Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(18):3774-3784. doi:10.7150/jca.21035 This issue Cite
Research Paper
Combined Inhibitions of Glycolysis and AKT/autophagy Can Overcome Resistance to EGFR-targeted Therapy of Lung Cancer
Mingtong Ye1, 3, 4, Sufan Wang1, 3, Ting Wan1, 3, Rui Jiang1, 3, Yun Qiu1, 3, Lei Pei1, 3, Nengzhi Pang1, 3, Yuanling Huang1, 3, Yufeng Huang2, Zhenfeng Zhang2 ![]() , Lili Yang1, 3
, Lili Yang1, 3 ![]()
1. Department of Nutrition, School of Public Health, Sun Yat-Sen University, Guangzhou, Guangdong, PR China;
2. Department of Radiology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, PR China;
3. Guangdong Provincial Key Laboratory of Food, Nutrition and Health, Guangzhou, Guangdong, PR China;
4. The First Women and Children's Hospital of Huizhou, Huizhou, Guangdong, PR China.
Received 2017-5-15; Accepted 2017-8-15; Published 2017-10-17
Abstract
Efficacy of EGFR-targeted tyrosine kinase inhibitors (TKIs), such as erlotinib, to treat human non-small cell lung cancers (NSCLCs) with activating mutations in EGFR is not persistent due to drug resistance. Reprogramming in energy (especially glucose) metabolism plays an important role in development and progression of acquired resistance in cancer cells. We hypothesize that glucose metabolism in EGFR-TKI sensitive HCC827 cells and erlotinib-resistant sub-line of HCC827 (which we name it as erlotinib-resistant 6, ER6 cells in this study) is different and targeting glucose metabolism might be a treatment strategy for erlotinib-resistant NSCLCs. In this study, we found increased glucose uptakes, significant increase in glycolysis rate and overexpression of glucose transporter 1 in ER6 cells compared to its parental cells HCC827. We also found AKT and autophagy of ER6 cells were more activated than HCC827 cells after glucose starvation. Combining glucose deprivation and AKT or autophagy inhibitor could synergize and overcome the acquired resistance against EGFR-targeted therapy for NSCLCs. Our data suggest that the combinations of inhibitors of AKT or autophagy together with glucose deprivation could be novel treatment strategies for NSCLC with acquired resistance to targeted therapy.
Keywords: Drug resistance, glucose transporter 1, glycolysis, non-small cell lung cancers, autophagy, glucose deprivation.
Introduction
It had been a successful history of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, such as erlotinib and gefitinib, to treat non-small cell lung cancer (NSCLC) patients. Nevertheless, the initial expectation to treat EGFR mutant lung cancer did not last long because of acquired resistance to the inhibitor developed inevitably after a median response duration of 9 to 13 months, despite initial dramatic and rapid response to EGFR tyrosine kinase inhibitor TKIs therapy [1]. Mechanisms of resistance to TKIs in NSCLC are complex. A major acquired resistant mechanism is EGFR-T790M mutation [2, 3], other resistant mechanisms involved overexpression of AXL or MET as reported [4-6].
The Warburg effect describes the increased utilization of glycolysis rather than oxidative phosphorylation in tumor cells for their energy requirements under physiological oxygen conditions. The Warburg effect has great association with invasion, clinical stages and prognosis of cancer and drug resistance of cancer. The microenvironment of tumor is characterized by hypoxia (low oxygen) and it has the potential to inhibit tumor cell differentiation [7]. In addition, the excessive lactate produced by glycolysis creates an acidic tumor microenvironment that will promote migration and invasion of tumor [8]. Glycolysis is able to generate biosynthetic precursors (e.g. nucleic acid and amino acid) and depresses apoptosis to facilitate proliferation of tumor [9, 10].
Glucose transporter family such as GLUT1, GLUT3 and GLUT4 and glycolytic enzymes such as hexokinase2 (HK2), the first rate-limiting enzyme in the glycolytic pathway, are reported to have close correlation with chemoresistance [11]. Metabolism reprogramming of cancer cells, for example, the Warburg effect mentioned above, is a way for cancer cells to survive, while targeting this pathway can also be applied to kill cancer cells. However, targeting metabolism changes, for instance, glucose addiction therapy for different cancers, remains a controversy [12]. A study demonstrated that increased glucose metabolism in tumor cells has been associated with resistance to TKIs treatment [13]. Another study showed that resistance to a TKI-axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma [14]. We have recently explored transcriptomic-metabolomic reprogramming in EGFR-mutant NSCLC early adaptive drug resistance and showed the early adaptive drug escape linked TGFβ2-bioenergetics-mitochondrial priming [15]. We have successfully established a series of erlotinib-resistant subclonal cells (ER1-6) originated from HCC827 cells through de-sensitizing the HCC827 cells in gradually increasing erlotinib concentrations until 10μM in the culture media, and reported AXL kinase as a novel resistance molecule in ER1-5 cells [4]. However, the resistance mechanism of ER6 cells against erlotinib is still not known after thoroughly exploring series of regular high-throughput sequencings including whole genome RNA-seq and whole exome NGS. We turned to focus on metabolomics mechanism study for the ER6 cells. In this study, we explored the status of glucose metabolism in ER6 cells in contrast to HCC827 cells, investigated the underlying mechanisms of the change of glucose metabolism, and evaluated the potential application of these metabolic changes for a potential therapy of NSCLC with EGFR mutations.
Materials and Methods
Reagents
The antibodies of NOX2(ab129068), COX-2, NOX-1, GAPDH and β-actin were purchased from abcam (Cambridge, MA, USA); the antibodies of AKT(pan), phospho-AKT(Ser473), LC3B, phospho-AMPK and AMPK were purchased from Cell Signaling Technologies (Beverly, MA, USA); the antibodies of MCT4(D-1)(sc-376140) , GLUT1, GLUT3, goat anti-rabbit and goat anti-mouse were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 2-DG(2-deoxy-D-glucose) (D109194), oligomycin (O111756), nevirapine (N129779), glucose, ATP, ADP and AMP were purchased from Aladdin chemicals (Shanghai, China). PMS(P9625) was purchased from Sigma-Aldrich (Shanghai, China); MTS Reagent (G1112) was purchased from Promega Corporation (WI, USA). Seahorse XF Cell Test Kit was purchased from Seahorse Agilent technologies (Beijing, China). RPMI-1640(8116322) and glucose-free RPMI 1640(1779211) were purchased from Gibco Thermo Fisher Scientific (Guangzhou, China). Hydroxychloroquine, MK2206 and Compound C were purchased from Selleck Chemicals (Houston, TX, USA). GSH/GSSG and ROS test kits were purchased from Beyotime Biotechnology (Shanghai, China). Acetonitrile and methanol (HPLC grade) were purchased from Oceanpak (Goteborg, Sweden).
Cell culture
All cell lines were maintained in RPMI-1640 medium supplemented with 10% or 2% fetal bovine serum and 1% Penicillin & Streptomycin in incubator under 37℃, 5% carbon dioxide condition. Glucose-free medium supplemented with 10% or 2% fetal bovine serum and 1% Penicillin & Streptomycin was used in the glucose deprivation assays.
Cell viability assay
Cells were plated to a 96-well plate in 100 microliter of full medium. When cell growth reached 50% confluence, fresh medium or medium supplemented with special drugs replaced original medium. Cells were washed with PBS for three times. One hundred microliter of fresh medium was added, then 20 microliter of MTS-PMS mixed solution was added into 96-well plate. After incubated in incubator under 37℃, 5% carbon dioxide condition for 1 to 4 hours, absorbance was measured in a microplate reader with the wave length of 490 nanometer.
Cell survival test
Cells were plated to a 6-well plate and 1 milliliter of full medium was added to each well. When cell growth reached 50% confluence, fresh medium or medium supplemented with special drugs replaced the old medium. Photos were taken under light microscope.
Protein expression assays
Protein was isolated from cells using RIPA with 10μM PMSF. Lysates were standardized for protein content and protein was separated by SDS-polyacrylamide gel electrophoresis and transferred onto NC membranes using the BIO-RAD system. The NC membranes were blocked with 5% non-fat milk in PBST for one hour and then washed out non-fat milk with PBST buffer. Primary antibody was diluted in PBST and incubated overnight at 4℃ condition. The following day, blots were washed with PBST buffer and incubated with goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibody (Santa Cruz) for one hour at room temperature. After washing in PBST buffer, the immune-reactive proteins were visualized using ECL (Thermo Fisher Scientific).
Extracellular flux analysis (OCR and ECAR)
The XF24 extracellular flux analyzer (Seahorse Biosciences Agilent technologies) is a fully integrated 24-well instrument that measures in real time the uptake and release of metabolic end products. Each XF24 assay well contains a disposable sensor cartridge, embedded with 24 pairs of fluorescent biosensors (oxygen and pH), coupled to fiber-optic wave guides. This technology was used to measure ECAR expressed in mpH/min and OCR in pmol/min in two kinds of cell lines. Each well plated with 50,000 cells and then cultured overnight in incubator within a 37℃, 5% carbon dioxide condition. OCR and ECAR analysis followed the instructions of Seahorse Analyzer. After measurement was completed, the 24-well plate was washed for 3 times with PBS carefully and then 6 mol/L NaOH was added to lysate cells at 70℃. Total protein for each well was quantified by BCA method.
Electron microscope study
All cells were collected with trypsin when cell growth reached 90% confluence. Then the cells were centrifuged at 1000g for 5 minutes at room temperature. Cell pellets were washed for three times with PBS. Then cells were fixed with glutaraldehyde fixation reagent, and analyzed by electron microscope core of Zhongshan medical school of Sun Yat-sen University.
ROS measurement
Cells were cultured in 24-well plate. When cell growth reached 80% confluence, cells were washed for three times with PBS and then 300μL of FBS-free RPMI-1640 medium with DCFH-DA was added. Then the 24-well plate was placed in incubator within a 37℃, 5% carbon dioxide condition for 20 minutes. After that, cells were washed for three times with 300μL FBS-free RPMI-1640 medium to get rid of extracellular DCFH-DA. Fluorescence intensity was measured in a fluorescence microplate reader with wave length of 488nm/525nm. Cell lysates were collected with 50μL of 6 mol/L NaOH at 70℃. Total protein for each well was quantified by BCA method.
GSH and GSSG measurement
When cell growth reached 80% confluence, cells were washed for three times with PBS and collected after trypsinization. The measurement procedures followed the guideline of manufacturer.
Measurement of energy metabolites, lactic acid and glucose by UPLC-TOF
Cells were cultured in 6-well plate and metabolites were collected when they reached 80% confluence. Cells were washed for three times with PBS, then 400μL of ice-cold 80% (v/v) methanol/water was added to 6-well plate (the concentration of the internal standard nevirapine is 0.02µmol/L). The metabolites were collected with cell scrapers. 6-well plate was washed for another 2 times with 200μL ice-cold 80% (v/v) methanol/water. Then cell lysates were combined to a 2mL Eppendorf tube and homogenized by a high throughput tissue lyser. The lysates were centrifuged at 12000g for 20 minutes at 4℃. Supernatants were collected and transferred into a new tube and solvents were evaporated at 30℃ in vacuum dryer. The metabolites were resolved in ice-cold 150μL chloroform/water (v/v, 2:1) and mixed for 30 seconds with vortex mixer. After 5 minutes suspension, the collection was centrifuged at 680g for 20 minutes at 4℃. The upper water solution was collected for LC-MS detection following the method as described previously (Trammell et al., 2013). Lactic acid in culture medium was analyzed by LC/MS method as described previously (Goudarzi M, et al., 2015), and used a platform as the same to metabolites measurement above. Lactic acid measurement was performed by gradient chromatography on a Poroshell 120 EC-C18 column (Agilent) with mobile phases (A) water and 0.1% formic acid, and (B) 100% acetonitrile. Glucose in culture medium was analyzed as described previously (Chen et al., 2012). Glucose analysis was performed by aqueous neutral phase gradient chromatography on a Diamond Hydride column (Microsolv) with mobile phases (A) 50% isopropanol, containing 0.025% acetic acid, and (B) 90% acetonitrile containing 5 mmol/L ammonium acetate. All raw data were collected and analyzed by Agilent MassHunter Qualitative software.
Statistical analysis
Mean values and SD were calculated by GraphPad Prism 5 software. The analysis of variance (ANOVA) was applied to identify statistical differences between experimental factors by SPSS17.0. P<0.05 was considered to have statistical significance.
Results
ER6 cells rely more on glycolysis to utilize glucose and have much less normal mitochondria than their parental cells HCC827
ER6 and HCC827 cells were both cultured overnight and were applied to Seahorse XF24 Extracellular Flux Analyzer (Seahorse Biosciences, Agilent Technologies) to measure oxygen consumption rate (OCR) and extracellular acid rate (ECAR). When glucose was added into XF24 assay plate under glycolysis measurement procedure, the increased rate of OCR and EACR of ER6 cells was higher than HCC827 cells, which indicates that ER6 cells have higher glycolysis potential and oxygen consumption when glucose was added (Figure 1A, 1B). When oligomycin, an ATP synthase inhibitor, was added, the OCR of HCC827 cells decreased while the OCR of ER6 cells did not change (Figure 1B), suggesting that there might be mitochondrial dysfunction in ER6 cells. Decreased OCR was accompanied by the increase of ECAR of HCC827 cells, while no change of ECAR in ER6 cells was observed (Figure 1A), which suggests that ER6 cells depend more on glycolysis than HCC827 cells to maintain survival.
ER6 cells rely more on glycolysis to utilize glucose and have much less normal mitochondria than their parental cells HCC827. A. ER6 and HCC827 cells were plated on Seahorse plate, and cultured for 24 hrs. Extracellular acidification rate analysis (ECAR) was performed according to the instructions of the Seahorse XF24 glycolysis measurement kit. Three chemicals were injected to culture medium according to the time schedule, that is, glucose (10mM), oligomycin (1μM), and 2-DG (2-Deoxy-D-glucose, 100mM). ** p<0.01 comparing to HCC827 group. B. Oxygen consumption rate (OCR) of ER6 and HCC827 cells. ** p<0.01 comparing to HCC827 group C. Morphology of HCC827 and ER6 cells under electron microscope. Panel a&b: images on 1700x magnitude, black arrows point to mitochondria. Panel c&d: images on 7800x magnitude, yellow arrows point to mitochondria.
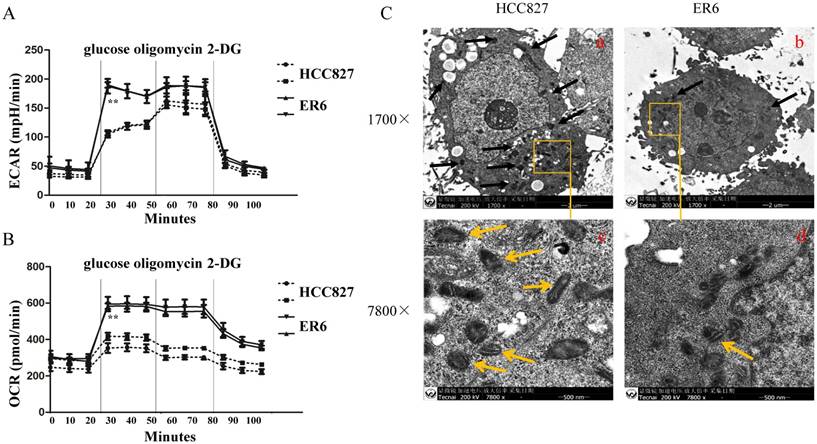
To further confirm our findings that ER6 cells depend on glycolysis to survive with mitochondria dysfunctions, we observed the number and structure of mitochondria of HCC827 and ER6 cells under transmission electron microscope (TEM). ER6 cells have far less mitochondria compared to HCC827 cells (Figure 1C-a, 1C-b, on the top). Moreover, the structure of most mitochondria in ER6 cells was not as intact as in HCC827 cells, which had become smaller and lost their normal ridges (as shown on the bottom of Figure 1C-c and 1C-d). This indicates that ER6 cells had to rely on glycolysis to supply energy due to the loss of numbers and structure of their mitochondria.
ER6 cells have higher oxidative stress and consume more glucose than their parental cells HCC827, possibly via the increased uptake of glucose through GLUT1
Considering the fact that ER6 cells have higher OCR with mitochondria dysfunction, this would cause proton leak and produce more reactive oxygen species (ROS). We hence measured total ROS of ER6 and HCC827 cells and found ER6 cells produce more ROS than HCC827 cells (Figure 2A). We also found that ER6 cells consumed more GSH than HCC827 cells (Figures 2B) to fight against oxidative stress.
ER6 cells have higher oxidative stress and consume more glucose than their parental cells HCC827, possibly via the increased uptake of glucose through GLUT1. A. Intracellular level of ROS of HCC827 and ER6 cells. ** p<0.01 comparing to HCC827 group. B. Ratio of intracellular GSH to GSSG, ** p<0.01 comparing to HCC827 group. C. Glucose abundance in the medium, normalized by the third day's total proteins. 1 mL full medium was added into 6-well plates, 50uL medium was collected in the first, second and the third day for detection. ** represents p<0.01 comparing to HCC827 group. D. Lactic acid abundance in the medium, normalized by total proteins in 48th hour. 1 mL full medium was added into 6-well plates, 50uL medium was collected in the second day. ** representsp<0.01 comparing to HCC827 group E&F. Protein expressions of GLUT1, MCT-4 and GLUT3 of ER6 and HCC827 cells analyzed by western blot.
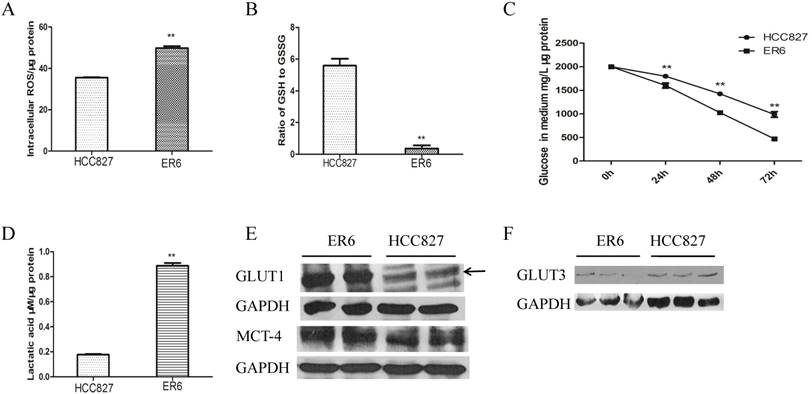
Based on the observation that the glycolysis rate of ER6 cells is higher than HCC827 cells, we analyzed relative glucose concentration in the medium by UPLC-TOF. It was observed that ER6 cells consumed glucose much quicker and produced more lactic acid than HCC827 cells (Figure 2C, 2D). Glucose was transported into the cells via glucose transporters. The expression of GLUT1 protein in ER6 cells was observed to be higher than HCC827 cells (Figure 2E). Nevertheless, the expression of another major glucose transporter GLUT3 had no difference between ER6 and HCC827 cells (Figure 2F). Meanwhile, monocarboxylate transport 4 (MCT-4), an important lactate transporter, overexpressed in ER6 cells (Figure 2D), which was in line with the rapid acid production of ER6 cells after adding 10mM glucose to the analysis medium (Figure 1A). Deprivation of glucose and applying GLUT1 specific inhibitor STF-31 are able to depress ROS production of ER6 cells (supplement Figure S1A, S1B). In summary, ER6 cells have higher glucose uptake by overexpression of GLUT1, higher glucose metabolism via glycolysis, and higher oxidative stress than HCC827 cells.
ER6 cells rely more on glucose to survive and to keep energy homeostasis than their parental cells HCC827
Based on the fact that ER6 cells have higher glycolysis rate and are able to consume glucose more rapidly than HCC827 cells, we started to question if glucose deprivation or metabolism inhibition could inhibit ER6 cells viability selectively. ER6 cells started to die in large scale on the second day in medium without glucose even though 10% fetal bovine serum (FBS) was supplied, while the growth of HCC827 just merely affected, which means ER6 cells are more susceptible to glucose deprivation (Figure 3A). We used 2-Deoxy-D-glucose(2-DG), a glycolysis inhibitor, to inhibit glucose metabolism in ER6 cells and HCC827 cells. ER6 cells are less viable than HCC827 cells at low level of 2-DG treatment (Figure 3B).
ER6 cells rely more on glucose to survive and to keep energy homeostasis than their parental cells HCC827. A. Images of cell survival from day 1-3 under optical microscopy (10×40X); Cells were cultured in medium with glucose supplemented with 10% FBS and 1% P/S (10%FBS G+ medium), or medium without glucose supplemented with 10% FBS and 1% P/S (10%FBS G- medium) for 3 days. B. Viability analysis of ER6 and HCC827 cells with supplement of 2-deoxy-D-glucose (2-DG) for 48 hrs. ** representsp<0.01 comparing to control group. C-E. Intracellular abundance of ATP(C), ADP(D) and AMP (E) in ER6 and HCC827 cells. ** representsp<0.01 comparing to corresponding HCC827 group; ***representsp<0.001 comparing to corresponding HCC827 group.
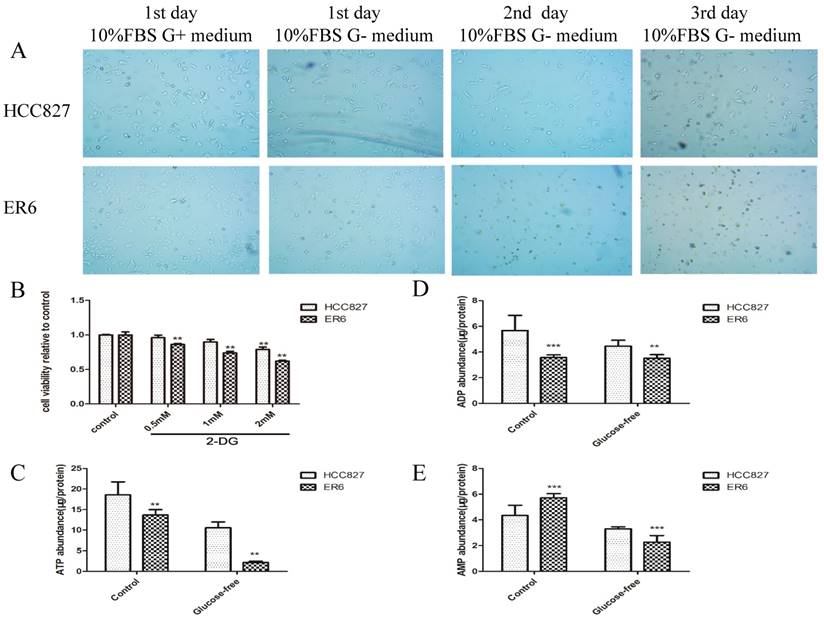
It is well known that the ATP production efficiency of glycolysis is much lower than tricarboxylic acid cycle. We have observed that ER6 cells have much higher glycolysis rate, so we wonder whether energy homeostasis in ER6 cells was destroyed. We analyzed the content of ATP, ADP and AMP of ER6 cells cultured in normal glucose medium or glucose-free medium. Interestingly, the content of ATP and ADP are lower in ER6 cells than that in HCC827 cells cultured in normal glucose medium (Figure 3C, 3D, control group), while the content of AMP is higher in ER6 cells than HCC827 cells cultured in normal glucose medium (Figure 3E, control group). In cells cultured in glucose free medium, ATP content of ER6 cells decreased hastily comparing with cells cultured in normal glucose medium (Figure 3C). This suggests that less viability of ER6 cells under inhibition of glycolysis might due to insufficient ATP production.
Combining glucose deprivation and autophagy inhibitor decreases viability of ER6 cells
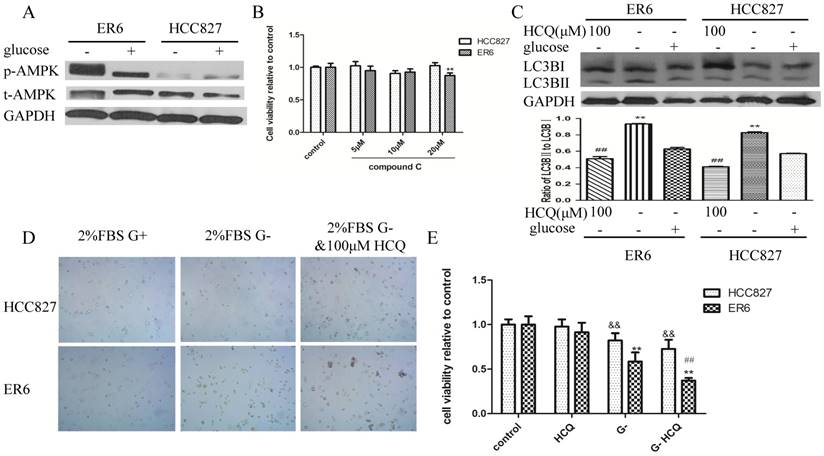
Adenosine 5-monophosphate (AMP)-activated protein kinase (AMPK) can be induced by glucose deprivation which can also cause autophagy [16, 17]. We wondered whether activated AMPK and autophagy were the underlying mechanisms to maintain energy homeostasis for ER6 cells in glucose-free medium. When glucose was deprived, ER6 cells activated AMPK significantly, while HCC827 cells did not (Figure 4A). We applied compound C, an inhibitor of AMPK phosphorylation, to depress activation of AMPK of ER6 cells in glucose-free medium. We found compound C was able to decrease the cell viability of ER6 cells (Figure 4B). Meanwhile, glucose deprivation induced autophagy in both ER6 and HCC827 cells (Figure 4C). Therefore we applied hydroxychloroquine (HCQ), an autophagy inhibitor, to block autophagy combined with glucose starvation (Figure 4C, 4D, 4E). Interestingly, we found that autophagy inhibitor could selectively decrease the viability of ER6 cells with glucose deprivation, while autophagy inhibitor per se had no effect on both ER6 and HCC827 cells (Figure 4E). Our data indicate that combination of autophagy inhibitor together with glucose deprivation could decrease cell viability of ER6 cells.
Combining glucose deprivation and autophagy inhibitor decreases viability of ER6 cells. A. Western blot analysis of protein levels of p-AMPK and t-AMPK after 6 hrs glucose deprivation in both ER6 and HCC827 cells. B. Treatment with compound C for 24 hrs in glucose-free medium only slightly decrease cell viability of ER6 cells. Control, medium without glucose supplemented with 2%FBS, and 1% P/S, ** represents p<0.01 comparing to ER6 control group. C. Western blot analysis of levels of LC3B in response to glucose deprivation and/or HCQ (hydroxychloroquine, 100μM) after 6hrstreatment in ER6 and HCC827cells. D. Images of cell growth under optical microscopy (10 ×40) after 12 hrs treatment with G+ medium (medium with glucose + 2%FBS; G- medium(medium without glucose + 2% FBS); G-+HCQ (100μM). E. Cell viability of ER6 and HCC827 cells treated with G+, HCQ, G-, or G-+HCQ after 12 hrs treatment. Control, medium with glucose +2%FBS; HCQ, medium with glucose+2%FBS+100 μM hydroxychloroquine; G-, medium without glucose +2% FBS; G- HCQ, medium with no glucose + 2% FBS + 100 μM hydroxychloroquine, ** represents p<0.01 comparing to ER6 control group, ## represents p<0.01 comparing to ER6 G- group, &&represents p<0.01 comparing to HC827 control group.
Combining glucose deprivation and AKT inhibitor decreases viability of ER6 cells
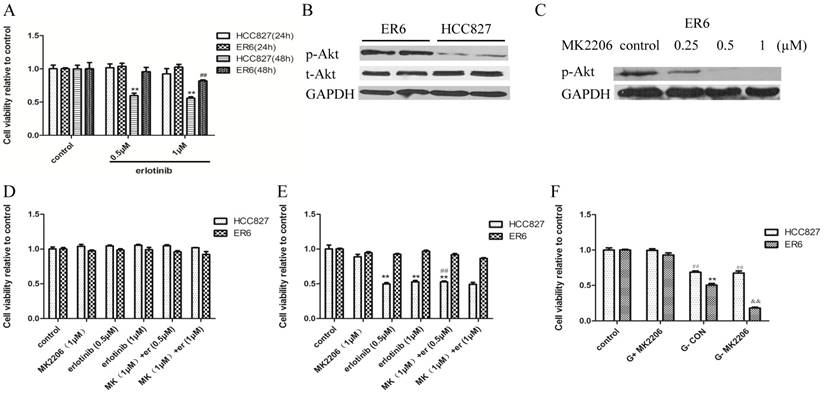
ER6 cells are resistant to erlotinib, we wondered whether glucose starvation can reverse ER6 cells' resistance to erlotinib. Unfortunately, we found that glucose starvation did not reverse ER6 cells' sensitivity to erlotinib significantly (Figure 5A).We found that AKT was highly phosphorylated in ER6 cells (Figures 5B). We therefore applied MK2206, an AKT inhibitor, to suppress AKT phosphorylation in ER6 cells (Figure 5C). Even though we did not found MK2206 and erlotinib had combined effects in normal glucose medium (Figure 5D, 5E), we found that inhibiting AKT in the condition of glucose deprivation was able to decrease viability of ER6 cells (Figure 5F). These data confirm that combing glucose deprivation and AKT inhibition decreases viability of ER6 cells.
Combining glucose deprivation and Akt inhibitor decreases viability of ER6 cells. A. Cell viability of ER6 and HCC827 cells treated with increasing concentration of erlotinib in glucose-free medium after 24 and 48 hrs treatment. ** represents p<0.01 comparing to HCC827(48h) control group. B. Western blot analysis of protein levels of p-Akt and t-Akt of ER6 and HCC827 cells. C. Dose-dependent inhibition of Akt activation in ER6 cells treated by p-Akt specific inhibitor, MK2206. D. Cell viability of ER6 and HCC827 cells treated with MK2206 and erlotinib after 24 hrs treatment. E. Cell viability of ER6 and HCC827 cells treated with MK2206 and erlotinib after 48hrs treatment. ** representsp<0.01 comparing to HCC827 control group; ## p>0.05 comparing to HCC827 erlotinib (0.5 μM) group. F. Cell viability of ER6 and HCC827 cells treated with MK2206 and glucose starvation after 24 hrs treatment. ** represents p<0.01 comparing to ER6 control group, ## represents p<0.01 comparing to HCC827 control group, && represents p<0.01 comparing to ER6 G-CON group. Control, medium with glucose supplemented with 2% FBS and 1% P/S; G-CON, medium without glucose supplemented with 2%FBS, and 1% P/S.
Discussion
In 1924, Otto Warburg reported that cancer cells used glycolysis more than mitochondrial oxidative phosphorylation (OXPHOS) to meet their energy requirements [11]. Over the decades, a better understanding to this phenomenon has developed. Recent reports have emerged that glycolysis also has a strong correlation with reprogramming in glycolytic activity of drug resistance to overcome chemotherapy in pancreatic adenocarcinoma, cancer-associated fibroblasts, breast cancer, lung cancer and prostate cancer [14,18-20]. Glucose transporter family such as GLUT1, GLUT3 and GLUT4 and some key glycolytic enzymes such as HK2, the first rate-limiting enzyme in the glycolytic pathway, are reported to have tight link with chemoresistance [11]. To have a better understanding of HCC827 cells and ER6 cells in metabolism, we measured oxygen consumption rate (OCR) and extracellular acid rate (ECAR), which indicate the activity of mitochondrial oxidative phosphorylation and glycolysis respectively. We observed that drug resistant ER6 cells had higher glycolysis rate and lower mitochondria potential ability than HCC827 cells. We found that GLUT1 overexpressed in ER6 cells than HCC827 cells. One study reported that multi-drug resistance (MDR) in human tumor cells overexpressed GLUT1 and had a slightly higher expression level of hexokinase 2 (HK2), GAPDH and lactate dehydrogenase (LDH) in the MDR cells, which all of the four proteins are the key glycolytic proteins [21]. Recently, data showed that acute myeloid leukemia (AML) drug resistant cell lines overexpressed GLUT1 and HK2 compared with parental cells and serum LDH level in AML patients was higher than healthy people [22]. Hence, these data support our finding that increasing glycolysis with GLUT1 overexpression is a mechanism to overcome chemotherapy for ER6 cells. Other mechanisms might also be related to the high glycolysis rate of ER6 cells. GLUT1 was reported to have close connection with glycolysis and glucose uptakes when it was up-regulated in plenty of malignancies [23, 24]. Although GLUT3, another member of glucose transporter family, was reported to be significantly increased in brain tumors [25], and was up-regulated in glioblastoma cells exposed to temozolomide, which led to acquired resistance to the drug [26], we did not find increased expression of GLUT3 in ER6 cells, which suggests that GLUT3 might not play a vital role in the drug resistance effect of ER6 cells. ER6 cells consumed glucose much quicker than HCC827 did, ER6 cells generated more lactic acid, the final production of glycolysis, than HCC827 cells. As expected, ER6 cells overexpressed lactate transporter MCT-4, which transports lactic acid to the culture medium. This is similar as reported in other's research that drug resistant cells with higher glycolysis produce more lactic acid and/or overexpressed MCT-4 [14, 27-30]. Our data indicated that ER6 cells up-regulated GLUT1 and MCT-4 to increase the uptake of glucose and transportation of lactic acid through the membrane, which was observed as increased glycolysis rate in ER6 cells.
Data showed that translocation of GLUT1 was attributed to the phosphorylation of Protein Kinase B (p-AKT), and resistance to axitinib could be inhibited by a specific inhibitor of p-AKT (MK2206) in pancreatic adenocarcinoma cells [14]. Another research indicated that p-AKT stimulates aerobic glycolysis in cancer cells [31] and MiR-128 could regulate glycolysis by inhibiting AKT phosphorylation [32]. All data above suggest that AKT phosphorylation might contribute to drug resistance of cancer cells via regulating glycolysis. In our data, we found ER6 cells overexpressed p-AKT, and MK2206 can achieve inhibition of expression of p-AKT dose dependently. However, inhibition of p-AKT was not able to inhibit cell viability of ER6 cells. Researches had shown that hypoxia-inducible factor 1α (HIF-1α) is associated with the increased expression of GLUT1 [33]. It suggested that ER6 cells with destructive mitochondria might up-regulate GLUT1 to overcome hypoxia microenvironment, and this effect might not via AKT signaling pathway.
We observed under transmission electron microscope (TEM) that ER6 cells have far less normal mitochondria than their parental cells HCC827. That is coincident to the phenomenon that ER6 cells are not sensitive to oligomycin, an ATP synthase inhibitor, in ECAR measurement procedure by Seahorse bioanalyzer. The primary metabolic function of mitochondria is oxidative phosphorylation to synthesize ATP for energy requirement. Otto Warburg first hypothesized that increased rates of aerobic glycolysis observed in a variety of tumor cell types might be due to an impaired respiratory capacity [34]. We confirmed that ER6 cells up-regulate their glycolysis capacity under the condition of mitochondria dysfunction to overcome chemotherapy. In addition, mitochondria are the primary source of intracellular ROS generation via the electron transport dysfunction and reacting with oxygen to generate superoxide where complex I and complex III are the main sources for this production [35]. We found that oxygen consumption rate of ER6 cells was higher than HCC827 cells and ER6 cells were not sensitive to oligomycin which was mentioned above. We also found the production of ROS in ER6 cells was increased, which might come from dysfunctional mitochondria. Oxygen was consumed in many other ways besides mitochondria, including cyclooxygenase 2 (COX-2), NADPH oxidase (NOX) 1 and 2, etc [36-38]. Unfortunately, we did not find apparent up-regulation of NADPH oxidase family, and we found the down-regulated expression of COX-2 in ER6 cells (supplement figure S1C, S1D, S1E) instead. In our study, ER6 cells have to rely more on glycolysis to maintain energy homeostasis due to mitochondrial dysfunction. Mitochondria dysfunction induces electron leak and consumes oxygen to generate ROS. Therefore, higher ROS production might be, or in part, the reason of higher oxygen consumption of ER6 cells than HCC827 cells. To further verify our hypothesis, we measured ROS after applying glucose deprivation and GLUT1 inhibitor STF-31. Both glucose deprivation and GLUT1 inhibition were able to reduce ROS production (supplement figure S1A, S1B). This is consistent to the data that glucose deprivation impaired mitochondrial oxygen consumption in HCC cells [39]. In summary, higher glycolysis with mitochondria dysfunction of ER6 cells generated more ROS than HCC827 cells and consumed more oxygen than HCC827 cells.
Some research showed that cancer cells with higher glycolysis rate were more sensitive to glycolysis inhibition by 2-DG [14, 39-43]. In addition, glucose deprivation was reported to have important anticancer effects [39, 44, 45]. Considering ER6 cells have higher glycolysis rate than HCC827 cells, we then applied 2-DG, or glucose starvation to both HCC827 and ER6 cells to measure their effects on cell survival. It was observed that ER6 cells were more sensitive to both glycolysis inhibitor 2-DG and glucose starvation than HCC827 cells. It was reported that inhibition of glycolysis inhibits energy production, and glucose deprivation impaired ATP generation in HepG2 cells [39]. To further uncover the energy equilibrium of ER6 cells and HCC827 cells after glucose starvation, we measured intracellular content of ATP, ADP and AMP cultured both in normal glucose medium and glucose-free medium. Energy generation efficiency of ER6 cells is lower than HCC827 cells both in normal glucose medium and glucose-free medium and ER6 cells rely more on glucose than HCC827. This confirms that deficiency of energy under glucose deprivation is one cause of decreased viability of ER6 cells.
Besides glucose deprivation, we also explored whether other factors regulating glucose metabolism can affect viability of ER6 cells. It was reported that there was anti-cancer synergy of dichloroacetate (DCA) and EGFR tyrosine kinase inhibitors in NSCLC cell lines [46]. DCA is an inhibitor for pyruvate dehydrogenase kinase (PDK) which is a critical enzyme inversely regulating glucose metabolism. In order to explore whether glucose deprivation can reverse the resistance to erlotinib of ER6 cells, we combined glucose deprivation and erlotinib to explore if similar synergy effects existed in ER6 cells. We did not find an obvious reversible outcome of ER6 cells compared to HCC827 in our study. This inconsistency might be due to the different effect of PDK inhibition to completely glucose deprivation, the former might be able to adaptively activate glycolysis. Data had shown that glucose up-regulated activation of the AKT pathway [47] and site specific activation of AKT protects cells from death induced by glucose deprivation [48]. This supports our data that combined glucose deprivation with AKT inhibitor could decrease viability of ER6 cells significantly.
In recent decades, autophagy raised great attention in cancer study. Basal autophagy levels are low in normal situation but are profoundly stimulated by calorie restriction. Cancer cells up-regulate autophagy when specific stimulations were given and can be more autophagy-dependent than normal cells [49]. Some studies proved that glucose deprivation could induce autophagy [50-53], and cancer cells can utilize autophagy to maintain survival during glucose starvation or other calorie restriction [49]. We found that glucose deprivation activated AMPK and autophagy for ER6 cells. Hence, we combined glucose deprivation with autophagy inhibitor (hydroxylchloroquine) and found that this can efficiently decrease viability of ER6 cells.
In summary, we found that ER6 cells have mitochondrial dysfunction and rely more on glycolysis for survival than HCC827 cells. We also demonstrated that combined treatment with autophagy/AKT inhibitor and glucose deprivation can selectively decrease viability of ER6 cells than its parental HCC827 cells. In conclusion, targeting glycolysis pathway and blocking autophagy/AKT, are potential strategies against acquired resistance to TKIs in NSCLCs' targeted therapies.
Abbreviations
TKIs: tyrosine kinase inhibitors; NSCLCs: non-small cell lung cancers; GLUT: glucose transporter; ECAR: extracellular acidification rate; OCR: oxygen consumption rate; 2-DG: 2-deoxygen glucose; GSH: glutathione; GSSG: oxidized glutathione; MCT-4: mono-carboxylate transporter 4; AKT: protein Kinase B; p-AKT: phosphorylation protein Kinase B; ROS: reactive oxygen species; AMPK: adenosine 5'-monophosphate (AMP)-activated protein kinase; p-AMPK: phosphorylation AMPK; LC3: microtubule-associated protein 1 light chain; HCQ: hydroxychloroquine.
Supplementary Material
Figure S1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.81370528 and 81573142 to Lili Yang; No.81372274 and 81461168028 to Zhenfeng Zhang), the Science and Technology Planning Project of Guangdong Province (2014A030313033 to Zhenfeng Zhang). We greatly appreciate Prof. Peng Huang (Cancer Center of Sun Yat-sen University) for the great help from his research team in Seahorse Analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol Therapeut. 2017;174:1-21
2. Kobayashi S, Boggon TJ, Dayaram T. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New Engl J Med. 2005;352:786-792
3. Ohashi K, Maruvka YE, Michor F. et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistant Disease. J Clin Oncol. 2013;31:1070-1080
4. Zhang Z, Lee JC, Lin L. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852-860
5. Wu X, Liu X, Koul S. et al. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546-9563
6. Engelman JA, Zejnullahu K, Mitsudomi T. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-1043
7. Kim Y, Lin Q, Glazer PM. et al. Hypoxic Tumor Microenvironment and Cancer Cell Differentiation. Curr Mol Med. 2009;9:425-434
8. Hirschhaeuser F, Sattler UGA, Mueller-Klieser W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011;71:6921-6925
9. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324:1029-1033
10. Tomiyama A, Serizawa S, Tachibana K. et al. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J Natl Cancer Inst. 2006;98:1462-1473
11. Bhattacharya B, Omar MFM, Soong R. The Warburg effect and drug resistance. Brit J Pharmacol. 2016;173:970-979
12. Granja S, Pinheiro C, Reis RM. et al. Glucose Addiction in Cancer Therapy: Advances and Drawbacks. Curr Drug Metab. 2015;16:221-242
13. Butler EB, Zhao Y, Munoz-Pinedo C. et al. Stalling the Engine of Resistance: Targeting Cancer Metabolism to Overcome Therapeutic Resistance. Cancer Res. 2013;73:2709-2717
14. Hudson CD, Hagemann T, Mather SJ. et al. Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death and Disease. 2014;5:e1160
15. Thiagarajan PS, Wu X, Zhang W. et al. Transcriptomic-metabolomic reprogramming in EGFR-mutant NSCLC early adaptive drug escape linking TGFβ2-bioenergetics-mitochondrial priming. Oncotarget. 2016;7:82013-82027
16. Wu S, Wei Y. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: Implication of the cell survival in mitochondrial diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2012;1822:233-247
17. Wu N, Zheng B, Shaywitz A. et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167-1175
18. Yu T, Yang G, Hou Y. et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2016;36:2131-2145
19. Ruprecht B, Zaal EA, Zecha J. et al. Lapatinib Resistance in Breast Cancer Cells Is Accompanied by Phosphorylation-Mediated Reprogramming of Glycolysis. Cancer Res. 2017;77:1842-1853
20. Aldonza MBD, Hong J, Lee SK. Paclitaxel-resistant cancer cell-derived secretomes elicit ABCB1-associated docetaxel cross-resistance and escape from apoptosis through FOXO3a-driven glycolytic regulation. Experimental & Molecular Medicine. 2017;49:e286
21. Milane L, Duan Z, Amiji M. Role of hypoxia and glycolysis in the development of multi-drug resistance in human tumor cells and the establishment of an orthotopic multi-drug resistant tumor model in nude mice using hypoxic pre-conditioning. Cancer Cell Int. 2011;11:3
22. Song K, Li M, Xu X. et al. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett. 2016;12:334-342
23. Arcila ME, Nafa K, Chaft JE. et al. EGFR Exon 20 Insertion Mutations in Lung Adenocarcinomas: Prevalence, Molecular Heterogeneity, and Clinicopathologic Characteristics. Mol Cancer Ther. 2013;12:220-229
24. Halasi M, Wang M, Chavan TS. et al. ROS inhibitor N-acetyl-l-cysteine antagonizes the activity of proteasome inhibitors. Biochem J. 2013;454:201-208
25. Flavahan WA, Wu Q, Hitomi M. et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373-1382
26. Le Calve B, Rynkowski M, Le Mercier M. et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia. 2010;12:727-739
27. Chaube B, Malvi P, Singh SV. et al. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget. 2015;6:37281-37299
28. Woo YM, Shin Y, Lee EJ. et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLOS ONE. 2015;10:e132285
29. Hardeman KN, Peng C, Paudel BB. et al. Dependence On Glycolysis Sensitizes BRAF-mutated Melanomas For Increased Response To Targeted BRAF Inhibition. Sci Rep. 2017;7:42604
30. Martel F, Guedes M, Keating E. Effect of polyphenols on glucose and lactate transport by breast cancer cells. Breast Cancer Res Treat. 2016;157:1-11
31. Elstrom RL, Bauer DE, Buzzai M. et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004;64:3892-3899
32. Yang J, Li J, Le Y. et al. PFKL/miR-128 axis regulates glycolysis by inhibiting AKT phosphorylation and predicts poor survival in lung cancer. Am J Cancer Res. 2016;6:473-485
33. Fan R, Hou W, Zhao Y. et al. Overexpression of HPV16 E6/E7 mediated HIF-1α upregulation of GLUT1 expression in lung cancer cells. Tumor Biol. 2016;37:4655-4663
34. Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Mitochondrion. 2013;4:755-762
35. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426-431
36. Srivastava S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin Transl Med. 2016;5:25
37. Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895-2899
38. Yu HA, Arcila ME, Rekhtman N. et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res. 2013;19:2240-2247
39. Zhou J, Tang Y, Xie ZJ. et al. AKT activation was not essential for hepatocellular carcinoma cell survival under glucose deprivation. Anticancer Drugs. 2017;28:427
40. Park GB, Chung YH, Kim D. 2-Deoxy-D-glucose suppresses the migration and reverses the drug resistance of colon cancer cells through ADAM expression regulation. Anti-Cancer Drug. 2017;28:410-420
41. Reyes R, Wani NA, Ghoshal K. et al. Sorafenib and 2-Deoxyglucose Synergistically Inhibit Proliferation of Both Sorafenib-Sensitive and -Resistant HCC Cells by Inhibiting ATP Production. Gene Expression. 2017;17:129-140
42. Huang A, Ju HQ, Liu K. et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016;377:149-157
43. Graham RM, Hernandez F, Puerta N. et al. Resveratrol augments ER stress and the cytotoxic effects of glycolytic inhibition in neuroblastoma by downregulating Akt in a mechanism independent of SIRT1. Experimental & Molecular Medicine. 2016;48:e210
44. Pasto A, Pagotto A, Pilotto G. et al. Resistance to glucose starvation as metabolic trait of platinum-resistant human epithelial ovarian cancer cells. Oncotarget. 2017;8:6433-6445
45. Lashinger LM, O Flanagan CH, Dunlap SM. et al. Starving cancer from the outside and inside: separate and combined effects of calorie restriction and autophagy inhibition on Ras-driven tumors. Cancer & Metabolism. 2016;4:18
46. Yang Z, Tam KY. Anti-cancer synergy of dichloroacetate and EGFR tyrosine kinase inhibitors in NSCLC cell lines. Eur J Pharmacol. 2016;789:458-467
47. Liu PP, Liao J, Tang ZJ. et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014;21:124-135
48. Gao M, Liang J, Lu Y. et al. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene. 2013;33:745-755
49. Guo JY, White E. Autophagy, Metabolism, and Cancer. Cold Spring Harbor Symposia on Quantitative Biology. 2017;81:73-78
50. Brisson L, Banski P, Sboarina M. et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell. 2016;30:418-431
51. Nuschke A, Rodrigues M, Wells AW. et al. Mesenchymal stem cells/multipotent stromal cells (MSCs) are glycolytic and thus glucose is a limiting factor of in vitro models of MSC starvation. Stem Cell Res Ther. 2016;7:179
52. Parker AL, Turner N, McCarroll JA. et al. βIII-Tubulin alters glucose metabolism and stress response signaling to promote cell survival and proliferation in glucose-starved non-small cell lung cancer cells. Carcinogenesis. 2016;37:787-798
53. Moruno-Manchón JF, Pérez-Jiménez E, Knecht E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem J. 2013;449:497-506
Author contact
![]() Corresponding authors: Zhenfeng Zhang, 250 Changgang Rd East, Guangzhou, 510260, China; E-mail: zhangzhfedu.cn; and Lili Yang, 74 zhongshan road2, Guangzhou, Guangdong, PR China; E-mail: yangll7sysu.edu.cn
Corresponding authors: Zhenfeng Zhang, 250 Changgang Rd East, Guangzhou, 510260, China; E-mail: zhangzhfedu.cn; and Lili Yang, 74 zhongshan road2, Guangzhou, Guangdong, PR China; E-mail: yangll7sysu.edu.cn