Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(18):3667-3674. doi:10.7150/jca.20750 This issue Cite
Review
Pancreatic Cancer in Lynch Syndrome Patients
Luis Bujanda1, Marta Herreros-Villanueva1, 2 ![]()
1. Department of Gastroenterology, Hospital Donostia/Instituto Biodonostia, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Universidad del País Vasco UPV/EHU, San Sebastián 20014, Spain;
2. Faculty of Life Sciences, Universidad Isabel I, Spain.
Received 2017-4-26; Accepted 2017-8-23; Published 2017-10-11
Abstract
Although colorectal cancer (CRC) is the most common cancer type in Lynch syndrome (LS) families, patients have also increased lifetime risk of other types of tumors. The accumulated risk of pancreatic cancer (PC) in LS patients is around 3.7% and developed tumors often present a characteristically medullary appearance with prominent lymphocytic infiltration. LS patients are considered in high risk for PC development as they present 8.6-fold increase compared with the general population.
Here we review PC cases reported in LS patients and current management guidelines. Literature data show that LS is clearly associated with PC and recent publications also demonstrated a connection with pancreatic neoplasic precursor lesions such as intraductal papillary mucinous neoplasms (IPMN) in these patients.
While screening techniques are well established for CRC detection, clear strategies are not yet uniform for PC. Magnetic resonance imaging (MRI) and/or endoscopic ultrasound every 1-2 years in MMR mutation carriers with PC in a first or second-degree relative is recommended.
Better pancreatic cancer detection strategies should be urgently defined due to the importance of early diagnosis in this disease.
Keywords: Colorectal cancer, Hereditary, Lynch syndrome, Pancreatic cancer, Risk.
Introduction
Pancreatic cancer (PC) is one of the neoplasias with worse prognosis and despite the increased efforts in research, progress in patient's survival has remained slow with a 5-year survival rate estimated in 2016 of 8%1. It is expected that PC death rate will increase being the second leading cause of cancer-related death by 20302.
Since pancreatic ductal adenocarcinoma (PDAC) is the predominant pancreatic tumor type, we will refer to this pathology as PC.
Majority of PC cases (>80%) are considered sporadic, due to sporadic mutations. The main modifiable risk factors are smoking, obesity, diabetes mellitus, non-O blood group, Helicobacter pylori infection and chronic pancreatitis. While all of these factors increase risk up to 2 fold, chronic pancreatitis could give up to 13.3 fold of higher risk3.
Hereditary pancreatic cancer (accounts for 5-10% of cases) is defined as a genetic syndrome with an identifiable gene mutation associated with an increased risk for PC. These mutations are principally BRCA2, p16, ATM, STK11, PRSS1/PRSS2, SPINKI, PALB2 and DNA mismatch repair genes (MMR)- each one associated to increased risk for PC development4. However, BRCA2 is probably the most common inherited disorder.
Considering pathologies, individuals should be considered to be at risk for PC if they have hereditary pancreatitis, cystic fibrosis or other PC-related syndromes. These other syndromes linked to PC risk include Lynch syndrome (LS), Peutz-Jeghers syndrome (PJS), Familial atypical multiple mole melanoma syndrome (FAMMM) and Li-Fraumeni syndrome.
Familial Pancreatic Cancer (FPC) is defined as a family with at least one pair of first-degree relatives (parent-child or sibling pair) with PC without an identifiable syndrome in the family5.
Lynch Syndrome Definition and Types
LS, formerly known as hereditary nonpolyposis colorectal cancer or HNPCC was firstly described by Warthin in 1913 and it is an autosomal dominant disease caused by germline mutation in MMR genes MLH1, MSH2, MSH6 and PMS2, or by germline mutation in EPCAM which causes epigenetic silencing of MSH2 6, 7.
LS is suspected on the basis of patient and family history, MMR protein expression pattern and Microsatellite instability (MSI) phenotype. The incidence of this syndrome has been postulated to be between 1:660 and 1:20008.
MMR mutation is generally inherited from one parent and patient has every cell in the body with one defective copy of the particular gene and a fully functional copy that maintains DNA repair in cells. A cell develops a DNA repair defect only when this functional copy of the gene becomes nonfunctional as a result of a random somatic mutation9.
There are two forms of LS: Type I, the one limited to colon specific syndrome and type II, that includes cancer in different anatomic sites such as endometry, ovary, pancreas, etc.
As determined in the ACG Clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes, LS should be considered in individuals whose tumors show evidence of MMR deficiency (without the presence of a BRAF mutation or MLH1 promoter hypermethylation), and those whose personal and/or family history fullfill the Amsterdam criteria, Bethesda Guidelines (Available from http://www.nccn.org/professionals/physician_gls/f_guidelines.asp) or who have a ≥5% risk of carrying a germline mutation based on available prediction models10, 11.
The primary strategy currently used in routine practice to identify LS patients is tumor sample evaluation for evidence of high level MSI (MSI-H) and/or DNA MMR deficiency (MMR-D)10, 11. Several studies have consistently demonstrated that universal tumor testing- mostly by using Polymerase chain reaction (PCR) for MSI testing and immunohistochemical staining for MMR protein expression of all CRC tumors is a more effective way to screen LS compared to previous guidelines based on Bethesda guidelines or Amsterdam criteria. However, successful results depend on availability of both, healthy and cancer relatives undergoing genetic testing12, 13.
LS accounts for approximately 2-5% of all colorectal cancers (CRC) diagnosis and is the most common cause of inherited colon cancer conferring a lifetime risk between 52-82% with a mean diagnosis age of 44 years. However, endometrial tumors occur with a similar frequency to colon cancer in woman with LS. LS also confers an increased risk of other types of tumors such as endometrial (lifetime risk 25-60%), ovarian (4-12%), gastric (6-13%), pancreatic (1.3-4%), ciliary tract and kidney cancer14-17. This means that LS patients frequently develop colorectal cancer before the age of 50 and approximately one-third of them develop another LS typical tumor within 10 years18.
In this review, we will focus on PC- cases reported, increased risk and current management guidelines.
Genetic association of LS and PC
LS is an autosomal dominant hereditary disorder due to mutation in a mismatch repair (MMR) genes. In PC cases derived from LS, mutations have been specifically described in genes encoding DNA reparation proteins: MLH1 (mutL homolog 1), MSH2, MSH6 (mutS homolog 2 y 6, respectively), PMS2 (PMS1 homolog 2, mismatch repair system component). Less frequently, PALB2 (partner and localizer of BRCA2) and EPCAM (Epithelial cell adhesion molecule) gene located upstream of MSH2 have been seen in LS patients diagnosed with PC. EPCAM deletions lead to a transcriptional read-through, hypermethylation and subsequent silencing MSH2 and are estimated to cause Lynch syndrome in ~20-25% of patients with MSH2-negative cancers, but no detectable MSH2 germline mutation6.
Mutations in the MLH1 and MSH2 genes have more effect on DNA repair than mutations in MSH6 or PALB2. Patients with an MLH1 or MSH2 mutation therefore have a substantially higher risk of tumors than patients with an MSH6 mutation. The risk of patients with a PMS2 mutation seems to be even lower than that of patients with an MSH6 mutation9.
As it occurs in other type of LS tumors, defect in the DNA mismatch repair system increase the error rate of replication by 100 to 1000 fold. Areas of the genome that contain repetitive sequences (microsatellites) are particularly susceptible to insertion and deletion of bases during the replication process. Errors produces in the DNA sequence are recognized by proteins such as MSH2 or MHS6 19.
Typically these alterations are germline mutations and genetic testing of patients should include germline mutation genetic testing for the MLH1, MSH2, MSH6, PMS2, and/or EPCAM genes or the altered gene(s) indicated by immunochemistry testing11.
MSI-H tumors have been associated with poor differentiation and the presence of wild type KRAS and p53 genes (frequently mutated in sporadic cases os PC). Moreover, patients with MSI-H tumors have longer overall survival compared to those with MSI-L or MSS tumors20. Additionally, some authors support that MSI and MMR loss of expression in LS is associated to medullary carcinomas of pancreas17, 21.
Next generation data could help to identify other mutations and epigenetic modifications occurring in tumor suppressor genes and oncogenes finally responsible for neoplastic transformation. Confirmatory statements about mutational status in genes frequently alternated in PC such as KRAS, SMAD4, TP53, CDKN2A are still needed.
Reported cases of PC in the literature
The accumulated risk of PC in LS patients is around 3.7%22 (it is 1.5% in general population)17, 23, 24. It has been described that pancreatic tumors developed by LS patients often have a characteristically medullary appearance21, 25, with prominent lymphocytic infiltration and MSI26.
When a search for ''Lynch syndrome & pancreatic cancer'' was performed, as shown in Figure 1, 106 records were identified from PubMed database. If research was done for terms ''hereditary nonpolyposis colorectal cancer & pancreatic cancer'' 79 records were found; however, all of them except one (non-specific) were duplicated of those included in the 106.
Flow diagram for selected studies
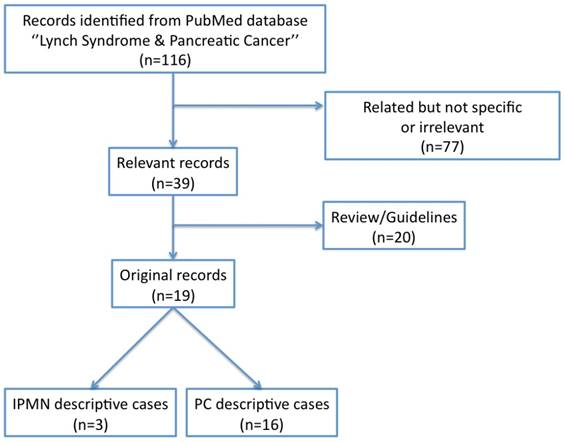
Considering the 106 cases after removing 71 ''related but not specific or irrelevant'' records, we found 17 review/guidelines, 3 IPMN-related cases and 15 relevant descriptive cases. Here, we present summary of the total 18 reporting PC cases in LS patients. A note with these studies is presented in Table 1.
Most relevant studies supporting evidence of PC in LS patients.
| Most relevant cases described in the literature | Genetic Evidence | Associated Disease | Country | Number LS patients / Number PC cases | # Reference |
|---|---|---|---|---|---|
| Lynch et al. 1985 | ns | PC | USA (Nebraska) | Family Case Report (3 PC) | 27 |
| Wei et al. 2002 | ns | Taiwan | 202 LS/1 PC | 28 | |
| Banville et al. 2006 | MSH2, MLH1 nonsense mutations | Ireland | Case Report | 21 | |
| Geary et al. 2008 | ns | United Kingdom | 130 LS/22PC | 29 | |
| Barrow et al. 2009 | ns | United Kingdom | 938 LS /2 PC | 16 | |
| Vergara-Fernández et al. 2009 | ns | Mexico | Case Report | 25 | |
| Gargiulo et al. 2009 | ns | Italy | *135 PC/19 LS | 30 | |
| Kastrinos et al. 2009 | MSH2, MLH1, MSH6 mutations | USA (Massachusets) | 147 LS families/47 PC | 17 | |
| Kempers et al. 2011 | EPCAM deletions | The Netherlands | 194 EPCAM deletion carriers/ 4 PC | 31 | |
| Lindor et al. 2011 | MSH2 (P349L) missense alteration | USA (Northern European ancestry) | Family Case Report (3 PC) | 32 | |
| Laghi et al. 2012 | MLH1 deficiency and MLH1 methylation | Italy & Germany | 203 LS/5PC | 34 | |
| Borelli et al. 2014 | MLH1 (c.2252_2253delAA) mutation | Italy | 67 LS/ 5 PC | 38 | |
| Salo-Mullen et al. 2015 | MHS2, MLH1, MSH6 mutations | USA (New York) | *159 PC/4 LS | 35 | |
| Catts et al. 2016 | MSH2, MLH1 mutations | USA (Delaware) | 16 FPC families / 2 LS | 36 | |
| Cajal et al. 2016 | MLH1 (c.2252_2253delAA) mutation | Italy & Spain | Case Report | 37 | |
| Sparr et al. 2009 | MSH2, MSH6 mutations | IPMN | USA (Massachusets) | Case Report | 41 |
| Flanagan et al. 2015 | MSH2 mutation | USA (Wasintong) | Case Report | 42 | |
| Lee et al. 2015 | MHS2, MSH6 mutation? | South Korea | Case Report | 43 | |
| Dymerska et a. 2017 | EPCAM mutation | Poland | Family Case Report (1 PC) | 33 |
Ns, not specified. PC, Pancreatic Cancer; IPMN, intraductal papillary mucinous neoplasms. * LS mutations (cases) were evaluated in PC patients.
Lynch et al. were the first in 1985 reporting an increased risk of developing PC in LS families27. They described a kindred with vertical transmission of cancer through 5 generations in which HNPCC and PC was shared.
Since 1985, several authors have described cases and incidence of PC around the world. Wei et al.28 in 2002 reviewed 10 Taiwanese families including a total of 202 individuals meeting Amsterdam criteria for LS and they found one PC case. Geary et al. in 2008 examined 982 cancers in 723 individuals who belonged to 130 families and found 22 PC, 14 of them diagnosed before the age of 6029. In this series, PC was the third more frequent after colorectal and endometrial cancer. In 2009, Gargiulo et al.30 noted that estimated frequency of LS was small since among 135 PC patients they only found 19 LS carriers. Kastrinos et al. analyzed 6342 individuals from 147 families with MMR gene mutations. Thirty-one families (21.1%) reported at least 1 PC case. Forty-seven pancreatic cancers were reported (21 men and 26 women), with no sex-related difference in age of diagnosis (51.5 vs 56.5 years for men and women, respectively). The cumulative risk of PC in these families with gene mutations was 1.31% up to age 50 years and 3.68% up to age of 70 years, which represents an 8.6-fold increase compared with the general population17. In 2011 Kempers et al.31 evaluated 194 carriers of an EPCAM deletion and four were found to have PC and Lindor et al. reported a family in which MSH2 P349L missense alteration cosegregatated with PC in 3 first degree relatives (FDR)32. Recently, in early 2017 Dymerska et al.33 reported the presence of a founder mutation (c.858+2478_*4507del) in eight Polish families. Laghi et al. in 2012 evaluated 338 consecutive PDAC in German and Italian centers concluding that MSI prevalence is negligible for sporadic PC. Differently, the prevalence of PC is 2.5% in LS patients34.
In 2015, Salo-Mullen et al. published a study in which they analyzed mutations in 159 PC patients identifying 24 pathogenic mutations including BRCA2, BRCA1, p16, PALB2 and LS35. Four different mutations in MSH2 and MLH1 were found in these LS patients and they were especially frequent in early new onset disease.
Catts et al.36 conducted a retrospective review of PC families in US from 2002 to 2013. They analyzed familial pancreatic cancer individuals (including FPC criteria subjects, individuals with only one first-degree relative affected by PC [first-degree families] and PC affected). 175 families completed a testing: 46 being FPC and 99 first-degree families. 16 out 46 FPC went LS testing and pathogenic mutations in MMR were identified in 2 of them: one in MLH1 and one in MSH2. 32 out the 99 first-degree families went LS testing and 4 of them had pathogenic mutation: three in MLH1 and one in MSH6. Authors concluded that in FPC, breast and ovarian cancer and then LS are the most prevalent syndromes leading to PC. Cajal et al.37 identified a mutation n c.2252_2253delAA, p.Lys751Serfs*3 en el exón 19 del gen MLH1 in an Italian patient. This mutation was previously noted as pathogenic in Korea, Denmark, United Kingdom, Germany and Australia and described by Borrelli et al.38 in a MLH1 mutation (c2253_2253delAA) that co-segregates with LS cancer in 11 unrelated families. All families had at least one colon cancer diagnosed before age of 50 and one case with multiple LS related tumors. Interestingly, a statistically significant association was found with higher frequency of PC compared to families with other MLH1 mutations.
While all the publications presented above reported an increased rate of pancreatic cancer in LS families and patients, another study published by Barrow et al.16 in 2009 observed no excess risk. Additionally, two additional studies in the literature reported in the eighties and nineties found no high correlation between LS and PC since in a study of 40 Finnish LS kindreds, only 6 out of 243 carriers with clinically or histologically cancer had PC39. Conversely, no PC was found in a series of 22 Dutch LS families with at least one person diagnosed with PC40.
Although LS has been clearly associated with PC (understood as Pancreatic Ductal Adenocarcinoma-PDAC), no clear connection has been established with pancreatic neoplasic precursor lesions in these patients. Only a few recent publications have demonstrated that intraductal papillary mucinous neoplasm (IPMN) could be LS related and consequently, these lesions should be carefully considered at patient's screening.
Firstly, in 2009, Sparr et al.41 described the case of a 61-year old woman with LS and multiple cancers including colorectal adenocarcinoma and IPMN. More recently, a case of a 58 year-old woman was reported by Flanagan et al. This LS patient (exon 1 deletion in MSH2 that was classified as a suspected deleterious change) was diagnosed firstly with colorectal cancer, then uterine and breast cancer and finally CT images showed a progression of main duct IPMN to invasive pancreatic ductal adenocarcinoma. Due to LS and previous history of cancer, distal pancreatectomy was indicated. However, MMR immunohistochemical stain was negative for MLH1, PMS2, MSH2 and MSH642. Finally, Lee et al. reported a case of a 49 year-old woman diagnosed with LS, past history of colon and endometrial carcinoma in which IPMN originated from the ideal heterotopic pancreas was surgically resected. It was positive for MLH1/PMS2 and negative for MSH2/MSH643.
All of this data suggest that IPMN are part of the spectrum of lesions found in LS.
Moreover, Acinar cell carcinoma (ACC)- a rare pancreatic malignancy has been documented by Liu et al.44 in 36 ACC cases, of which 5 were MMR deficient and 2 out of these 5 were LS. ACC was also reported by Karamurzin et al.45 in a single case.
Strategies for PC in LS patients
It is well known that screening general population for PC is not cost-effective and screening strategy has been only confined to High Risk (HR) Population- defined as subjects with 6-8 times higher risk of having PC compared with age-matched controls. This HR group, as established now, includes individuals with 2 first-degree relatives with PC which is the cohort currently screened for familial PC46. Additionally, and still controversial is to include new onset diabetes subjects older than 50 years, which as demonstrated by Chari et al.47 have 6-8 fold higher probability of being diagnosed with PC, as HR. This last scenario is nowadays more accepted in US than in Europe or any other country.
For healthy individuals who have a germline mutation in a gene linked to PC risk, as it is the case of LS, the priority is to manage and reduce PC risk due to the importance of early diagnosis in this disease. However, in this case of healthy LS individuals, factual risk and clear strategies are not yet uniform and well-defined.
If, as demonstrated by some authors, patients with LS have an 8.6-fold increased risk of developing PDAC compared to the general population17, they should then considered as HR subjects and consequently, enter clearly into screening programs.
In one side, it has been purposed in 2011 by the International Cancer of the pancreas screening (CAPS) consortium that LS patients and one first-degree relative with PC should be candidates for screening48. On the other side, it is indicated in the summary of recommendations by Syngal et al. about how to manage extracolonic neoplasia screening in LS patients: ''Screening beyond population-based recommendations for cancers of the urinary tract, pancreas, prostate, and breast is not recommended unless there is a family history of the specific cancers (conditional recommendation, low quality of evidence)''. In this case (family history demonstrated), it can be considered a magnetic resonance imaging (MRI) and/or endoscopic ultrasound every 1-2 years in MMR mutation carriers with PC in a first 48 or second-degree relative11 depending on the recommendation guides (Table 2).
Guidelines for PC screening in LS patients.
| Management strategy for PC Screening | Technique | Guidelines/Author recommending | Year |
|---|---|---|---|
| ''LS individuals with at least one affected FDR with PC'' | MRI/EUS/CT/ERCP* | CAPS International cancer of pancreas screening/Canto et. al. | 2011 |
| ''LS-MMR mutation carriers with PC in a FDR'' (conditional recommendation; very low quality of evidence) | MRI/EUS | ACG Clinical Guideline/Syngal et. al | 2015 |
CT, Computed tomography; ERCP, Endoscopic retrograde cholangiopancreatography; LS, Lynch syndrome; FDR, First degree relative; MSI, Magnetic resonance imaging; PC, Pancreatic cancer; * Initial screening should include (multiple answers allowed): EUS 83.7%, MRI 73.5%, CT 26.5%, abdominal ultrasound 14.3%, ERCP 2.0%. B2 When previous screening did not detect an abnormality that met criteria for shortening of the interval or surgical resection, follow-up screening should include (multiple answers allowed): EUS 79.6% MRI 69.4%, CT 22.4%, abdominal ultrasound 4.1%, ERCP 2.0%
The frequency and age to begin surveillance are not yet well defined. Most centers utilize endoscopic ultrasound and/or MRI surveillance programs, both of which detect pancreatic lesions better than CT49. Past screening efforts, mostly using HR- FPC, PJS and FAMMM patients have demonstrated diagnostic yields from 1.1 to 50%, depending on their definition of yield3. However, since it is well-known that in general terms there is not effective screening test for this malignancy, some authors considerer PC a ''rare LS related tumor'' and no specific screening measures other than abdominal ultrasound and general physical examination is recommended9.
In general it can be said that literature contains little reliable information on screening for other tumors than colorectal and ovarian/endometrio in LS patients. Prospective data indicated that screening colonoscopies beginning in the early 20s can markedly reduce colorectal cancer incidence and colorectal cancer-related mortality in individuals with LS50. For women with LS, risk-reducing hysterectomy and salpingo-oophorectomy can drastically reduce the risk of endometrial and ovarian cancers51. Being PC less frequent than these other tumor types, more investigation to reach consensus is needed. PancPro (https://www4.utsouthwestern.edu/breasthealth/cagene) is a clinical prediction model that uses personal and family history data, including prior genetic testing results, to estimate an individual's future risk of developing pancreatic cancer.
Discussion and conclusion
PC is a leading cause of cancer death and very few patients are candidates for the only curative option-the surgery. Screening of HR patients amenable to surgical resection should be a priority.
It must be recognized that based on the literature, Peutz-Jeghers syndrome (132 fold), FAMMM syndrome (47 fold) or hereditary pancreatitis (69 fold) confer a bigger increase in PC risk compared to LS (8.6 fold)3. This is, although PC is not one of the top cancer types in LS patients (lower incidence than colorectal and endometrial tumors), still these patients are considered in high risk- as they present 8.6-fold increase compared with the general population.
As it has been presented above, most of the studies published in the literature (including patients from USA, Europe and Asia) demonstrate a good association of PC in LS kindreds. This review summarizes the studies that should serve to support evidence of PC risk in LS patients.
Accepting this evidence and the clinical need for PC screening in LS individuals, only a few guidelines describe further management strategies. They recommend screening LS individuals MMR carriers with at least one PC case in a FDR. Unfortunately, adequate screening technique for PC does not currently exist and further research must be done and options for these patients include MRI and EUS. However, optimal interval for screening and the management of PC precursor lesions is still controversial52. Moreover, there is an urgent need to find new biomarkers that could help to screen HR individuals53, including LS patients. How clinicians should fully address PC screening and management is still unclear as many uncertainties in PC and LS remain.
Recent reviews highlight the increasingly recognized role of genetics in the development of PC and associated syndromes. Given the poor prognosis of PC, the development of next-generation genome sequencing will show additional alterations that combined with more powerful screening and surveillance tools may lead to an improved survival54.
Abbreviations
CRC, Colorectal cancer
FPC, Familial pancreatic cancer
HR, High risk
LS, Lynch syndrome
IPMN, intraductal papillary mucinous neoplasm
MMR, DNA Mismatch repair
MSI, Microsatellite instability
PC, Pancreatic cancer
PDAC, Pancreatic ductal adenocarcinoma
Author contributions
Herreros-Villanueva M and Bujanda L designed, wrote and edited the manuscript. All authors approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30 [PMID: 26742998 DOI: 10.3322/caac.21332]
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21 [PMID: 24840647 DOI: 10.1158/0008-5472.CAN-14-0155]
3. Becker AE, Hernandez YG, Frucht H, Lucas AL. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World J Gastroenterol. 2014;20:11182-98 [PMID: 25170203 DOI: 10.3748/wjg.v20.i32.11182]
4. Yeo TP. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin Oncol. 2015;42:8-18 [PMID: 25726048 DOI: 10.1053/j.seminoncol.2014.12.002]
5. Brand RE, Lerch MM, Rubinstein WS, Neoptolemos JP, Whitcomb DC, Hruban RH, Brentnall TA, Lynch HT, Canto MI. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56:1460-9 [PMID: 17872573 DOI: 10.1136/gut.2006.108456]
6. Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet. 2009;41:112-7 [PMID: 19098912 DOI: 10.1038/ng.283]
7. Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, Penzone P, Lombardi J, Dunn P, Cohn DE, Copeland L, Eaton L, Fowler J, Lewandowski G, Vaccarello L, Bell J, Reid G, de la Chapelle A. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810-7 [PMID: 16885385 DOI: 10.1158/0008-5472.CAN-06-1114
8. Jarvinen HJ, Renkonen-Sinisalo L, Aktan-Collan K, Peltomaki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol. 2009;27:4793-7 [PMID: 19720893 DOI:10.1200/JCO.2009.23.7784]
9. Steinke V, Engel C, Buttner R, Schackert HK, Schmiegel WH, Propping P. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. Dtsch Arztebl Int. 2013;110:32-8 [PMID: 23413378 DOI: 10.3238/arztebl.2013.0032]
10. Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, Levin TR, Lieberman DA, Robertson DJ, Syngal S, Rex DK. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014;109:1159-79 [PMID:25070057 DOI: 10.1038/ajg.2014.186]
11. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:223-62 quiz 263. [PMID: 25645574 DOI: 10.1038/ajg.2014.435]
12. Ladabaum U, Wang G, Terdiman J, Blanco A, Kuppermann M, Boland CR, Ford J, Elkin E, Phillips KA. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med. 2011;155:69-79 [PMID: 21768580 DOI: 10.7326/0003-4819-155-2-201107190-00002]
13. Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol. 2013;31:2554-62 [PMID: 23733757 DOI:10.1200/JCO.2012.46.8454]
14. Yurgelun MB. Next-generation strategies for hereditary colorectal cancer risk assessment. J Clin Oncol. 2015;33:388-93 [PMID: 25559814 DOI: 10.1200/JCO.2014.58.9895]
15. Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, Levin TR, Lieberman DA, Robertson DJ, Syngal S, Rex DK. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. 2014;147:502-26 [PMID: 25043945 DOI: 10.1053/j.gastro.2014.04.001]
16. Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, Hill J, Evans DG. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet. 2009;75:141-9 [PMID: 19215248 DOI: 10.1111/j.1399-0004.2008.01125.x]
17. Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, Bandipalliam P, Stoffel EM, Gruber SB, Syngal S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790-5 [PMID:19861671 DOI:10.1001/jama.2009.1529]
18. Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1-18 [PMID:19659756 DOI: 10.1111/j.1399-0004.2009.01230.x]
19. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816-9 [PMID: 8484122 ]
20. Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, Imai K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001;61:3139-44 [PMID: 11306499]
21. Banville N, Geraghty R, Fox E, Leahy DT, Green A, Keegan D, Geoghegan J, O'Donoghue D, Hyland J, Sheahan K. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol. 2006;37:1498-502 [PMID: 16996571 DOI:10.1016/j.humpath.2006.06.024]
22. Leoz ML, Sanchez A, Carballal S, Ruano L, Ocana T, Pellise M, Castells A, Balaguer F, Moreira L. [Hereditary gastric and pancreatic cancer predisposition syndromes]. Gastroenterol Hepatol. 2016;39:481-93 [PMID: 26916701 DOI: 10.1016/j.gastrohep.2015.11.009]
23. Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L, Burn J, Liu B, Kinzler KW, Vogelstein B. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997;6:105-10 PMID: 9002677
24. Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Jarvinen HJ, Myrhoj T, Sunde L, Wijnen JT, Lynch HT. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444-9 [PMID: 18398828 DOI: 10.1002/ijc.23508]
25. Vergara-Fernandez O, Zamora-Valdes D, Rodriguez-Zentner HA, Tapia H, Sanchez-Fernandez N, Gamboa-Dominguez A, Medina-Franco H, Chan-Nunez C. [Pancreatic-duodenectomy for invasive colon cancer in a patient with Lynch syndrome. Case report.]. Rev Gastroenterol Mex. 2009;74:374-8 [PMID: 20423772]
26. Grover S, Syngal S. Hereditary pancreatic cancer. Gastroenterology. 2010;139:1076-80 1080 e1-2. [PMID:20727885 DOI: 10.1053/j.gastro.2010.08.012]
27. Lynch HT, Voorhees GJ, Lanspa SJ, McGreevy PS, Lynch JF. Pancreatic carcinoma and hereditary nonpolyposis colorectal cancer: a family study. Br J Cancer. 1985;52:271-3 [PMID: 4027169]
28. Wei SC, Wang MH, Shieh MC, Wang CY, Wong JM. Clinical characteristics of Taiwanese hereditary non-polyposis colorectal cancer kindreds. J Formos Med Assoc. 2002;101:206-9 [PMID: 12051017]
29. Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne SJ, Fisher S, Hodgson SV. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer. 2008;7:163-72 [PMID:17939062 DOI:10.1007/s10689-007-9164-6]
30. Gargiulo S, Torrini M, Ollila S, Nasti S, Pastorino L, Cusano R, Bonelli L, Battistuzzi L, Mastracci L, Bruno W, Savarino V, Sciallero S, Borgonovo G, Nystrom M, Bianchi-Scarra G, Mareni C, Ghiorzo P. Germline MLH1 and MSH2 mutations in Italian pancreatic cancer patients with suspected Lynch syndrome. Fam Cancer. 2009;8:547-53 [PMID: 19728162 DOI: 10.1007/s10689-009-9285-1]
31. Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, Schackert HK, Steinke V, Holinski-Feder E, Morak M, Kloor M, Buttner R, Verwiel ET, van Krieken JH, Nagtegaal ID, Goossens M, van der Post RS, Niessen RC, Sijmons RH, Kluijt I, Hogervorst FB, Leter EM, Gille JJ, Aalfs CM, Redeker EJ, Hes FJ, Tops CM, van Nesselrooij BP, van Gijn ME, Gomez Garcia EB, Eccles DM, Bunyan DJ, Syngal S, Stoffel EM, Culver JO, Palomares MR, Graham T, Velsher L, Papp J, Olah E, Chan TL, Leung SY, van Kessel AG, Kiemeney LA, Hoogerbrugge N, Ligtenberg MJ. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12:49-55 [PMID: 21145788 DOI: 10.1016/S1470-2045(10)70265-5]
32. Lindor NM, Petersen GM, Spurdle AB, Thompson B, Goldgar DE, Thibodeau SN. Pancreatic cancer and a novel MSH2 germline alteration. Pancreas. 2011;40:1138-40 [PMID: 21926548 DOI: 10.1097/MPA.0b013e318220c217]
33. Dymerska D, Golebiewska K, Kuswik M, Rudnicka H, Scott RJ, Billings R, Plawski A, Borun P, Siolek M, Kozak-Klonowska B, Szwiec M, Kilar E, Huzarski T, Byrski T, Lubinski J, Kurzawski G. New EPCAM founder deletion in Polish population. Clin Genet. 2017 Mar3 31. [PMID: 28369810 DOI: 10.1111/cge.13026]
34. Laghi L, Beghelli S, Spinelli A, Bianchi P, Basso G, Di Caro G, Brecht A, Celesti G, Turri G, Bersani S, Schumacher G, Rocken C, Grantzdorffer I, Roncalli M, Zerbi A, Neuhaus P, Bassi C, Montorsi M, Scarpa A, Malesci A. Irrelevance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS One. 2012;7:e46002. [PMID: 23029359 DOI: 10.1371/journal.pone.0046002]
35. Salo-Mullen EE, O'Reilly EM, Kelsen DP, Ashraf AM, Lowery MA, Yu KH, Reidy DL, Epstein AS, Lincoln A, Saldia A, Jacobs LM, Rau-Murthy R, Zhang L, Kurtz RC, Saltz L, Offit K, Robson ME, Stadler ZK. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015;121:4382-8 [PMID: 26440929 DOI: 10.1002/cncr.29664]
36. Catts ZA, Baig MK, Milewski B, Keywan C, Guarino M, Petrelli N. Statewide Retrospective Review of Familial Pancreatic Cancer in Delaware, and Frequency of Genetic Mutations in Pancreatic Cancer Kindreds. Ann Surg Oncol. 2016;23:1729-35 [PMID: 26727920 DOI: 10.1245/s10434-015-5026-x]
37. Cajal AR, Pinero TA, Verzura A, Santino JP, Solano AR, Kalfayan PG, Ferro A, Vaccaro C. [Founder mutation in Lynch syndrome]. Medicina (B Aires). 2016;76:180-2 [PMID: 27295708]
38. Borelli I, Casalis Cavalchini GC, Del Peschio S, Micheletti M, Venesio T, Sarotto I, Allavena A, Delsedime L, Barberis MA, Mandrile G, Berchialla P, Ogliara P, Bracco C, Pasini B. A founder MLH1 mutation in Lynch syndrome families from Piedmont, Italy, is associated with an increased risk of pancreatic tumours and diverse immunohistochemical patterns. Fam Cancer. 2014;13:401-13 [PMID: 24802709 DOI: 10.1007/s10689-014-9726-3]
39. Aarnio M, Mecklin JP, Aaltonen LA, Nystrom-Lahti M, Jarvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64:430-3 [PMID: 8550246]
40. Vasen HF, den Hartog Jager FC, Menko FH, Nagengast FM. Screening for hereditary non-polyposis colorectal cancer: a study of 22 kindreds in The Netherlands. Am J Med. 1989;86:278-81 [PMID: 2919609]
41. Sparr JA, Bandipalliam P, Redston MS, Syngal S. Intraductal papillary mucinous neoplasm of the pancreas with loss of mismatch repair in a patient with Lynch syndrome. Am J Surg Pathol. 2009;33:309-12 [PMID: 18987546 DOI: 10.1097/PAS.0b013e3181882c3d]
42. Flanagan MR, Jayaraj A, Xiong W, Yeh MM, Raskind WH, Pillarisetty VG. Pancreatic intraductal papillary mucinous neoplasm in a patient with Lynch syndrome. World J Gastroenterol. 2015;21:2820-5 [PMID: 25759555 DOI: 10.3748/wjg.v21.i9.2820]
43. Lee SH, Kim WY, Hwang DY, Han HS. Intraductal papillary mucinous neoplasm of the ileal heterotopic pancreas in a patient with hereditary non-polyposis colorectal cancer: A case report. World J Gastroenterol. 2015;21:7916-20
44. Liu W, Shia J, Gonen M, Lowery MA, O'Reilly EM, Klimstra DS. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: frequency and clinical significance. Pancreas. 2014;43:1264-70 [PMID: 25058881 DOI: 10.1097/MPA.0000000000000190]
45. Karamurzin Y, Zeng Z, Stadler ZK, Zhang L, Ouansafi I, Al-Ahmadie HA, Sempoux C, Saltz LB, Soslow RA, O'Reilly EM, Paty PB, Coit DG, Shia J, Klimstra DS. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol. 2012;43:1677-87 [PMID: 22516243 DOI: 10.1016/j.humpath.2011.12.012]
46. Kenner BJ, Chari ST, Maitra A, Srivastava S, Cleeter DF, Go VL, Rothschild LJ, Goldberg AE. Early Detection of Pancreatic Cancer-a Defined Future Using Lessons From Other Cancers: A White Paper. Pancreas. 2016;45:1073-9 [PMID: 27518362 DOI: 10.1097/MPA.0000000000000701]
47. Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M, Petersen GM. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95-101 [PMID: 18061176 DOI: 10.1053/j.gastro.2007.10.040]
48. Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, Nio Y, Schulick RS, Bassi C, Kluijt I, Levy MJ, Chak A, Fockens P, Goggins M, Bruno M. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62:339-47 [PMID: 23135763 DOI: 10.1136/gutjnl-2012-303108]
49. Canto MI, Hruban RH, Fishman EK, Kamel IR, Schulick R, Zhang Z, Topazian M, Takahashi N, Fletcher J, Petersen G, Klein AP, Axilbund J, Griffin C, Syngal S, Saltzman JR, Mortele KJ, Lee J, Tamm E, Vikram R, Bhosale P, Margolis D, Farrell J, Goggins M. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142:796-804 quiz e14-5. [PMID: 22245846 DOI:10.1053/j.gastro.2012.01.005]
50. Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829-34 [PMID: 10784581]
51. Schmeler KM, Lynch HT, Chen LM, Munsell MF, Soliman PT, Clark MB, Daniels MS, White KG, Boyd-Rogers SG, Conrad PG, Yang KY, Rubin MM, Sun CC, Slomovitz BM, Gershenson DM, Lu KH. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261-9 [PMID:16421367 DOI: 10.1056/NEJMoa052627]
52. Grover S, Jajoo K. Screening for Pancreatic Cancer in High-risk Populations. Gastroenterol Clin North Am. 2016;45:117-27 [PMID: 26895684 DOI: 10.1016/j.gtc.2015.10.001]
53. Herreros-Villanueva M, Bujanda L. Non-invasive biomarkers in pancreatic cancer diagnosis: what we need versus what we have. Ann Transl Med. 2016;4:134. [PMID: 27162784 DOI: 10.21037/atm.2016.03.44]
54. Sohal DPS, Willingham FF, Falconi M, Raphael KL, Crippa S. Pancreatic Adenocarcinoma: Improving Prevention and Survivorship. Am Soc Clin Oncol Educ Book. 2017;37:301-310 [PMID: 28561672 DOI: 10.14694/EDBK_175222]
Author contact
![]() Corresponding author: Marta Herreros-Villanueva, Ph.D., Department of Gastroenterology, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Hospital Donostia/Instituto Biodonostia, Universidad del País Vasco UPV/EHU, San Sebastián, Spain. martahvh1978com. Telephone: +034-943-006012. Fax: +034-943-006407.
Corresponding author: Marta Herreros-Villanueva, Ph.D., Department of Gastroenterology, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Hospital Donostia/Instituto Biodonostia, Universidad del País Vasco UPV/EHU, San Sebastián, Spain. martahvh1978com. Telephone: +034-943-006012. Fax: +034-943-006407.