerobic glycolysis. HKII, a key mediator of glycolysis, is another main target of miRNAs. miR-143 directly inhibits the expression of HKII and regulates cancer glycolysis [98-102]. The miR-143 level inversely correlates with HKII protein expression in several cancers, including head and neck squamous cell carcinoma (HNSCC) [100], breast cancer [102], glioma [99], and lung cancer [98]. The absence of miR-143-mediated repression of HKII may contribute to the shift toward aerobic glycolysis in tumors [101] and enhance stemness of glioblastoma stem-like cells [99]. miR-143 can be downregulated by mTOR activation [98] or by miR-155 [102], which also stimulates HKII transcription via activating the signal transducer and activator of transcription 3 (STAT3). In addition to miR-143, miR-98 and miR-199a-5p directly targets HKII [103, 104], and miR-29b downregulates HKII/PKM2 through directly targeting Akt [105]. Expression of these miRNAs is downregulated in several cancers. miR-378* induces glycolytic shift in breast cancer cells via the PGC-1β/ERRγ transcription pathway [106].
Non-coding RNAs target glucose transporters and glycolytic enzymes. The downregulation of several miRNAs in some tumors facilitates aerobic glycolysis and promotes the development and progression of the tumors. HIF-1α is a primary target of non-coding RNAs. miRNA absence, or lncRNA-mediated HIF-1α stabilization, enhances HIF-1α activity, contributing to the Warburg effect. PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase.
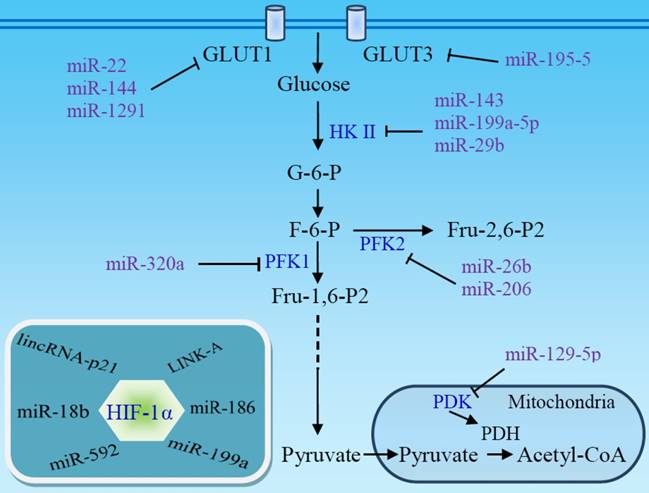
In addition to HKII, other glycolytic enzymes and signal molecules are miRNA targets. miR-320a regulates PFK1 expression and, consequently, its lactate production [107]. miR-26b and miR-206 downregulate PFK2-driven glycolysis [108, 109]. A set of miRNAs targets LDHA and regulates glycolysis in colorectal cancer [110]. miR-129-5p blocks glycolysis to retard hepatocarcinogenesis via targeting mitochondrial pyruvate dehydrogenase kinase 4 (PDK4) [111]. miR-448 promotes glycolytic metabolism in gastric cancer by downregulating KDM2B, a reader for methylated CpGs [112]. miR-21 acts as a molecular switch to regulate aerobic glycolysis in bladder cancer cells [113].
Along with targeting glucose transporters and glycolytic enzymes, many miRNAs exert their functions by targeting HIF-1α, a master regulator of glycolysis (Fig 4). It has been reported that miR-18b [114], miR-186 [115], miR-199a [116, 117], and miR-592 [118] inhibit aerobic glycolysis through directly targeting HIF-1α in several cancer types. Low expression of these miRNAs facilitates aerobic glycolysis and promotes the development and progression of the tumors. The upregulation of HIF-1α under hypoxic conditions, in turn, suppresses miRNA expression and promotes glycolysis [103, 117]. miR-150 targets VHL, a specific E3 ligase for HIF-1α, and promotes the Warburg effect in glioma [119].
Long non-coding RNA (lncRNA) is also an important player in the regulation of the Warburg effect [120, 121]. lncRNA-p21 is hypoxia-responsive and is essential for hypoxia-enhanced glycolysis. It binds to HIF-1α and VHL, disrupting VHL-HIF-1α interaction and VHL-mediated HIF-1α ubiquitination, resulting in HIF-1α accumulation [120]. Long intergenic non-coding RNA for kinase activation (LINK-A), a cytoplasmic lncRNA, mediates BRK-dependent HIF-1α phosphorylation, leading to HIF-1α stabilization under normoxic conditions [121]. LINK-A-dependent normoxic HIF-1α signaling promotes breast cancer glycolysis reprogramming and tumorigenesis [121].
Sirtuin family proteins and deacetylation
Sirtuins are a highly conserved family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases that regulate a large number of cellular processes [122]. Growing evidence demonstrates that sirtuins are involved in the regulation of cancer metabolism [123, 124]. Of the seven mammalian sirtuins (SIRT1-7), SIRT1, SIRT3, and SIRT6 have been implicated in the regulation of glucose utilization [125, 126].
The histone deacetylase SIRT6 has been identified as a tumor suppressor that regulates aerobic glycolysis in cancer cells. Deficiency of SIRT6 in mice results in severe hypoglycemia [127]. SIRT6 acts as a histone H3K9 deacetylase to function as a co-repressor of HIF-1α and Myc and control the expression of multiple glycolytic genes [128, 129]. HIF-1α activity and glycolysis are increased in SIRT6-deficient cells [128, 129]. Lack of SIRT6 can lead to tumor formation even without activation of known oncogenes [129].
SIRT3 is the major deacetylase within the mitochondrial matrix and works as a tumor suppressor by inhibiting the Warburg effect [130, 131]. SIRT3 regulates the stability of HIF-1α via lowering cellular ROS levels [130, 131]. Absence of SIRT3 increases cellular ROS, leading to stabilization of HIF-1α and metabolic reprogramming [131, 132]. In contrast, SIRT3 overexpression represses glycolysis and proliferation in breast cancer cells [131]. The SIRT3-mediated alterations in ROS are attributed to deacetylation and activation of isocitrate dehydrogenase 2 (IDH2) and superoxide dismutase 2 (SOD2) [133]. In addition, SIRT3 deacetylates glutamate oxaloacetate transaminase 2 (GOT2) to inhibit its binding to malate dehydrogenase 2 (MDH2), consequently preventing the malate-aspartate shuttle in the mitochondrial intermembrane space [134]. The malate shuttle is able to restore cytosolic NAD+, which is essential for a high rate of glycolysis. SIRT3 also deacetylates and activates pyruvate dehydrogenase A1 (PDHA1) and PDH phosphatase 1 (PDP1) of the PDH complex (PDC), promoting the conversion of pyruvate to acetyl-CoA for OXPHOS [135, 136]
It has been reported that SIRT1 stimulates the expression of glycolysis genes and the tumor cell proliferation in pancreatic neoplastic lesions [137]. A SIRT1-mTOR/HIF-1α glycolytic pathway is required for differentiation of myeloid-derived suppressor cells into the M1 phenotype [138].
Other regulators
Although the roles of the several mentioned master controllers are critical to the Warburg effect, other regulators are also involved in the glycolytic shift in cancer cells. Wnt signaling-mediated PDK1 expression promotes glycolysis and tumor growth [139]. CUE domain-containing protein 2 (CUEDC2) facilitates aerobic glycolysis and tumorigenesis via upregulating the GLUT3 and LDHA [140]. Pro-inflammatory cytokine interleukin-22 facilitates aerobic glycolysis in colon cancer cells via c-Myc and STAT3-mediated up-regulation of HKII [141]. Carboxyl terminus of Hsc70-interacting protein (CHIP), an E3 ligase, inhibits aerobic glycolysis progression of ovarian carcinomas through CHIP-mediated PKM2 degradation [142]. iNOS/NO promotes glycolysis via inducing PKM2 nuclear translocation [143]. Mitochondrial calcium uptake 1 (MICU1) increases aerobic glycolysis and chemoresistance in ovarian cancer [144]. Epidermal growth factor (EGF) promotes aerobic glycolysis, inducing epithelial-mesenchymal transition (EMT) and cancer stem-like cell properties in human oral carcinoma cells [145]. Toll-like receptor 3 signaling [146] and serotonin signaling [147] also trigger metabolic reprogramming of cancer cells. Molecular chaperone TNF receptor-associated protein 1 (TRAP1) [148], focal adhesion kinase (FAK) [149], plasma membrane-associated protein Caveolin 1 [150-152], α/β-hydrolase domain-containing 5 (Abhd5) [153], Krüppel-like factor 4 (KLF4) [154, 155], Ecdysoneless [156], and Jumonji C domain-containing dioxygenase (JMJD5) [157] are associated with the glycolytic switch in tumors. Some viruses or virus-encoded proteins can induce aerobic glycolysis in tumors [158-160].
Conclusions
The triggering of the Warburg effect is a complex process with the involvement of multiple regulators (Table 1) [161]. HIF-1α is a master activator. In tumorigenesis, overproduced or mutated growth factors activate transcription factors HIF-1α, NFκB, and c-Myc via the RTKs-PI3K-Akt-mTOR pathway, leading to the expression of glucose transporters and glycolytic enzymes. Oncogene activation and tumor suppressor inactivation during carcinogenesis modify the key signal molecules of the PI3K-Akt-mTOR pathway and downstream HIF-1α activity, promoting glycolytic flux and tumor development. Oncoproteins may also activate sirtuins, a protein deacetylase family, directly suppressing the transcription of glycolytic enzymes or inhibiting HIF-1α and c-Myc expression. Hypoxia and the ROS accumulation and energy depletion resulting from rapid tumor growth further stimulate HIF-1α activity or regulate the production of glycolytic enzymes and glucose transporter through energy sensor AMPK. Absence of miRNAs or lncRNA dysfunction during carcinogenesis promotes aerobic glycolysis via targeting glycolytic enzymes or regulating HIF-1α. A crucial question is whether the Warburg effect is the cause or the effect of cancer. There is no doubt that aerobic glycolysis is a hallmark of tumor metabolism, and is essential to tumor survival and growth. An important focus of study is the stage in tumorigenesis at which reprogramming of glucose metabolism is initiated. Research has revealed that the expression of glycolytic enzymes is modified in the precancerous stage of some tumors [162, 163]. The imaging data also indicated that elevated glycolysis may occur at early-stages of neoplasia and critically contribute to cancer initiation [164, 165]. It has been reported that 14-3-3ζ-mediated upregulation of LDHA in early stage precancerous breast epithelial cells promotes glycolysis, contributing to breast cancer initiation [166]. We have found enhanced expression of several enzymes involved in glycolysis in high grade cervical intraepithelial neoplasia, a typical precancerous lesion of the cervix (Yu et al., unpublished data). This implies that the reprogramming of glucose metabolism occurs at an early stage of carcinogenesis. Additional studies are needed to shed light on this topic.
The major players in the glycolytic switch and their main features
| Regulators | Downstream molecules | Effects | References |
|---|---|---|---|
| Akt | GLUT1, HK II, PDK1, PFK2 | + | [32, 33, 35-37] |
| AMPK | HIF-1α | +, - | [86] |
| c-Myc | Glucokinase, GLUT1, HKII, LDHA, MCTs, PFK, PK, PKM2 | + | [14, 69-75] |
| HIF-1α | GLUT1, HK II, PDK, PKM2 | + | [14-17] |
| K-Ras | GLUT1 | + | [48] |
| lncRNA | HIF-1α, VHL | + | [120, 121] |
| miRNAs | Akt, GLUT1, GLUT3, HIF-1α, HKII, LDHA, PDK4, PFK, PFKFB3, PKM2 | +, - | [94-105, 107-111] |
| mTOR | GLUT3, HIF-1α, c-Myc, NFκB, PKM2 | + | [39-42] |
| p53 | AMPK, GLUT1, GLUT3, GLUT4, G6PD, miR143, PGM, Pten, RRAD, TIGAR, TSC2 | - | [55-60, 62-65] |
| SIRT1, SIRT3, SIRT6 | HIF-1α, Myc, PDHA1, PDP1 | -, + | [128-132, 135-137] |
Increased glycolysis in tumor cells provides a potential target for tumor therapy. Actually, disrupting glycolysis does interfere with tumor growth [167, 168]. Glucose transporters, monocarboxylate transporters, and critical glycolytic enzymes such as HK II, LDHA, PFK, and PKM2 have been proposed as potential targets. Several small molecules including lonidamine, 2-deoxyglucose (2-DG), dichloroacetate, and 3-bromopyruvate (3-BP) have been clinically tested, but many candidates are still under experimental studies [161].
Acknowledgements
This study was funded by the National Natural Science Foundation of China (No. 31670788 and No. 81172485), the Ph.D. Program Foundation of Ministry of Education of China (No. 20130171110007), and Open Fund of Guangdong Key Laboratory of Pharmaceutical Functional Genes (No. 2014B030301028).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nolop KB, Rhodes CG, Brudin LH, Beaney RP, Krausz T, Jones T. et al. Glucose utilization in vivo by human pulmonary neoplasms. Cancer. 1987;60:2682-9
2. Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res. 2004;10:6661-8
3. Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002;364:309-15
4. Martin M, Beauvoit B, Voisin PJ, Canioni P, Guerin B, Rigoulet M. Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. J Bioenerg Biomembr. 1998;30:565-78
5. Pasdois P, Deveaud C, Voisin P, Bouchaud V, Rigoulet M, Beauvoit B. Contribution of the phosphorylable complex I in the growth phase-dependent respiration of C6 glioma cells in vitro. J Bioenerg Biomembr. 2003;35:439-50
6. Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393-418
7. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R. et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51
8. de Souza AC, Justo GZ, de Araujo DR, Cavagis AD. Defining the molecular basis of tumor metabolism: a continuing challenge since Warburg's discovery. Cell Physiol Biochem. 2011;28:771-92
9. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152
10. Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol. 2010;8:88
11. Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. J Exp Med. 2012;209:211-5
12. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64
13. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54-61
14. Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51-6
15. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705-13
16. Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P. et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035-47
17. Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996;56:2468-71
18. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177-85
19. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187-97
20. Grandjean G, de Jong PR, James BP, Koh MY, Lemos R, Kingston J. et al. Definition of a Novel Feed-Forward Mechanism for Glycolysis-HIF1alpha Signaling in Hypoxic Tumors Highlights Aldolase A as a Therapeutic Target. Cancer Res. 2016;76:4259-69
21. Abu-Remaileh M, Aqeilan RI. Tumor suppressor WWOX regulates glucose metabolism via HIF1alpha modulation. Cell Death Differ. 2014;21:1805-14
22. Pang B, Zheng XR, Tian JX, Gao TH, Gu GY, Zhang R. et al. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1alpha signaling. Oncotarget. 2016;7:45134-43
23. Zhang W, Tong D, Liu F, Li D, Li J, Cheng X. et al. RPS7 inhibits colorectal cancer growth via decreasing HIF-1alpha-mediated glycolysis. Oncotarget. 2016;7:5800-14
24. Shi S, Xu J, Zhang B, Ji S, Xu W, Liu J. et al. VEGF Promotes Glycolysis in Pancreatic Cancer via HIF1alpha Up-Regulation. Curr Mol Med. 2016;16:394-403
25. Wan W, Peng K, Li M, Qin L, Tong Z, Yan J. et al. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1alpha. Oncogene. 2017;36:3868-77
26. Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S. et al. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res. 2010;16:857-66
27. Schonenberger D, Harlander S, Rajski M, Jacobs RA, Lundby AK, Adlesic M. et al. Formation of Renal Cysts and Tumors in Vhl/Trp53-Deficient Mice Requires HIF1alpha and HIF2alpha. Cancer Res. 2016;76:2025-36
28. Liu Q, Wang L, Wang Z, Yang Y, Tian J, Liu G. et al. GRIM-19 opposes reprogramming of glioblastoma cell metabolism via HIF1alpha destabilization. Carcinogenesis. 2013;34:1728-36
29. Qin Y, Zhu W, Xu W, Zhang B, Shi S, Ji S. et al. LSD1 sustains pancreatic cancer growth via maintaining HIF1alpha-dependent glycolytic process. Cancer Lett. 2014;347:225-32
30. Robey RB, Hay N. Is Akt the "Warburg kinase"?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25-31
31. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR. et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892-9
32. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr. et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281-6
33. Li W, Peng C, Lee MH, Lim D, Zhu F, Fu Y. et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73:6938-50
34. Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z. et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098-111
35. Zhuo B, Li Y, Li Z, Qin H, Sun Q, Zhang F. et al. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem Biophys Res Commun. 2015;464:401-6
36. Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272:17269-75
37. Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV. et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell. 2016;30:257-72
38. Shimura T, Noma N, Sano Y, Ochiai Y, Oikawa T, Fukumoto M. et al. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother Oncol. 2014;112:302-7
39. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X. et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129-34
40. Zha X, Hu Z, Ji S, Jin F, Jiang K, Li C. et al. NFkappaB up-regulation of glucose transporter 3 is essential for hyperactive mammalian target of rapamycin-induced aerobic glycolysis and tumor growth. Cancer Lett. 2015;359:97-106
41. Shukla SK, Gunda V, Abrego J, Haridas D, Mishra A, Souchek J. et al. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget. 2015;6:19118-31
42. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T. et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726-39
43. Ran C, Liu H, Hitoshi Y, Israel MA. Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res. 2013;73:1831-43
44. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G. et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J Biol Chem. 2015;290:17495-504
45. Salani B, Ravera S, Amaro A, Salis A, Passalacqua M, Millo E. et al. IGF1 regulates PKM2 function through Akt phosphorylation. Cell Cycle. 2015;14:1559-67
46. Ji H, Lee JH, Wang Y, Pang Y, Zhang T, Xia Y. et al. EGFR phosphorylates FAM129B to promote Ras activation. Proc Natl Acad Sci U S A. 2016;113:644-9
47. Lee JH, Ji H, Lu Z. FAM129B activates Ras and promotes aerobic glycolysis. Cell Cycle. 2016;15:1391-2
48. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H. et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555-9
49. Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W. et al. ARF6, induced by mutant Kras, promotes proliferation and Warburg effect in pancreatic cancer. Cancer Lett. 2017;388:303-11
50. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70
51. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y. et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399-412
52. Hutton JE, Wang X, Zimmerman LJ, Slebos RJ, Trenary IA, Young JD. et al. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol Cell Proteomics. 2016;15:2924-38
53. Liang Y, Liu J, Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013;3:9
54. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O. et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650-3
55. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627-33
56. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611-8
57. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G. et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177-85
58. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M. et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310-6
59. Zhang C, Liu J, Wu R, Liang Y, Lin M, Chan CS. et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5:5535-46
60. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R. et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107-20
61. Liu S, Yan B, Lai W, Chen L, Xiao D, Xi S. et al. As a novel p53 direct target, bidirectional gene HspB2/alphaB-crystallin regulates the ROS level and Warburg effect. Biochim Biophys Acta. 2014;1839:592-603
62. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948-57
63. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S. et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043-53
64. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451-60
65. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L. et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014;8:1461-74
66. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X. et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935
67. Dando I, Cordani M, Donadelli M. Mutant p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life. 2016;68:722-6
68. Huang Q, Li J, Xing J, Li W, Li H, Ke X. et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014;61:859-66
69. Valera A, Pujol A, Gregori X, Riu E, Visa J, Bosch F. Evidence from transgenic mice that myc regulates hepatic glycolysis. FASEB J. 1995;9:1067-78
70. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M. et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797-800
71. Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA. et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658-63
72. He TL, Zhang YJ, Jiang H, Li XH, Zhu H, Zheng KL. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med Oncol. 2015;32:187
73. Gan L, Xiu R, Ren P, Yue M, Su H, Guo G. et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene. 2016;35:3037-48
74. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364-8
75. Luan W, Wang Y, Chen X, Shi Y, Wang J, Zhang J. et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015;6:13006-18
76. Mongiardi MP, Savino M, Falchetti ML, Illi B, Bozzo F, Valle C. et al. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget. 2016;7:33257-71
77. Tateishi K, Iafrate AJ, Ho Q, Curry WT, Batchelor TT, Flaherty KT. et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin Cancer Res. 2016;22:4452-65
78. Wang X, Duan W, Li X, Liu J, Li D, Ye L. et al. PTTG regulates the metabolic switch of ovarian cancer cells via the c-myc pathway. Oncotarget. 2015;6:40959-69
79. Lee GY, Chun YS, Shin HW, Park JW. Potential role of the N-MYC downstream-regulated gene family in reprogramming cancer metabolism under hypoxia. Oncotarget. 2016;7:57442-51
80. Xu X, Li J, Sun X, Guo Y, Chu D, Wei L. et al. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget. 2015;6:26161-76
81. Sharma BK, Kolhe R, Black SM, Keller JR, Mivechi NF, Satyanarayana A. Inhibitor of differentiation 1 transcription factor promotes metabolic reprogramming in hepatocellular carcinoma cells. FASEB J. 2016;30:262-75
82. Zhang P, Cao L, Fan P, Mei Y, Wu M. LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep. 2016;17:1204-20
83. Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S. et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433-5
84. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251-62
85. Hardie DG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res. 2015;21:3836-40
86. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113-24
87. Laderoute KR, Calaoagan JM, Chao WR, Dinh D, Denko N, Duellman S. et al. 5'-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J Biol Chem. 2014;289:22850-64
88. Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D. et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053
89. Song HT, Qin Y, Yao GD, Tian ZN, Fu SB, Geng JS. Astrocyte elevated gene-1 mediates glycolysis and tumorigenesis in colorectal carcinoma cells via AMPK signaling. Mediators Inflamm. 2014;2014:287381
90. Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR. et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2014;33:5251-61
91. Liu P, Ye F, Xie X, Li X, Tang H, Li S. et al. mir-101-3p is a key regulator of tumor metabolism in triple negative breast cancer targeting AMPK. Oncotarget. 2016;7:35188-98
92. Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ. et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016;23:649-62
93. Chen B, Li H, Zeng X, Yang P, Liu X, Zhao X. et al. Roles of microRNA on cancer cell metabolism. J Transl Med. 2012;10:228
94. Chen B, Tang H, Liu X, Liu P, Yang L, Xie X. et al. miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015;356:410-7
95. Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumour Biol. 2016;37:6855-60
96. Yamasaki T, Seki N, Yoshino H, Itesako T, Yamada Y, Tatarano S. et al. Tumor-suppressive microRNA-1291 directly regulates glucose transporter 1 in renal cell carcinoma. Cancer Sci. 2013;104:1411-9
97. Fei X, Qi M, Wu B, Song Y, Wang Y, Li T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012;586:392-7
98. Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao Y. et al. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol Chem. 2012;287:23227-35
99. Zhao S, Liu H, Liu Y, Wu J, Wang C, Hou X. et al. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013;333:253-60
100. Peschiaroli A, Giacobbe A, Formosa A, Markert EK, Bongiorno-Borbone L, Levine AJ. et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene. 2012;32:797-802
101. Gregersen LH, Jacobsen A, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer. 2012;12:232
102. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S. et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012;31:1985-98
103. Guo W, Qiu Z, Wang Z, Wang Q, Tan N, Chen T. et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology. 2015;62:1132-44
104. Zhu W, Huang Y, Pan Q, Xiang P, Xie N, Yu H. MicroRNA-98 Suppress Warburg Effect by Targeting HK2 in Colon Cancer Cells. Dig Dis Sci. 2017;62:660-8
105. Teng Y, Zhang Y, Qu K, Yang X, Fu J, Chen W. et al. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget. 2015;6:40799-814
106. Eichner LJ, Perry MC, Dufour CR, Bertos N, Park M, St-Pierre J. et al. miR-378( *) mediates metabolic shift in breast cancer cells via the PGC-1beta/ERRgamma transcriptional pathway. Cell Metab. 2010;12:352-61
107. Tang H, Lee M, Sharpe O, Salamone L, Noonan EJ, Hoang CD. et al. Oxidative stress-responsive microRNA-320 regulates glycolysis in diverse biological systems. FASEB J. 2012;26:4710-21
108. Du JY, Wang LF, Wang Q, Yu LD. miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol Rep. 2015;33:1890-8
109. Ge X, Lyu P, Cao Z, Li J, Guo G, Xia W. et al. Overexpression of miR-206 suppresses glycolysis, proliferation and migration in breast cancer cells via PFKFB3 targeting. Biochem Biophys Res Commun. 2015;463:1115-21
110. Wang J, Wang H, Liu A, Fang C, Hao J, Wang Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget. 2015;6:19456-68
111. Han H, Li W, Shen H, Zhang J, Zhu Y, Li Y. microRNA-129-5p, a c-Myc negative target, affects hepatocellular carcinoma progression by blocking the Warburg effect. J Mol Cell Biol. 2016. [Epub ahead of print]. doi: 10.1093/jmcb/mjw010
112. Hong X, Xu Y, Qiu X, Zhu Y, Feng X, Ding Z. et al. MiR-448 promotes glycolytic metabolism of gastric cancer by downregulating KDM2B. Oncotarget. 2016;7:22092-102
113. Yang X, Cheng Y, Li P, Tao J, Deng X, Zhang X. et al. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour Biol. 2015;36:383-91
114. Chen Y, Zhang Z, Luo C, Chen Z, Zhou J. MicroRNA-18b inhibits the growth of malignant melanoma via inhibition of HIF-1alpha-mediated glycolysis. Oncol Rep. 2016;36:471-9
115. Liu L, Wang Y, Bai R, Yang K, Tian Z. MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1alpha regulation. Oncogenesis. 2016;5:e224
116. Ding G, Huang G, Liu HD, Liang HX, Ni YF, Ding ZH. et al. MiR-199a suppresses the hypoxia-induced proliferation of non-small cell lung cancer cells through targeting HIF1alpha. Mol Cell Biochem. 2013;384:173-80
117. Li B, He L, Zuo D, He W, Wang Y, Zhang Y. et al. Mutual Regulation of MiR-199a-5p and HIF-1alpha Modulates the Warburg Effect in Hepatocellular Carcinoma. J Cancer. 2017;8:940-9
118. Jia YY, Zhao JY, Li BL, Gao K, Song Y, Liu MY. et al. miR-592/WSB1/HIF-1alpha axis inhibits glycolytic metabolism to decrease hepatocellular carcinoma growth. Oncotarget. 2016;7:35257-69
119. Li SJ, Liu HL, Tang SL, Li XJ, Wang XY. MicroRNA-150 regulates glycolysis by targeting von Hippel-Lindau in glioma cells. Am J Transl Res. 2017;9:1058-66
120. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014;53:88-100
121. Lin A, Li C, Xing Z, Hu Q, Liang K, Han L. et al. The LINK-A lncRNA activates normoxic HIF1alpha signalling in triple-negative breast cancer. Nat Cell Biol. 2016;18:213-24
122. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
123. Jeong SM, Haigis MC. Sirtuins in Cancer: a Balancing Act between Genome Stability and Metabolism. Mol Cells. 2015;38:750-8
124. Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15:608-24
125. Martinez-Pastor B, Mostoslavsky R. Sirtuins, metabolism, and cancer. Front Pharmacol. 2012;3:22
126. Zhong L, Mostoslavsky R. Fine tuning our cellular factories: sirtuins in mitochondrial biology. Cell Metab. 2011;13:621-6
127. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315-29
128. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD. et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280-93
129. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185-99
130. Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986-96
131. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19:416-28
132. Haigis MC, Deng CX, Finley LW, Kim HS, Gius D. SIRT3 is a mitochondrial tumor suppressor: a scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012;72:2468-72
133. Yu W, Denu RA, Krautkramer KA, Grindle KM, Yang DT, Asimakopoulos F. et al. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J Biol Chem. 2016;291:3268-79
134. Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M. et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015;34:1110-25
135. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. 2014;53:534-48
136. Ozden O, Park SH, Wagner BA, Yong Song H, Zhu Y, Vassilopoulos A. et al. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med. 2014;76:163-72
137. Pinho AV, Mawson A, Gill A, Arshi M, Warmerdam M, Giry-Laterriere M. et al. Sirtuin1 stimulates the proliferation and the expression of glycolysis genes in pancreatic neoplastic lesions. Oncotarget. 2016;7:74768-78
138. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X. et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014;74:727-37
139. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP. et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014;33:1454-73
140. Zhong X, Tian S, Zhang X, Diao X, Dong F, Yang J. et al. CUE domain-containing protein 2 promotes the Warburg effect and tumorigenesis. EMBO Rep. 2017;18:809-25
141. Liu Y, Xiang F, Huang Y, Shi L, Hu C, Yang Y. et al. Interleukin-22 promotes aerobic glycolysis associated with tumor progression via targeting hexokinase-2 in human colon cancer cells. Oncotarget. 2017;8:25372-83
142. Shang Y, He J, Wang Y, Feng Q, Zhang Y, Guo J. et al. CHIP/Stub1 regulates the Warburg effect by promoting degradation of PKM2 in ovarian carcinoma. Oncogene. 2017;36:4191-4200
143. Li L, Zhu L, Hao B, Gao W, Wang Q, Li K. et al. iNOS-derived nitric oxide promotes glycolysis by inducing pyruvate kinase M2 nuclear translocation in ovarian cancer. Oncotarget. 2017;8:33047-63
144. Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S. et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun. 2017;8:14634
145. Xu Q, Zhang Q, Ishida Y, Hajjar S, Tang X, Shi H. et al. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget. 2017;8:9557-71
146. Matijevic Glavan T, Cipak Gasparovic A, Verillaud B, Busson P, Pavelic J. Toll-like receptor 3 stimulation triggers metabolic reprogramming in pharyngeal cancer cell line through Myc, MAPK, and HIF. Mol Carcinog. 2017;56:1214-26
147. Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM. et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology. 2017;153:277-91
148. Yoshida S, Tsutsumi S, Muhlebach G, Sourbier C, Lee MJ, Lee S. et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci U S A. 2013;110:E1604-12
149. Zhang J, Gao Q, Zhou Y, Dier U, Hempel N, Hochwald SN. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene. 2016;35:1926-42
150. Ha TK, Chi SG. CAV1/caveolin 1 enhances aerobic glycolysis in colon cancer cells via activation of SLC2A3/GLUT3 transcription. Autophagy. 2012;8:1684-5
151. Ha TK, Her NG, Lee MG, Ryu BK, Lee JH, Han J. et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012;72:4097-109
152. Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J. et al. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 2013;73:1900-11
153. Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X. et al. Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep. 2014;9:1798-811
154. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J. et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014;20:4370-80
155. Minami K, Taniguchi K, Sugito N, Kuranaga Y, Inamoto T, Takahara K. et al. MiR-145 negatively regulates Warburg effect by silencing KLF4 and PTBP1 in bladder cancer cells. Oncotarget. 2017;8:33064-77
156. Dey P, Rachagani S, Chakraborty S, Singh PK, Zhao X, Gurumurthy CB. et al. Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin Cancer Res. 2012;18:6188-98
157. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF. et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:279-84
158. Ma T, Patel H, Babapoor-Farrokhran S, Franklin R, Semenza GL, Sodhi A. et al. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi's sarcoma. Angiogenesis. 2015;18:477-88
159. Lo AK, Dawson CW, Young LS, Ko CW, Hau PM, Lo KW. Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J Pathol. 2015;237:238-48
160. Zhang R, Su J, Xue SL, Yang H, Ju LL, Ji Y. et al. HPV E6/p53 mediated down-regulation of miR-34a inhibits Warburg effect through targeting LDHA in cervical cancer. Am J Cancer Res. 2016;6:312-20
161. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7:38908-26
162. Ribback S, Cigliano A, Kroeger N, Pilo MG, Terracciano L, Burchardt M. et al. PI3K/AKT/mTOR pathway plays a major pathogenetic role in glycogen accumulation and tumor development in renal distal tubules of rats and men. Oncotarget. 2015;6:13036-48
163. Ribback S, Calvisi DF, Cigliano A, Sailer V, Peters M, Rausch J. et al. Molecular and metabolic changes in human liver clear cell foci resemble the alterations occurring in rat hepatocarcinogenesis. J Hepatol. 2013;58:1147-56
164. Varone A, Xylas J, Quinn KP, Pouli D, Sridharan G, McLaughlin-Drubin ME. et al. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 2014;74:3067-75
165. Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ. et al. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab. 2011;14:131-42
166. Chang CC, Zhang C, Zhang Q, Sahin O, Wang H, Xu J. et al. Upregulation of lactate dehydrogenase a by 14-3-3zeta leads to increased glycolysis critical for breast cancer initiation and progression. Oncotarget. 2016;7:35270-83
167. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3
168. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425-34
Author contact
![]() Corresponding authors: Li Yu, Department of Pathology, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People's Republic of China. Tel.: 86-20-87755766-8864, Fax: 86-20-87331780, E-mail: yuli5sysu.edu.cn; Shangwu Chen, Department of Biochemistry, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, People's Republic of China, Tel.: 86-20-39332958, Fax: 86-20-39332950, E-mail: lsschshwsysu.edu.cn.
Corresponding authors: Li Yu, Department of Pathology, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People's Republic of China. Tel.: 86-20-87755766-8864, Fax: 86-20-87331780, E-mail: yuli5sysu.edu.cn; Shangwu Chen, Department of Biochemistry, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, People's Republic of China, Tel.: 86-20-39332958, Fax: 86-20-39332950, E-mail: lsschshwsysu.edu.cn.