Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(19):3916-3932. doi:10.7150/jca.20779 This issue Cite
Research Paper
Modification of proteolytic activity matrix analysis (PrAMA) to measure ADAM10 and ADAM17 sheddase activities in cell and tissue lysates
Toshie Yoneyama1,6, Michael Gorry1,6, Miles A Miller9, Autumn Gaither-Davis5, Yan Lin3, Marcia L. Moss7, Linda G. Griffith8, Douglas A. Lauffenburger8, Laura P. Stabile4, James G. Herman5, Nikola L. Vujanovic1,2,6 ![]()
1. Department of Pathology, University of Pittsburgh Cancer Institute, Pittsburgh, PA;
2. Department of Immunology, University of Pittsburgh Cancer Institute, Pittsburgh, PA;
3. Department of Biostatistics, University of Pittsburgh Cancer Institute, Pittsburgh, PA;
4. Department of Pharmacology and Chemical Biology, University of Pittsburgh Cancer Institute, Pittsburgh, PA;
5. Department of Medicine, University of Pittsburgh Cancer Institute, Pittsburgh, PA;
6. VAPHS, Pittsburgh, PA;
7. BioZyme Inc, Apex, NC;
8. Department of Biologic Engineering, Massachusetts Institute of Technology;
9. Center for Systems Biology, Massachusetts General Hospital and Harvard Medical School, Boston, MA
Received 2017-4-27; Accepted 2017-8-6; Published 2017-10-23
Abstract
Increases in expression of ADAM10 and ADAM17 genes and proteins have been evaluated, but not validated as cancer biomarkers. Specific enzyme activities better reflect enzyme cellular functions, and might be better biomarkers than enzyme genes or proteins. However, no high throughput assay is available to test this possibility. Recent studies have developed the high throughput real-time proteolytic activity matrix analysis (PrAMA) that integrates the enzymatic processing of multiple enzyme substrates with mathematical-modeling computation. The original PrAMA measures with significant accuracy the activities of individual metalloproteinases expressed on live cells. To make the biomarker assay usable in clinical practice, we modified PrAMA by testing enzymatic activities in cell and tissue lysates supplemented with broad-spectrum non-MP enzyme inhibitors, and by maximizing the assay specificity using systematic mathematical-modeling analyses. The modified PrAMA accurately measured the absence and decreases of ADAM10 sheddase activity (ADAM10sa) and ADAM17sa in ADAM10-/- and ADAM17-/- mouse embryonic fibroblasts (MEFs), and ADAM10- and ADAM17-siRNA transfected human cancer cells, respectively. It also measured the restoration and inhibition of ADAM10sa in ADAM10-cDNA-transfected ADAM10-/- MEFs and GI254023X-treated human cancer cell and tissue lysates, respectively. Additionally, the modified PrAMA simultaneously quantified with significant accuracy ADAM10sa and ADAM17sa in multiple human tumor specimens, and showed the essential characteristics of a robust high throughput multiplex assay that could be broadly used in biomarker studies. Selectively measuring specific enzyme activities, this new clinically applicable assay is potentially superior to the standard protein- and gene-expression assays that do not distinguish active and inactive enzyme forms.
Keywords: Cancer biomarker, Sheddase activity, ADAM10, ADAM17, Fluorogenic peptide substrates, Proteolytic activity matrix analysis, Cell lysate, Tissues, lysate, Gene knockout, Gene silence, Gene restoration, Protease inhibitors
Introduction
ADAM10 and ADAM17 are transmembrane metallopeptidases of ADAM (A Disintegrin And Metalloprotease) gene-superfamily that have important roles in normal cell functions and pathogenesis of inflammatory, cardiovascular, skin and malignant diseases [1]. Primary functions of ADAM10 and ADAM17 are proteolytic cleavage on the cell surface and release (shedding) of functional extracellular domains (ectodomains) of membrane-anchored (transmembrane) proteins that mediate epidermal growth and inflammation.
ADAM17, also referred to as tumor necrosis factor alpha converting enzyme (TACE) and CD156q, is a major sheddase of the epidermal growth factors (EGFs) TGF-α, amphiregulin (AR) and heparin-binding EGF (HB-EGF), and of the central proinflammatory and immunoregulatory cytokine tumor necrosis factor alpha (TNF) [1-5]. ADAM17 also sheds several cell-adhesion molecules, TNF superfamily receptors, cytokines and chemokines [5-12]. Due to the enzyme activity-generated biologically active molecules, ADAM17 mediates vital functions such as development and modeling of tissues and organs, wound healing, inflammation and regulation of immune responses [5, 7, 12, 13]. ADAM17 knockout mice show perinatal lethality and defects similar to those found in EGF receptor or ligand deficient mice, including defective development of the eyelids, hair, whiskers, heart valves, and mammary glands [1]. ADAM17-knockout mice also display impaired thymic epithelium formation and T-cell development, as well as defective B-cell follicle formation and B-cell maturation, and thus resemble to lymphotoxin- and TNF-signaling deficient mice [1].
Complementary and partially overlapping with ADAM17, ADAM10, also known as CDw156, CD156c and α-secretase, sheds and/or activates the EGF, HB-EGF, betacellulin, amyloid precursor protein, and several receptors and ligands, including Notch receptor and ligands [1, 14-18]. ADAM10 and Notch deficient mice have similar phenotypes, including defects in embryonal somitogenesis, neurogenesis and vasculogenesis, indicating a critical role of ADAM10 in Notch signaling [1, 14]. Importantly, deregulation of Notch signaling promotes cancer progression [19, 20]. These and other published data [1, 21] show that ADAM10 and ADAM17 play key roles in cancer growth, survival, invasion and metastasis, and indicate that they might be important cancer biomarkers and therapy targets.
Increases in expression of ADAM10 and ADAM17 genes and/or proteins were frequently found in cancer tissues, but were either not robust or not associated with poor prognosis and, consequently, not validated as cancer biomarkers [1, 21-34]. Gene- and protein-expression biomarker assays measure without distinction both inactive and active enzyme forms. However, only specific enzyme active forms and their proteolytic activities are directly related to enzymatic cellular functions. Therefore, the enzyme activities might be better cancer biomarkers than the mere quantity of enzyme genes or proteins. However, the specific enzyme-activity biomarker assays are not currently available to test this possibility.
Clinically applicable enzyme-activity tests should be sensitive, specific, quantitative, high throughput and able to assess cellular enzymes in solid tumors. Previous seminal studies have defined the proteolytic activity matrix analysis (PrAMA), which integrates the assessment of enzymatic processing of multiple fluorogenic peptide substrates containing a fluorescence-resonance energy transfer (FRET) donor and a quencher, and a mathematical-modeling based computation [35]. The original PrAMA infers individual enzyme activities of MPs, including ADAM10sa and ADAM17sa, in mixtures of relatively small varieties of known recombinant and unknown natural enzymes expressed on live cells. Compared to the otherwise non-specific FRET substrate-based enzyme-activity assays, PrAMA is more specific, multiplex and quantitative. It could be a clinically translatable biomarker assay, if it could efficiently measure specific cellular enzyme activities in solid tissues.
To measure the cell-surface enzyme activities in solid tissues, three methods could be potentially applied. The assay could be performed using isolated live cells, purified cell membranes, or soluble extracts (lysates). Live cells or cell membranes are obtained from fresh tissues, and their isolations are complex, laborious and could affect the enzymes of interest. Consequently, the enzyme activity assays with such isolated live cells or cell membranes are not fully accurate and are impractical for clinical use. In contrast, the preparation of tissue lysates could be effectuated with both fresh and cryopreserved tissues, and is simple, rapid and, therefore, practical for clinical application. However, tissue lysates contain not only cell-membrane bound MPs but also a large variety of potent cytoplasmic and nuclear “promiscuous” proteases that can process a broad spectrum of substrates, including those selected for ADAM10 and ADAM17. The activities of intracellular proteases could adversely affect PrAMA specificity. To specifically measure ADAM10sa and ADAM17sa in tissue lysates, the non-MP “promiscuous” proteases might need to be inhibited in tested specimens. A simple way to do that could be to perform the enzyme-activity assessment of the lysates in the presence of broad-spectrum non-MP protease inhibitors.
In the present study, we modified PrAMA by assessing enzymatic activities in cell and tissue lysates supplemented with broad-spectrum non-MP protease inhibitors and by analyzing the proteolytic activity data by newly applying systematic PrAMA mathematical modeling to maximize specificity of the assay for ADAM10sa and ADAM17sa. The modified PrAMA measured with a significant accuracy ADAM10sa and ADAM17sa in cultured cells and tumors, and showed the essential characteristics of a robust high throughput multiplex biomarker assay that could be applied in practical medicine.
Materials and Methods
Reagents
Seven internally quenched FRET-peptide enzyme substrates, including PEPDAB005, PEPDAB008, PEPDAB010, PEPDAB011, PEPDAB013, PEPDAB014 and PEPDAB022, were purchased from BioZyme Inc (Apex, NC). The substrates are based on the cleavage sites in specific proteins known to be processed by different ADAM family members, and are moderately specific [36-38] (Fig. 1A). Trypsin was obtained from GIBCO-Life Technologies (Grand Island, NY). Recombinant ADAM8, ADAM10, ADAM12, ADAM17, MMP1, MMP3, MMP7 and MMP9 were obtained from R&D Systems (Minneapolis, MN). Phycoerythrin-conjugated mouse anti-human ADAM10 and ADAM17, rat anti-mouse ADAM10 and corresponding isotype control monoclonal antibodies (mAbs) were obtained from R&D Systems. Human ADAM10 and ADAM17 siRNA (pools of three target specific siRNA), control (scrambled siRNA), siRNA transfection medium, and siRNA transfection reagent were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Lipofectamine 2000 was purchased from ThermoFisher Scientific (Pittsburgh, PA). Control (empty) and human ADAM10-cDNA clone plasmids were obtained from OriGene (Rockville, MD). EDTA-free broad-spectrum non-MP protease inhibitor cocktails, including Roche tablets (Roche Applied Science, Indianapolis, IN) and ThermoFisher Scientific Halt 100 X solution, were utilized to inhibit non-MP enzymes in cell and tissue lysates. GI254023X, a specific small molecule inhibitor of ADAM10sa was obtained from Sigma Aldrich (Sent Louis, MO). 10 mM stock solutions of PEPDAB substrates and GI254023X were prepared in DMSO and stored frozen at -80oC. Enzyme-free, PBS-based, GIBCO cell-dissociation solution was purchased from ThermoFisher Scientific, and utilized to detach cultured adherent cells.
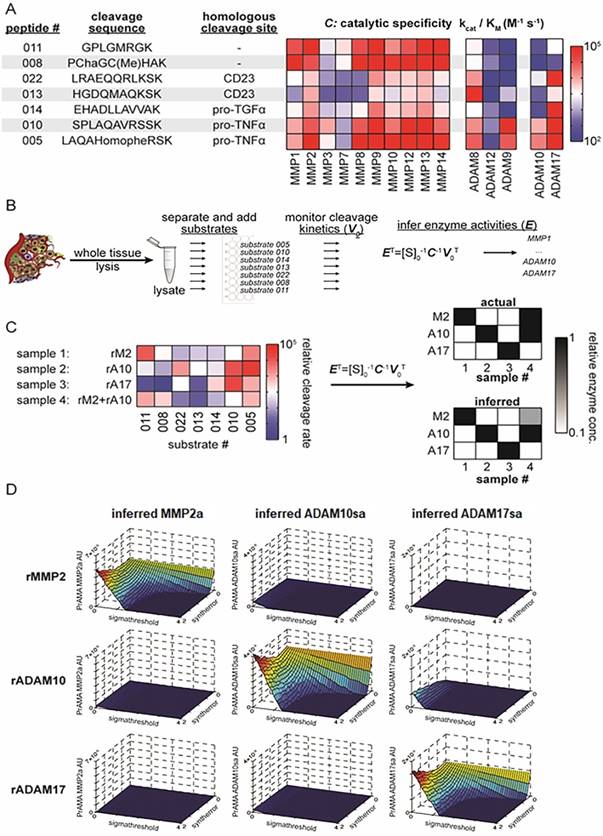
PrAMA approach for solid tissue analysis. (A) Summary table and heat-map show 7 FRET-based polypeptide substrates with cleavage sequences (HomoPhe, homophenylalanine; Cha, 3-cyclohexylalanine), endogenous-protein protease substrates from which the peptides were designed, and the catalytic efficiencies with which various recombinant proteases cleave the substrates (for more information see ref. 35). (B) Workflow illustrates PrAMA for cell and tissue lysates. (C) Proof-of-principle is shown for PrAMA applied to known solutions, and one combination, of recombinant enzymes (abbreviations: rM2, recombinant MMP2; rA10, recombinant ADAM10; rA17, recombinant ADAM17). Normalized cleavage rates for each of the 4 solutions were measured across the 7 substrates (left), and data were interpreted using known enzyme-substrate preferences [as in (A)] to infer which recombinant enzymes were present in the mixture. Actual mixture composition (top right) and PrAMA results (bottom right) are presented. (D) Surface plots depict three-dimensional “systematic PrAMA” inference as a function of the two parameters sensitivity (Syntherror) and specificity (Sigmathreshold). Processing data of seven substrates obtained with recombinant MMP2, ADAM10, and ADAM17 were analyzed by PrAMA across varying combinations of Syntherror and Sigmathreshold parameters to reveal how these two parameters influence PrAMA sensitivity and specificity. The three rows of surface-plots correspond to the analyzed individual three recombinant enzyme solutions (rMMP2, rADAM10 and rADAM17), and the three columns of surface-plots correspond to the three individual PrAMA-inferred enzyme activities from these solutions (MMP2a, ADAM10sa and ADAM17sa). The color scale ranges from red to blue, which reflects the surface heights as labeled on the vertical axis. The three-dimensional surface plots shown in the figure depict a representation of the two-dimensional “systematic PrAMA” shown in Figs. 3C-3H. In the latter cases (Figs. 3C-H) and the rest of presented data, Syntherror is held constant (0.5) across a range of Sigmathreshold values. The experimental details follow those described for Fig. 1C.
Cell Lines
Immortalized wild-type (ADAM10+/-, clone 37) and ADAM10 knockout (ADAM10-/-, clone 8T2) mouse embryonic fibroblasts (MEFs) were donated by Dr. Carl Blobel (Weill Medical College, Cornell University, New York, NY) [39], and wild-type (ADAM17+/+) and ADAM17 knockout (ADAM17-/-) MEFs were provided by Dr. Peter Dempsey (University of Colorado Medical School, Aurora, CO) [40]. The H441 lung carcinoma cell line was obtained from ATCC (Manassas, VA). MEFs were cultured in DMEM cell-culture medium supplemented with 10% fetal calf serum (FCS), while H441 cells were cultured in RPMI-1640 cell-culture medium supplemented with 10% FCS (all from GIBCO-Life Technologies). The cell lines were maintained in culture by their passage at 70% confluency using trypsinization. Five passages of these cells were maximally performed, after starting these cultures from their frozen stocks stored in liquid nitrogen. For experimental use, single-cell suspensions of viable cells were obtained using the GIBCO non-enzymatic cell-dissociation solution following the company recommended protocol. After detachment, cells were washed twice in DMEM or RPMI-1641 supplemented with 0.1% bovine serum albumin (BSA, GIBCO), to restore divalent cations that are essential for the activity of metalloproteinases.
Tissues
Six snap-frozen non-small cell lung carcinoma (NSCLC, 4 adeno, 2 squamous cell carcinoma) primary tumor tissues were obtained surgically from stage IA-IIB lung-cancer patients under an IRB-approved protocol (No. REN16070229/IRB9502100). Tumors were verified by histopathology, de-identified, stored and provided by the University of Pittsburgh Cancer Institute (UPCI) Lung Cancer (LC) SPORE Tissue Bank (TB).
Transfection of ADAM10-/- MEFs with Human ADAM10 cDNA
Two hundred thousand of ADAM10-/- MEFs per 2 mL of antibiotic-free DMEM medium supplemented with 10 % FCS were seeded in a Corning 6-well plate (ThermoFisher Scientific) and cultured for 24 h. After this culture, 3 μL of Lipofectamine 2000 were mixed with 197 μL of serum-free/antibiotic-free DMEM medium and combined with 200 μL of the medium alone or the medium containing 1 μg of empty plasmid or human ADAM10-cDNA plasmid (OriGene), incubated at room temperature for 30 min, and supplemented with 600 μL of DMEM. The cultured ADAM10-/- MEFs were washed twice with DMEM and covered with 1 mL of the Lipofectamine and/or plasmid suspensions, and incubated for 5 h at 37oC. The media containing Lipofectamine and/or plasmids were replaced with 2 mL of antibiotic-free DMEM medium supplemented with 10% FCS, and the cells were further cultured for 18 h.
Transfection of H441 Cells with Human ADAM10 and ADAM17 siRNA
Two hundred thousand H441 cells per 2 mL of antibiotic-free RPMI-1640 medium supplemented with 10 % FCS were seeded in a Corning 6-well plate and cultured for 24 h. Following the culture, 6 μL of siRNA transfection reagent was mixed with 100 μL of siRNA transfection medium in tube A, and 2 μL (20 pM) of control or human ADAM10 or human ADAM17 siRNA were mixed with 100 μL of siRNA transfection medium in tube B. The contents of tubes A and B were mixed, and the resulting mixture incubated at room temperature for 30 min. After this incubation, 800 μL of siRNA transfection medium were combined with the mixtures of siRNA transfection reagents and siRNA, and the resulting solution added to H441 cells washed with transfection medium. H441 cells and siRNA were co-cultured for 24 h. The medium containing siRNA was replaced with antibiotic-free RPM1-1640 medium supplemented with 10% FCS, and transfected cells were continued to culture for additional 24 h.
Stimulation of MEFs with PMA and Ionomycin
Wild-type, ADAM10-/-, ADAM10-/- transfected with ADAM10 cDNA plasmid, and ADAM17-/- MEFs were stimulated with 1 μM PMA (phorbol 12-myristate 13-acetate) and Ionomycin (eBioscience, San Diego, CA) for 2 h, before their assessment.
Preparation of Cell and Tissue Lysates
All procedures were performed at 0oC to 4oC. Single cell suspensions of viable cells were obtained from their cultures using the GIBCO cell dissociation buffer. Detached cells were washed twice in RPMI-1640 or DMEM medium and three times in PBS. Snap-frozen tissues were thawed on ice and washed three times in PBS. Cell and tissue lysates were prepared using a lysis buffer composed of 50 mM Tris-HCl/150 mM NaCl buffer supplemented with 0.1% SDS, 1% sodium deoxycholate, 1% Nonidet P40, 1% Triton X-100, and 1% EDTA-free broad-spectrum protease inhibitor cocktail (Roche). One million cells were lysed in 50 μl of the lysis buffer by 1-h incubation on ice and occasional vigorous vortexing. 50 mg of tissues were lysed by homogenization in 500 μl of the lysis buffer using the tissue grinder system (ThermoFisher Scientific), and subsequent 1-h incubation on ice and occasional vigorous vortexing of the homogenized tissue. The lysates were centrifuged at 16,000 g for 20 min, their supernatants were collected, protein concentrations determined using Quick Start Bradford Dye Reagent (Bio-Rad, Hercules, CA), aliquoted and stored at -80oC.
Enzyme-linked Immunosorbent Assay
Human ADAM10 and ADAM17 enzyme-linked immunosorbent assay (ELISA) kits were obtained from Antibodies Online Inc. (Atlanta, GA) and R&D, respectively. ELISAs were performed using 10 μg of the lysates.
Metalloprotease Activity Assay
The assay was performed in mixtures of 1 to 10 μg of cell or tissue lysate proteins or 10 ng of recombinant enzymes, and 10 μM PEPDAB substrates. The assay buffer was consisted of 25 mM Tris-HCl buffer pH 8, supplemented with 0.1 mg/mL BSA, 0.0006% Brij-35, 1% glycerol, EDTA-free Roche-tablet and ThermoFisher Scientific-Halt broad-spectrum protease inhibitor cocktails in the concentrations indicated in Results. Halt cocktail contains the following inhibitors (and their targets): AEBSF-HCl (inhibits serine proteases), Aprotinin (serine proteases), Bestatin (aminopeptidases), E64 (cysteine proteases), Leupeptin (serine/cysteine proteases), and Pepstatin A (aspartic acid proteases). To examine the inhibition of ADAM10sa in the cell and tissue lysates, 1 μM GI254023X [41] was pre-incubated with the lysates for 1 h at 21oC and 1 h at 37oC, before mixing the lysates and PEPDAB substrates. Total processing of substrates was obtained by their incubation with 1% trypsin. The enzymatic reaction was induced by the incubation of lysate/substrate mixtures in 96-well LUMITRAC 200 white-wall flat-bottom immunology plates (Sigma Aldrich, St. Louis, Mo) at 37oC for 0.5 to 4 h. The fluorescence was measured in a TECAN infinite 200 pro fluorimeter (Technical Communities, Inc., San Bruno, CA) using 485 nm excitation and 530 nm emission wavelengths. The experiments were performed in duplicates. The obtained data were computed using the following formulas: 1) Experimental fluorescence intensity gain (EFIG) = Experimental sample mean fluorescence intensity (ESMFI) - Background sample mean fluorescence intensity (BSMFI); 2) Trypsin-induced fluorescence intensity gain (TIFIG) = Trypsin sample mean fluorescence intensity (TSMFI) - BSMFI) (Suppl. Fig. 1); 3) pM processed substrates = EFIG x 106/TIFIG. Data are presented as means ± SD.
Proteolytic Activity Matrix Analysis
Proteolytic Activity Matrix Analysis (PrAMA) is an integrated experimental mathematical modeling analysis that interprets dynamic signals from panels of moderately specific fluorogenic polypeptide protease substrates (Fig. 1A) to deduce a profile of specific MP proteolytic activities [35]. Deconvolution of signals from complex mixtures of proteases is accomplished by using prior data on individual MP cleavage signatures (Fig. 1A, matrix - C: catalytic specificity) for the substrate panel measured with purified enzymes. In essence, the method relies upon the assumption that the total observed substrate cleavage is the linear sum of cleavage from all enzyme activities in a complex biological mixture. As a consequence of this assumption, a vector of specific enzyme activities, E, may be inferred from a signature of substrate cleavages, V0, by solving the following equation:
ET = [S]0-1 C-1 V0T
where [S]0 is the concentration of substrate, and C comprises a matrix of kinetic parameters that describe the efficiency (kcat/KM) with which particular enzymes cleave the substrates [34] (Figs. 1B, 1C). PrAMA is performed with adjustment of two parameters, “Syntherror” and “Sigmathreshold” which defines synthetic errors and statistical significant thresholds, respectively, which are used to obtain accuracy. These two parameters effectively control assay sensitivity and specificity, respectively [35]. The analysis was performed using combinations of 10 to 40 various systematically graded Syntherror and Sigmathreshold parameters (the lowest 0.05 to 0.1, the highest 1.0 to 2.0). The analysis, named “systematic PrAMA”, searched for 100% sensitivity and 100% specificity combinations of Syntherror/Sigmathreshold parameters, which showed no cross-reactivity of recombinant ADAM10 and/or ADAM17 (Fig. 1D), wild-type and gene knockout cells, gene knockout and gene restored cells, and control and gene silenced cells. The three-dimensional PrAMA surface plots depict a landscape of how the two parameters sensitivity and specificity (Syntherror and Sigmathreshold) influence the inference results (Fig. 1D). To simplify PrAMA interpretation, especially when comparing across multiple experiments, subsequent PrAMA results used a fixed high-sensitivity Syntherror parameter (typically, 0.5) that was determined based on Fig. 1D and previous optimization [35] data, and systematically graded Sigmathreshold parameters to maximize both sensitivity and specificity. This procedure thus yielded a two-dimensional “systematic PrAMA” showing how robust protease activity inference was to the significance threshold parameter, Sigmathreshold. Ten thousand bootstrapping repetitions were typically performed for each dataset analyzed with standard errors across bootstrapping replicates. The computation was performed using Matlab 2009a MathWorks (Natick, MA).
Flow Cytometry of Cell Surface Enzymes
Suspensions of viable cells were obtained from adherent-cell cultures using the GIBCO cell dissociation buffer. Detached cells were washed twice with PBS supplemented with 0.1% FCS and 0.01% Azide, and stained with fluorochrome-conjugated isotype-control, anti-human ADAM10 and ADAM17 extracellular-domain, or anti-mouse ADAM10 extracellular-domain mAbs. The stained cells were analyzed by single-color flow cytometry using the Accuri C6 cytometer (BD Biosciences, San Jose, CA, CA) and FlowJo v 10 (FlowJo, LLC Ashland, OR) software.
Statistics
Data were statistically evaluated using the SPSS (version 10.0 SPSS Inc., Chicago, IL) program package. Data are reported as means ± SD or SE. Statistical significance of data was assessed using the Student's t test. p values ≤0.05 were considered significant.
Results
PrAMA measures Specific Sheddase Activities of Recombinant ADAM10 and ADAM17 in the Presence of non-MP Protease Inhibitors
To efficiently measure ADAM10 and ADAM17 activities, PEPDAB005, 010, 014, 013 and 022 FRET substrates were chosen based on their known ability to be efficiently cleaved with relative selectivity by ADAM sheddases. Two additional substrates, PEPDAB008 and 011, which exhibit greater preference for MMPs, were also used to account for non-ADAM sheddase metalloproteinase activity [35-38] (Fig. 1A). Although the FRET substrates PEPDAB005 and 010 contain the TNF cleavage sites, and PEPDAB014 and 022 have the TGFα and CD23 cleavage sites, respectively, they are not only variably susceptible to ADAM17 and ADAM10, but also to ADAM8, ADAM9, ADAM12, and to a variety of other MPs [35-38] (Suppl. Fig. 2, Fig. 1A). Based on the characteristic differential processing patterns of the moderately specific substrates with the recombinant MPs, PrAMA was modeled to more selectively measure individual enzyme activities in a limited complexity mixtures of natural MPs that are expressed on the cell surface of live cells [35]. Fig. 1B shows the PrAMA workflow described here. As a validating proof-of-principle, PrAMA is able to correctly identify individual recombinant enzymes and a mixture thereof based on their observed cleavage patterns across the 7 substrates used in this work (Figs. 1C, 1D). While such recombinant enzyme experiments and many more described previously [35] are encouraging, measurements of specific enzyme activities in whole tissue presents additional challenges.
Broad-spectrum Roche and Halt non-MP protease inhibitors decrease large proportions of cell-lysate enzyme activities, but do not affect rADAM10 and rADAM17 sheddases activities. Tris-based reaction buffer solutions (150 μL) of 10 μM PEPDAB005, 2 μg of H441-cell lysate (A, B), 10 ng of rADAM10 (C, D), 10 ng of rADAM17 (E, F) and indicated concentrations of Roche and Halt inhibitors were distributed into wells of 96-well LUMITRAC-200 plates. Additionally, the effects of different concentrations of DMSO on rADAM10 and rADAM17 processing of PEPDAB005 were tested (G, H). Negative-control wells contained the assay buffer supplemented only with 10 μM of PEPDAB005, and positive-control (total substrate processing) wells contained the assay buffer supplemented with 1 μM of PEPDAB005 and 1% trypsin. Plates were incubated at 37oC for 4h. The fluorescence was measured with a TECAN infinite 200 pro fluorimeter, using an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The presented data are pM means of duplicate measurements ± SD (A, C, E, G) and % inhibition (B, D, F, H) of PEPDAB005 processing at 3 h of incubation. (A, B) Roche and Halt inhibitors significantly inhibited in a dose-dependent manner H441-cell lysate-mediated processing of PEPDAB005 (p=0.002-0.0001). Halt was more effective (p=0.012 to p=0.0012). Processing of PEPDAB005 by rADAM10 (C, D) and rADAM17 (E, F) was slightly (non-significantly) inhibited with 0.25%, 0.5 or 1%, but significantly inhibited with 2% and especially 3% of the protease inhibitors (rADAM10: p=0.048 to p=0.019; rADAM17: p=0.05 to p=0.0038). Halt cocktail did so more efficiently than the Roche cocktail (p=0.026 to p=0.013). (G, H) DMSO did not inhibit rADAM10 or rADAM17 enzyme activity.
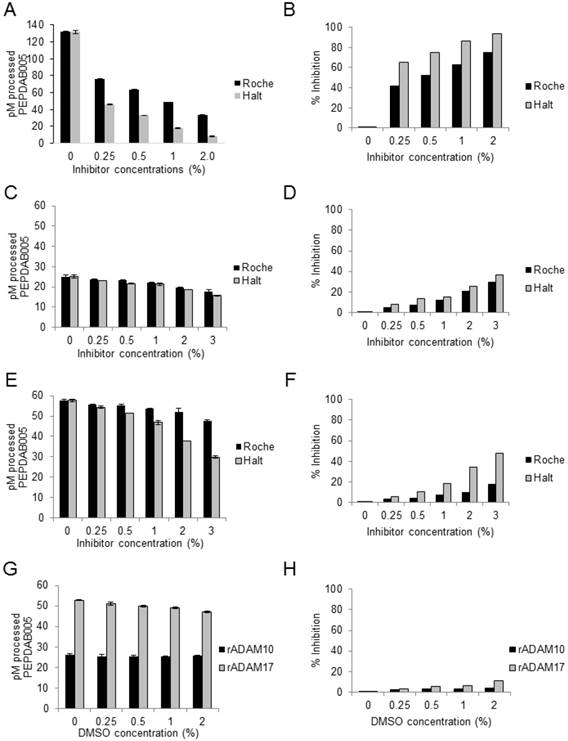
To specifically measure ADAM10sa and ADAM17sa in tissue lysates containing not only the cell-surface but also a plethora of cytoplasmic and nuclear “promiscuous” proteases, it might be necessary to selectively inhibit the non-MP proteases without affecting the enzymes of interest. We examined whether the cocktails of broad-spectrum non-MP protease inhibitors Roche tablet (Roche) and ThermoFisher Scientific Halt (Halt) affect enzyme activities of human NSCLC-H441 cell lysates and rADAM10 and rADAM17 (Fig. 2). Both cocktail inhibitors strongly inhibited in a dose-dependent manner (p=0.002 to p<0.0001) the cell-lysate processing of the prototypic ADAM10/ADAM17 substrate PEPDAB005, though Halt did so more effectively (p=0.012 to p=0.0012) (Figs. 2A, 2B). The company-recommended Roche and Halt concentrations of 0.5% to 1% decreased substrate processing in the cell lysate by 50% to 60% and 69% to 82%, respectively. The remaining cell-lysate enzyme activity after this inhibition should be mediated mostly by unaffected MPs, including ADAM10 and ADAM17. To confirm this possibility, we tested whether the protease inhibitors affect rADAM10 and rADAM17 processing of PEPDAB005 in a Tris-based assay buffer (Figs. 2C-2F). At 0.5% and 1% concentrations, Roche and Halt inhibitors induced low, nonsignificant suppression (5-20%, p≥0.07) of rADAM10 (Figs. 2C, 2D) and rADAM17 (Figs. 2E, 2F) activities, but they suppressed 30% to 50% of the enzyme activities, when administered at 2% to 3% concentrations (rADAM10: p=0.048 to p=0.019; rADAM17: p=0.05 to p=0.0038). Again, Halt appeared to be more potent than Roche, especially against rADAM17 (p=0.026 to p=0.013). The solvent of Halt inhibitors, DMSO, had no effect on the enzyme activities in any of the tested concentrations (Figs. 2G, 2H), indicating that the significant suppressive effects of higher concentrations of the protease inhibitors were caused by the inhibitors themselves, and not by the vehicle.
Combining the two non-MP protease-inhibitor cocktails could target a broader spectrum of and have a greater inhibiting efficiency on non-MP proteases. Consequently, this strategy could increase the specificity of PrAMA analyses of ADAM10sa and ADAM17sa in cell and tissue lysates. However, the combined use of inhibitors could also have an adverse effect on ADAM10sa and ADAM17sa. The combined Roche/Halt cocktails at a concentration of 0.5% only slightly, non-significantly, suppressed processing of PEPDAB005 by rADAM10 (10%) and rADAM17 (15%), and did not affect processing of PEPDAB010, PEPDAB08, PEPDAB013 or PEPDAB014 (Figs. 3A, 3B). In contrast, the combined inhibitors at a concentration of 1% significantly suppressed processing of PEPDAB005 with rADAM10 (43%, p=0.0049) and rADAM17 (40%, p=0.0046), and increased processing of PEPDAB010 with rADAM17 (23%, p=0.0038), but had no effect on the processing of the other tested substrates with either enzyme. These findings indicate that higher concentrations of protease inhibitors could modify the susceptibility of PEPDAB005 and PEPDAB010 to processing with, but not activity of rADAM10 or rADAM17.
Combined solutions of 0.5% Roche and 0.5% Halt inhibitors do not affect recombinant ADAM10-mediated processing of PEPDAB substrates or PrAMA-ADAM10 sheddase activity. Processing of 5 substrates with rADAM10 (A) and rADAM17 (B) in the absence and presence of 0.5% or 1% combined inhibitors is presented. Data are pM means of duplicate measurements ± SD of processed substrates. The decreases of PEPDAB005 processing with rADAM10 and rADAM17 and the increases of PEPDAB010 processing with rADAM17 in the presence of 1% Roche/Halt inhibitors are significant (p=0.0049, p=0.0046 and p=0.0038, respectively). PrAMA-ADAM10sa data of rADAM10 processing of PEPDAB substrates in the absence (C) and presence of 0.5% (D) or 1% (E) combined inhibitors are presented. PrAMA-ADAM17sa data of rADAM17-processing of substrates in the absence (F) and presence of 0.5% (G) or 1% (H) combined inhibitors are presented. The experiments in (A) and (B) were performed using similar conditions to those presented in Fig. 1. PrAMA was performed using a fixed 0.5 Syntherror parameter and systematically increased Sigmathreshold parameters from 0.0 to 2.0. Data are PrAMA ADAM10sa (C, D, E) and PrAMA ADAM17sa (F, G, H) arbitrary units (AU). PrAMA standard errors were <5%.The arrow-had line in (D) indicates that the PrAMA specificity Sigmathreshold parameters ≥ 1.0 detects only true-positive rADAM10-ADAM10sa without any false-positive ADAM17sa.
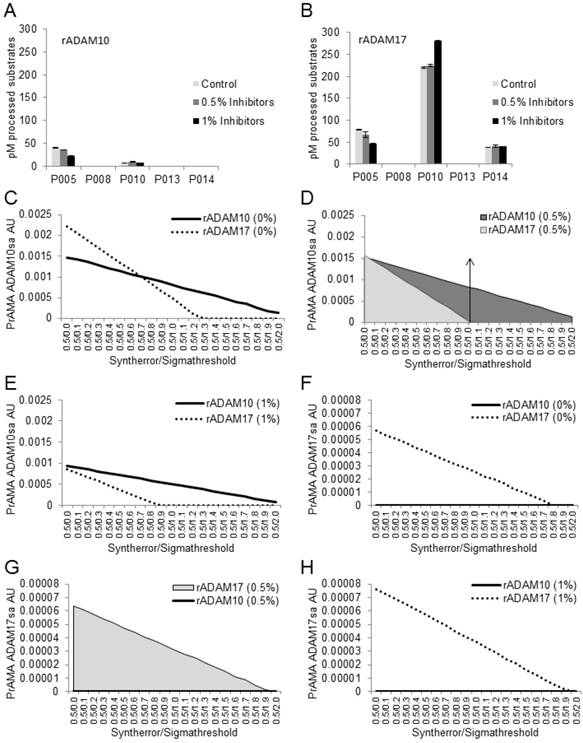
Next, we tested whether the observed changes of the substrate processing in the presence of combined Roche/Halt protease inhibitors could affect PrAMA (Figs. 3C-3H). The data presented in Figs. 3A-3B were analyzed using the PrAMA fixed sensitivity parameter Syntherror (0.5) and the systematically increased specificity parameter Sigmathreshold from 0.0 to 2.0, to maximize the assay specificity. PrAMA of rADAM10 measured true-positive ADAM10sa without false-positive ADAM17sa using Syntherror/ Sigmathreshold 0.5/1.2-2.0 (Fig. 3C), and 0.5/1.0-2.0 (Fig. 3D) or 0.5/0.8-2.0 (Fig. 3E) scripts in the absence (0%) and presence of 0.5% or 1% inhibitors, respectively. Importantly, the separation of rADAM10 true-positive ADAM10sa and false-positive ADAM17sa was better with protease inhibitors, and the sensitivity of ADAM10sa PrAMA was unaffected in the presence of 0.5% inhibitors. In contrast, the sensitivity of ADAM10sa PrAMA was significantly decreased in the presence of 1% inhibitors. On the other hand, PrAMA of rADAM17 showed high true-positive ADAM17sa completely separated from undetectable false-positive ADAM10sa using the Syntherror/Sigmathreshold 0.5/0.0-1.8 scripts, regardless of whether the inhibitors were present or absent in the assay buffer. However, the sensitivity of PrAMA ADAM10sa appeared to be more than two-log units higher than that of the PrAMA ADAM17sa. These data show that Tris-based assay buffer supplemented with 0.5% combined Roche/Halt protease-inhibitor cocktails strongly suppress non-MP proteases without affecting recombinant or natural ADAM10sa and ADAM17sa. They also show that the high specificity PrAMA Syntherror/Sigmathreshold 0.5/≥1.0 scripts measure only the true-positive and distinguish accurately recombinant ADAM10sa and ADAM17sa, indicating that these PrAMA scripts might be optimal for the examination of the natural ADAM10sa and ADAM17sa in cell and tissue lysates. Validation of the modified PrAMA for examination of natural ADAM10sa and ADAM17sa in cell and tissue lysates was further performed as described below.
Modified PrAMA detects ADAM10sa and ADAM17sa Presence in Wild-type Cells and Absence in Gene-knockout Cells
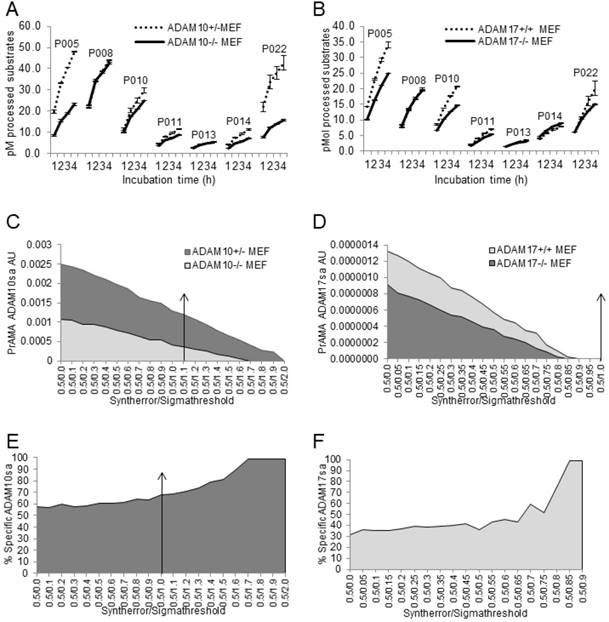
PrAMA has demonstrated the ability to detect ADAM10sa and ADAM17sa presence and absence on the surface of live wild-type and ADAM10-/- and ADAM17-/- MEFs, respectively [35]. We examined whether the modified PrAMA could similarly measure the endogenous enzyme activities in lysates of these cells (Fig. 4). ADAM10+/-, ADAM10-/-, ADAM17+/+ and ADAM17-/- MEFs were stimulated with PMA/Ionomycin, lysed and their lysates examined for enzyme activities in the presence of non-MP inhibitors (0.5% Roche/Halt) using 7 PEPDAB substrates, to increase PrAMA specificity [35] (Suppl. Fig. 3). The wild-type MEF lysates showed significantly higher enzyme activities than the corresponding knockout MEF lysates against PEPDAB005, 010, 011, 014 and 022 substrates (Figs. 4A, 4B; ADAM10+/- vs ADAM10-/- MEFs ADAM17+/+ vs ADAM17-/- MEFs: p<0.0001 and p<0.001, p=0.0056 and p<0.001, p=0.0016 and p=0.0025, p=0.0016 and p=0.009, and p=0.006 and p=0.004, respectively). However, the ADAM10-/- MEF lysate processed less PEPDAB005, 014 and 022, and more PEPDAB010 than the ADAM17-/- MEFs lysate. PrAMA detected relatively high levels of true-positive ADAM10sa in ADAM10+/- MEF lysates and relatively low levels of false-positive ADAM10sa in both ADAM10+/- and ADAM10-/- MEF lysates (Figs. 4C, 4E). Increases of PrAMA specificity by systematically increasing Syntherror/Sigmathreshold scripts from 0.5/0.0 to 0.5/1.7 led to gradual decreases of both true-positive and false-positive ADAM10sa, and increases of the proportion of true-positive ADAM10sa from 57% to 100%, respectively. Importantly, the PrAMA scripts between 0.5/1.7 and 0.5/1.9 detected only true-positive ADAM10sa in wild-type MEF without any false-positive ADAM10sa in knockout MEF. To detect ADAM17sa in MEF, we had to use lower specificity PrAMA (Syntherror/Sigmathreshold <0.5/0.95). The lower specificity PrAMA detected relatively low levels of true-positive ADAM17sa in ADAM17+/+ MEF lysate and significant levels of false-positive ADAM17sa in both ADAM17+/+ and ADAM17-/- MEF lysates (Figs. 4D, 4F). However, the systematic increases of PrAMA specificity by increasing Syntherror/Sigmathreshold scripts from 0.5/0.0 to 0.5/0.85 led to decreases of both true-positive and false-positive ADAM17sa, and increases of the proportion of true-positive ADAM17sa from 31% to 100%, respectively. This time, the absolute (100%) specificity was obtained using the PrAMA 0.5/0.8 script. These data show that the modified PrAMA can detect and measure with significant accuracy ADAM10sa and ADAM17sa in wild-type MEFs, and their absence in the gene knockout MEFs. They also show that wild type MEFs contain different amounts of ADAM10sa and ADAM17sa and, therefore, different PrAMA specificities has to be applied to specifically measure them.
Modified PrAMA detects ADAM10sa and ADAM17sa presence in wild-type and absence in ADAM10-/- and ADAM17-/- MEF lysates, respectively. ADAM10+/-, ADAM10-/- (A, C, E), ADAM17+/+ and ADAM17-/- (B, D, F) MEFs were activated with PMA/Ionomycin and lysed. The 150 μL Tris solutions of 2 μg cell lysates, 10 μM PEPDAB substrates and 0.5% Roche/Halt protease inhibitors were incubated for 4 h at 37oC, and the developed fluorescence was recorded hourly using TECAN fluorimeter. The presented experiments are representative of 5 performed. The enzymatic activity data are shown as pM means of duplicate measurements ± SD of the processed substrates (A, B). The decreased processing of PEPDAB005, 010, 011, 014 and 022 with knockout MEF lysates is significant (ADAM10+/- vs ADAM10-/- MEFs and ADAM17+/+ vs ADAM17-/- MEFs: p<0.0001 and p<0.001, p=0.0056 and p<0.001, p=0.0016 and p=0.0025, p=0.0016 and p=0.009, and p=0.006 and p=0.004, respectively). Systematic PrAMA data using Syntherror/Sigmathreshold parameters 0.5/0.0 to 0.5/2.0 and 0.5/0.0 to 0.5/1.0 are presented as ADAM10sa AU (C) and ADAM17sa AU (D), respectively. Standard errors of PrAMA data were 2.5% to 5.8%. True-positive ADAM10sa and ADAM17sa in ADAM10+/- and ADAM17+/+MEFs, respectively, are presented as % of specific enzyme activities in wild-type MEFs, containing both the true-positive and false-positive activities, relative to ADAM10-/- (E) and ADAM17-/- (F) ME Fs containing only the false-positive activities, respectively.
Modified PrAMA detects Restoration of ADAM10 in ADAM10-/- MEFs
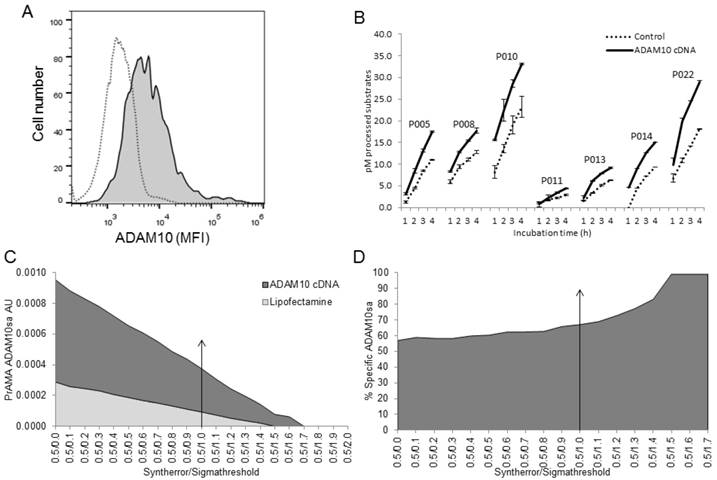
To confirm and extend the above findings, we investigated whether the modified PrAMA could detect newly produced ADAM10sa in ADAM10-/- MEFs following the enzyme-gene restoration. ADAM10-/- MEFs were treated either with Lipofectamine alone or Lipofectamine/ human ADAM10-cDNA containing plasmid, incubated for 24 h, and activated with PMA/Ionomycin. Controls also included untreated and Lipofectamine/empty plasmid transfected cells (data not shown). These cells were examined by flow cytometry for the surface expression of human ADAM10 protein, and their lysates were tested by the modified PrAMA for the presence of ADAM10sa (Fig. 5). ADAM10-cDNA transfected ADAM10-/- MEFs showed the expression of human ADAM10 protein on the cell surface (Fig. 5A), and their lysates contained significantly increased enzyme activities relative to the lysates of Lipofectamine alone treated cells (Fig. 5B; PEPDAB010: p=0.002; PEPDAB005, 011, 014 and 022: p<0.001 to p<0.0001). Consequently, PrAMA detected relatively high levels of true-positive ADAM10sa in the lysate of ADAM10-cDNA transfected ADAM10-/- MEFs, and low levels of false-positive ADAM10sa in the lysates of both Lipofectamine alone treated and ADAM10-cDNA transfected ADAM10-/- MEFs (Figs. 5C, 5D). Systematic increases of PrAMA specificity (Syntherror/Sigmathreshold scripts from 0.5/0.0 to 0.5/1.5) led to gradual decreases of both true-positive and false-positive ADAM10sa, and increases of the proportion of true-positive ADAM10sa in ADAM10-cDNA transfected cells from 58% to 100%, respectively. The highest (100%) specificity ADAM10sa was also obtained with the PrAMA script 0.5/1.6. These findings show that the modified PrAMA can accurately detect and measure newly produced ADAM10sa in ADAM10-/- MEF following transfection of human ADAM10 gene.
Modified PrAMA detects ADAM10sa in ADAM10-/- MEFs following restoration of ADAM10 gene. ADAM10-/- MEFs were transfected with Lipofectamine/human ADAM10-cDNA or treated with Lipofectamine alone and incubated for 24 h. In some experiments, empty plasmid-transfected ADAM10-/- MEFs were used as an additional control. (A) ADAM10-/- MEFs transfected with human ADAM10-cDNA express human ADAM10 on cell surface. Lipofectamine treated (empty histogram) and Lipofectamine/human ADAM10-cDNA transfected (filled histogram) cells were stained with PE-conjugated anti-human ADAM10 and examined by flow cytometry. Cells were also stained with PE-conjugated anti-mouse ADAM10 or isotype-control mAbs, and their mean fluorescence intensities (MFIs) were similar to that of the cells treated with Lipofectamine alone and stained with PE-conjugated anti-human ADAM10 mAb (data not shown). Data are from one of two similar experiments performed. (B) Lysates of ADAM10-/- MEFs transfected with human ADAM10-cDNA contain increased enzyme activities as assessed with all 7 PEPDAB substrates. Lipofectamine alone treated and Lipofectamine/human ADAM10-cDNA transfected ADAM10-/- MEFs were lysed. The 150 μL Tris solutions containing 2 μg cell lysates, 10 μM PEPDAB substrates and 0.5% Roche/Halt inhibitors were incubated for 4 h at 37oC, and the developing fluorescence was recorded every hour. Data are representative of three experiments performed. They are pM means of duplicate measurements ± SD of processed substrates. The increases of PEPDAB processing with the lysates of ADAM10-cDNA transfected cells are significant (PEPDAB010: p= 0.002; PEPDAB005, 008, 011, 013, 014 and 022: p<0.001 to p<0.0001). (C) PrAMA-ADAM10sa AU was obtained by analyzing the enzyme activity data at 4 h of incubation using the systematically increased Syntherror/Sigmathreshold parameters from 0.5/0.0 to 0.5/2.0. Standard errors of PrAMA data were 1.3% to 2.6%. (D) True-positive ADAM10sa restoration in ADAM10-/- MEFs is presented as % of the specific enzyme activity in ADAM10-cDNA transfected MEFs, containing both the true-positive and false-positive activities, relative to Lipofectamine-alone treated ADAM10-/- MEFs, containing only the false-positive activity.
Modified PrAMA detects in Human Cancer Cells ADAM10sa and ADAM17sa Decreases after Gene Silencing
Next, we examined whether the modified PrAMA could specifically measure down-modulation of ADAM10sa and ADAM17sa in the lysates of the prototypic human NSCLC cells H441 transfected with ADAM10 and ADAM17 siRNA, respectively (Fig. 6). Untreated H441 cells showed a 26-fold higher levels of cell-surface ADAM10 (Fig. 6A, Suppl. Fig. 4A) than ADAM17 protein (Fig. 6B, Suppl. Fig. 4B), as determined by flow cytometry, but 5-fold lower levels of ADAM10 than ADAM17 protein in the cell lysate, as measured by ELISA (Suppl. Figs. 4C). The H441 cell lysate mediated significant and characteristic processing of PEPDAB substrates (Fig. 6C), and exhibited presence of significant amounts of ADAM10sa (Fig. 6D) and ADAM17sa (Fig. 6E). Proportional equations of rADAM10-ADAM10sa and rADAM17-ADAM17sa with the cell-lysate ADAM10sa and ADAM17sa, respectively, indicated that H441-cell lysate contained more ADAM10sa than ADAM17sa, which correlated better with the cell-surface expression than with the cell-lysate quantities of the enzyme proteins.
Modified PrAMA detects decreases of ADAM10sa and ADAM17sa in human cancer cells after silencing of the corresponding enzyme genes. H441 cells were transfected with human ADAM10 siRNA, ADAM17 siRNA or scrambled siRNA or were treated with transfection reagents alone for 48 h. In some experiments, control cells were also untreated. (A, B) Cells were stained with PE-conjugated IgG control mAb, anti-human ADAM10 (A) or anti-human ADAM17 mAb (B) and analyzed by flow cytometry. Empty histograms represent MFI of H441 cells stained with isotype control mAb. Dark gray histograms represent MFI of H441 cells treated with scrambled siRNA and stained with PE-conjugated anti-ADAM10 (A) or anti-ADAM17 (B) mAbs. Light grey histograms represent MFI of H441 cells treated with ADAM10 (A) or ADAM17 (B) siRNA and stained with PE-conjugated anti-ADAM10 or anti-ADAM17 mAbs, respectively. Data are from a representative experiment of 8 similar performed (Suppl. Figs. 4A, 4B). In the presented experiment, ADAM10 and ADAM17 siRNA induced 90% and 45% decreases of ADAM10 and ADAM17 protein expression on H441 cell surface, respectively. (C) After performing transfection, 2 μg of cell lysates were tested for processing PEPDABs in the presence of 0.5% Roche/Halt protease inhibitors. Data are from one of seven similar experiments performed. They are pM means of duplicate measurements ± SD of processed substrates. Processing of PEPDAB substrates was differently decreased in H441 cells transfected with ADAM10 or ADAM17 siRNA (PEPDAB005: p=0.0023 and p=0.0077; PEPDAB010: p=0.012 and p=0.017; PEPDAB011: p=0.048 and p=0.06; and PEPDAB014: p=0.07 and p=0.0.019, respectively). The substrate processing data obtained at 4 h of incubation were analyzed using the systematically increased Syntherror/Sigmathreshold parameters from 0.5/0.0 to 0.5/2.0. The resulted PrAMA ADAM10sa (D) and ADAM17sa (E) AU are shown. PrAMA standard errors were 1.3% to 8.9%. Decreases of ADAM10sa and ADAM17sa in ADAM10 (F) and ADAM17 (G) siRNA transfected H441 cells, respectively, are presented as % of the specific enzyme activities in the siRNA transfected cells relative to transfection reagent-treated cells. Proportion-equation analysis of PrAMA-ADAM10sa and PrAMA-ADAM17sa of rADAM10 and rADAM17 vs the scrambled siRNA-transfected H441-cell lysates, respectively, showed that 10 μg of H441-cell lysate contained 17.0 ng of ADAM10sa and 1.65 ng of ADAM17sa.
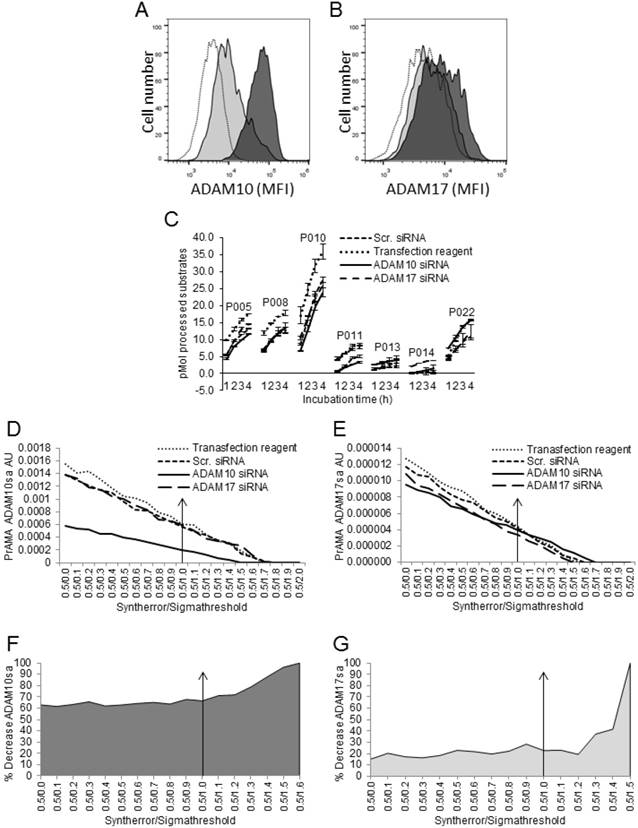
Silencing ADAM10 or ADAM17 in H441 cells with ADAM10 and ADAM17 siRNA led to 70% to 95% and 35% to 70% decreases of ADAM10 and ADAM17 protein expression on the cell surface, respectively (Figs. 6A, 6B; Suppl. Figs. 4A, 4B). Subsequently, processing of PEPDAB substrates was variably decreased (Fig. 6C; ADAM10 siRNA: PEPDAB005, p=0.0023; PEPDAB010, p=0.012; PEPDAB011, p=0.048; PEPDAB014, p=0.07. ADAM17 siRNA: PEPDAB005, p=0.0077; PEPDAB010, p=0.017; PEPDAB011, p=0.06; PEPDAB014, p=0.0.019). Consequently, ADAM10sa (Figs. 6D, 6F) and ADAM17sa (Figs. 6E, 6G) were reduced in the cell lysates from 67% (Syntherror/Sigmathreshold 0.5/1.0 script) to 88% (0.5/1.5 script) and from 28% (0.5/1.0 script) to 42% (0.5/1.4 script), respectively. These changes of ADAM10sa and ADAM17sa correlated with those of the cell-surface levels of ADAM10 and ADAM17 proteins. In addition, the high similarity of the false-positive ADAM10sa in MEFs and the remaining ADAM10sa in H441 cells after the effective gene silencing indicated that the false-positive ADAM10sa was low in H441 cells, and that the high-specificity PrAMA (i.e., Syntherror/ Sigmathreshold ≥0.5/1.4) mostly measured in these cancer cells the true-positive enzyme activities. Therefore, the modified PrAMA can accurately measure ADAM10sa and ADAM17sa in human cancer cells.
Modified PrAMA detects GI254023X-induced Inhibition of ADAM10sa in Human Cancer-Cell and Tumor-tissue Lysates
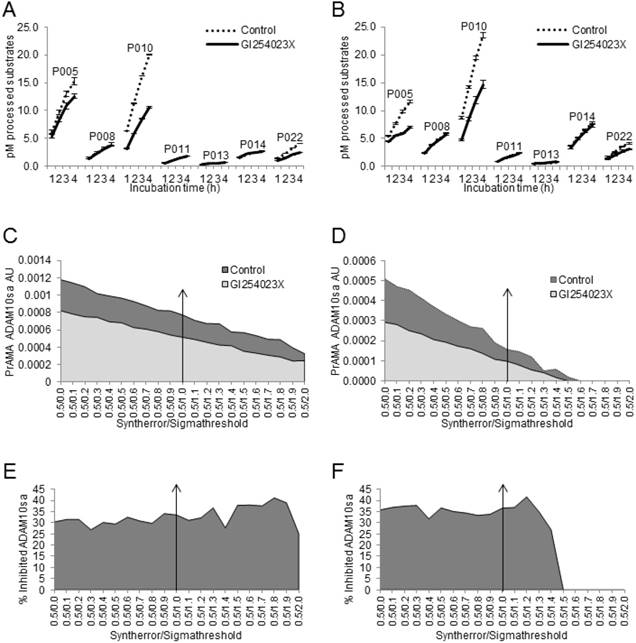
Finally, we tested whether the modified PrAMA could measure the GI254023X-induced suppression of ADAM10sa [41] in human cancer-cell and tumor-tissue lysates (Fig. 7). NSCLC H441-cell and NSCLC T2495-tumor-tissue lysates were diluted with Tris-based reaction buffer supplemented with 0.5% Roche/Halt protease inhibitors and either vehicle (Control) or 1 μM G1254023X, and examined for the processing of PEPDAB substrates and presence of ADAM10sa and ADAM17sa. GI254023X significantly inhibited the lysate-mediated processing of PEPDAB005, 010 and 022 (H441: p=0.017, 0.0041 and 0.0058; T2495: p=0.05, 0.040, 0.08, respectively), and the inhibition of PEPDAB005 processing was more pronounced in T2495-tissue lysate than H441-cell lysate (Figs. 7A, 7B). The high specificity PrAMA (Syntherror/Sigmathreshold 0.5/1.2 and 0.5/1.4 scripts) detected 37% and 42% decreases of ADAM10sa in GI254023X supplemented H441-cell (Figs. 7C, 7E) and T2495-tumor lysates (Figs. 7D, 7F), respectively. These findings confirm the gene silencing data, and indicate that modified PrAMA can accurately measure the ADAM activities not only in cells but also in solid tissues.
Modified PrAMA detects decreases of ADAM10sa in human-cell and -tissue lysates exposed to ADAM10 inhibitor. H441-cell (A) and T2495-tissue (B) lysates (2 μg) were supplemented with 0.5% Roche/Halt protease inhibitors, vehicle (Control) or GI254023X (1 μM), and PEPDAB substrates (10 μM), and incubated for 4 h. The developing fluorescence was measured hourly. The presented data are pM means of duplicate measurements ± SD of processed substrates, and are representative of ten experiments performed. The GI254023X-mediated inhibitions of PEPDAB005, 010 and/or 022 processing with the lysates are significant (H441: p=0.017, 0.0041 and 0.0058; T2495: p=0.05, 0.04 and 0.08, respectively). The substrate processing data were analyzed using the systematically increased Syntherror/Sigmathreshold parameters from 0.5/0.0 to 0.5/2.0. The obtained data at 4 h incubation are presented as PrAMA ADAM10sa AU of H441-cell (C) and T2495-tissue (D) lysates. PrAMA standard errors were 1.2% to 9%. Decreases of ADAM10sa in H441-cell (E) and T2495-tissue (F) lysates are presented as % of the specific enzyme activities in the control lysates relative to the GI254023X treated lysates.
Modified PrAMA simultaneously measures ADAM10sa and ADAM17sa in Multiple Specimens of Human Tumors
The presented data show that the modified PrAMA can measure with significant sensitivity, specificity and accuracy not only the recombinant human but also the natural (endogenous) mouse and human ADAM10sa and ADAM17sa in mouse and human cell lysates. They also indicate that this test could specifically measure the enzyme activities in tumor lysates. To confirm these findings and test the potential clinical applicability of the enzyme-activity assay, we simultaneously examined ADAMA10 and ADAM17 protein levels and specific sheddase activities in five human tumor tissues obtained from NSCLC patients (Fig. 8). We found that all the lysates contained significant, but various quantities of ADAM10 and ADAM17 proteins, having approximately 13-fold more ADAM17 than ADAM10, as measured by ELISA (Fig. 8A).
Modified PrAMA efficiently measures ADAM10sa and ADAM17sa in multiple human tumor-tissue specimens. (A) NSCLC tumor lysates contain less ADAM10 than ADAM17 protein. ADAM10 and ADAM17 were quantified in lysates of 5 human NSCLC tumor tissues using ELISAs. Presented data are means pg/10 μg lysates of ADAM10 and ADAM17 proteins ± SD of 5 tumor tissues. (B) NSCLC tissue lysates process high amounts of PEPDAB05, 008 and 010, moderate amounts of PEPDAB014 and 022, and low amounts of PEPDAB011 and 013. Duplicates of 150 μL of Tris-based reaction buffer supplemented with NSCLC tumor-tissue lysates (10 μg/replicate), 0.5% Roche/Halt protease inhibitors and 10 μM of PEPDABs were incubated at 37oC, and fluorescence quantified hourly for 4 h. Data are pM means of duplicate measurements ± SD of processed substrates with the 5 tissue lysates. (C, E) PrAMA ADAM10sa and (D, F) PrAMA ADAM17sa are robust but quantitatively different. The 4 h substrate processing data were analyzed using the systematically increased Syntherror/Sigmathreshold scripts from 0.5/0.0 to 0.5/2.0. Presented data are of the individual tissue lysates. PrAMA standard errors were 2.7% to 5.4%. Proportion-equation analysis of rADAM10 and rADAM17, and the tissue lysate PrAMA-ADAM10sa and PrAMA-ADAM17sa, respectively, showed that 10 μg of these tissue lysates contained 20.0 ng of ADAM10sa and 1.25 ng of ADAM17sa.
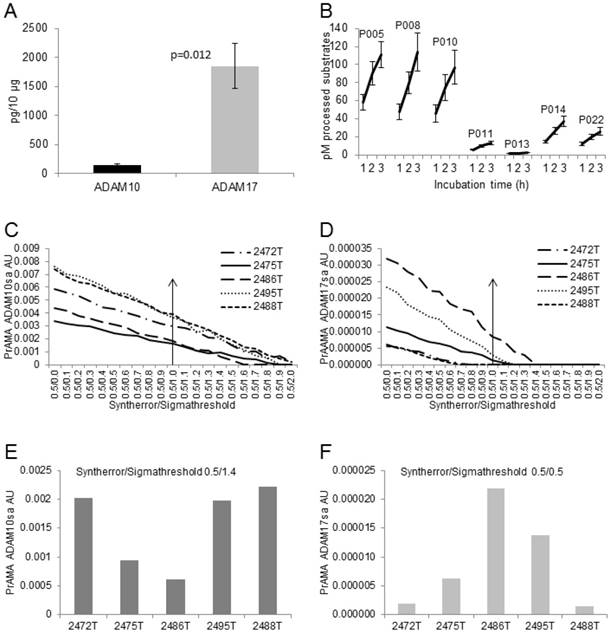
Mimicking a small-scale clinical laboratory testing, we simultaneously examined in duplicates the enzyme activities of the five tumor lysates using 7 PEPDAB substrates. The preparation of dilutions of lysates and substrates and their loading into 96-well assay-plate took 0.5 h. The incubations and measurements of the fluorescence required 4 h. The data analysis required additional 3 h. The obtained enzyme-activity duplicate data were robust and with less than 5% standard deviation. The resulted PrAMA ADAM10sa and ADAM17sa data were also robust and similar to those obtained with the lysates of H441 cells and T2495 tumor tissue (Figs. 7, 8), indicating their consistency. We found that the five tumor-tissue lysates characteristically processed substrates, showing a high efficiency against PEPDAB005, 008 and 010, a moderate efficiency against PEPDAB014 and 022, and a low efficiency against PEPDAB011 and 013. However, the levels of enzyme activities of the lysate specimens notably varied, as supported by the large standard deviations of their means (Fig. 8B). Consistent with the substrate processing data, the high specificity PrAMA (Syntherror/Sigmathreshold scripts ≥0.5 /1.4) measured relatively large, but various amounts of ADAM10sa in the tumor lysates (Figs. 8C, 8E). On the other hand, the high specificity PrAMA could not detect any ADAM17sa in these tumor lysates (Fig. 8D). However, the lower specificity PrAMA (Syntherror/Sigmathreshold <0.5/0.5 scripts) also measured significant, but various amounts of ADAM17sa (Figs. 8D, 8F), which inversely correlated with those of ADAM10sa in the same tumor lysates. These findings indicate that ADAM10sa was more abundant than ADAM17sa in these tumor tissues. In addition, the amounts of the enzyme proteins and sheddase activities inversely correlated, indicating that high levels of inactive and low levels of active ADAM10 and ADAM17 were present in NSCLC tumor lysates. Taking together, the modified PrAMA, alike a standard high through-put clinical-laboratory test, can examine simultaneously and with significant sensitivity, specificity and accuracy ADAM10sa and ADAM17sa in multiple tumor-tissue specimens.
Discussion
Clinically applicable tests that could measure specific enzyme activities are unavailable [1]. PrAMA has been recently developed as a high throughput real-time assay to specifically measure the individual activities of relatively small variety of plasma-membrane integral MPs of live cells [35]. Herein, we introduce a clinically applicable modification of PrAMA to specifically measure ADAM10sa and ADAM17sa in cells and solid tissues. The modification includes selective measurements of MP activities in cell and tissue lysates supplemented with non-MP protease inhibitors, and maximization of ADAM10sa and ADAM17sa specificity by performing PrAMA while systematically changing the specificity Sigmathreshold parameter.
The optimal concentration of Roche/Halt non-MP protease inhibitors of 0.5% suppressed 60% to 80% of enzyme activities in cell and tissue lysates without affecting recombinant or natural ADAM10sa and ADAM17sa. Thus, the selective inhibition of non-MP proteases enabled differential processing of multiple peptide substrates by rADAM10 and rADAM17; lysates of wild-type and ADAM10 or ADAM17 knockout MEFs, ADAM10 knockout and ADAM10 restored MEFs, control and ADAM10 or ADAM17 silenced cancer cells; and control and ADAM10sa-inhibited cancer-cell and tumor-tissue lysates. The observed quantitative differences of substrate processing and related PrAMA indicated that 35% to 60% of the remaining enzyme activities in the lysates supplemented with non-MP protease inhibitors were mediated by ADAM10sa and ADAM17sa. The findings indicate that the selective suppression of non-MP proteases leads to decreases of the enzyme complexity, increases of ADAM10sa and ADAM17sa proportions and, consequently, increases of PrAMA accuracy while measuring the sheddase activities in cell and tissue lysates.
The original PrAMA measured with a significant accuracy the individual MP activities in the limited complexity mixtures of the recombinant or cell-surface enzymes using the single Syntherror 0.5/Sigmathreshold 1.0 parameter [35]. The high-sensitivity Syntherror parameter 0.5 could be also used in the modified PrAMA, and was able to quantify relatively small amounts of the recombinant (0.5 ng) and natural (0.5 μg lysate) ADAM10sa and ADAM17sa. However, using the original PrAMA Sigmathreshold (specificity) parameter 1.0, the modified-PrAMA could not measure with significant specificities ADAM10sa and ADAM17sa in cell and tissue lysates. This could be caused by higher complexities and variabilities of MP mixtures in cell and tissue lysates than on the cell surface. To accurately measure ADAM10sa and ADAM17sa in the lysates, we customized the maximal specificity PrAMA for each cell type or tumor-tissue specimen tested by performing the analysis via systematic increases of the specificity Sigmathreshold parameter. The systematic PrAMA showed simultaneous and gradual decreases of false-positive and increases of true-positive ADAM10sa and ADAM17sa proportions, and defined the applicable maximal specificity PrAMA. Thus modified PrAMA could measure mainly or exclusively true-positive ADAM10sa and ADAM17sa of the recombinant, MEF, cancer-cell and tumor-tissue enzymes using the specificity Sigmathreshold parameters 1.0 and 0.0, 1.7 and 0.8, 1.4 and 1.4, and 1.4 and 0.8, respectively. Therefore, different amounts of ADAM10sa and ADAM17sa present in different cells and tissues could be measured with a significant accuracy using the PrAMA Syntherror/Sigmathreshold parameters 0.5/1.4 to 0.5/1.7 and 0.5/0.8 to 0.5/1.4, respectively.
More ADAM10 than ADAM17 protein was found expressed on the cell surface of NSCLC cells. Inversely, more ADAM17 than ADAM10 protein was found in the cell and tumor lysates comprising both cell-surface and cytoplasmic molecules. Correlating with the cell-surface protein expression, more ADAM10sa than ADAM17sa was found in the lysates. These findings confirm that the active forms of ADAM10 and ADAM17 are selectively expressed on the cell surface, where they perform sheddase activities, whereas the inactive pro-enzymes are primarily distributed inside of cells, where they are produced and/or temporarily stored. The findings also indicate that, in the presence of non-MP protease inhibitors, PrAMA selectively measured the cell-surface ADAM10sa and ADAM17sa in cell and tissue lysates.
Testing recombinant enzymes, PrAMA-ADAM10sa specificity was found to be lower than PrAMA-ADAM17sa specificity. This might be caused by the higher specificity of PEPDAB cleavage sites for ADAM17sa than ADAM10sa. Inversely, when cell and tissue lysates were tested, PrAMA-ADAM10sa specificity was found to be higher than PrAMA-ADAM17sa specificity. These differences of PrAMA specificities for recombinant and natural enzymes might be caused by the high and similar levels of recombinant ADAM10sa and recombinant ADAM17sa, and high ADAM10sa and low ADAM17sa levels in the lysates. These findings and considerations indicate that quantities of active enzymes could influence not only PrAMA sensitivity but also PrAMA specificity.
In conclusion, we describe herein development of a PrAMA modification as a potential biomarker assay to specifically measure ADAM10sa and ADAM17sa in cell and tissue lysates by assessing their proteolytic activities in the presence of non-MP inhibitors and by maximizing the assay specificity via systematic changes of PrAMA-Sigmathreshold parameters. The modified PrAMA efficiently and with significant sensitivity and specificity measures the presence, absence, restoration and inhibition of ADAM10sa and/or ADAM17sa in cell and tumor-tissue lysates; and shows the essential characteristics of a robust high throughput clinically valuable assay that can specifically and accurately measure the sheddase activities in multiple specimens of solid tumors. The modified PrAMA is a unique biomarker assay. It is different from the original PrAMA that specifically measures live-cell MP activities in the absence of the protease inhibitors and using a single specificity parameter to infer the individual MPs from substrate-processing data [35]. Additionally, the modified PrAMA is especially different from and likely superior as a biomarker assay to standard gene- and protein-expression assays measuring without discrimination both inactive and active enzyme forms [21-34], sharply contrasting the modified PrAMA that selectively measures the specific enzyme proteolytic activities - the only enzyme forms directly relevant to cellular functions. Further studies are needed to determine the biomarker ability of the modified PrAMA.
Abbreviations
ADAM, A Disintegrin And Metalloproteinase; ADAM10sa, ADAM10 sheddase activity; ADAM17sa, ADAM17 sheddase activity; rADAM10, recombinant ADAM10; rADAM17, recombinant ADAM17; MP, Metalloproteinase; FRET, Fluorescence-Resonance Energy Transfer; PrAMA, Proteolytic Activity Matrix Analysis; TNF, Tumor Necrosis Factor; TACE, Tumor necrosis factor Alpha Converting Enzyme; EGF, Epidermal Growth Factor; AR, Amphiregulin; HB-EGF, Heparin-Binding EGF, EGFR, EGF receptor; MEF, mouse embryonic fibroblast.
Acknowledgements
This study was supported by VA Merit Award Grant 1-I01-BX000993-01/VUJ-ONCA-053-12S (N. L. Vujanovic), and in part by NIH P30CA047904 for using the UPCI shared resources Flow Cytometry Facility. We thank to Dr. Carl Blobel, and Dr. Peter Dempsey for generously providing the ADAM10 and ADAM17 knockout MEFs, respectively. We also thank Dr. Jill Siegfried, Dr. William Bigbee and the UPCI Lung Cancer SPORE for providing NSCLC tumor specimens; and to Lazar Vujanovic for reviewing the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? European J. Cell Biol. 2011;90:527-35
2. Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34:1-5
3. Black RA, Rauch CT, Kozlosky CJ. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-[alpha] from cells. Nature. 1997;385:729-33
4. Moss ML, Jin SL, Milla ME. et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733-6
5. Lee DC, Sunnarborg SW, Hinkle CL. et al. TACE/ADAM17 Processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci. 2003;995:22-38
6. Horiuchi K, Miyamoto T, Takaishi H. et al. Cell surface colony-stimulating aftor 1 can be cleaved by TNF-alpha converting enzyme or endocytosed in a clathrin-dependent manner. J Immunol. 2007;179:6715-24
7. Peschon JJ, Slack JL, Reddy P. et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281-4
8. Rovida E, Paccagnini A, Del Rosso M. et al. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol. 2001;166:1583-9
9. Sahin U, Blobel CP. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Letters. 2007;581:41-4
10. Sahin U, Weskamp G, Kelly K. et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769-79
11. Sunnarborg SW, Hinkle CL, Stevenson M. et al. Tumor necrosis factor-alpha ocnverting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem. 2002;277:12838-45
12. Black RA, Doedens JR, Mahimkar R. et al. Substrate specificity and inducibility of TACE (tumour necrosis factor alpha-converting enzyme) revisited: the Ala-Val preference, and induced intrinsic activity. Biochem Soc Symposia. 2003;70:39-52
13. Vujanovic NL. Role of TNF superfamily ligands in innate immunity. Immunol Res. 2011;50:159-74
14. Hartmann D, de Strooper B, Serneels L. et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615-24
15. Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact mediated axon repellent. Science. 2000;289:1360-5
16. Janes PW, Saha N, Barton WA. et al. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;23:291-304
17. Sahin U, Weskamp G, Kelly K. et al. Distinct role for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769-79
18. Reiss K, Maretzky T, Ludwig A. et al. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signaling. EMBO J. 2005;24:742-52
19. Ranganthan P, Weaver KL, Capobianco AJ. Notch signaling in solid tumors a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338-51
20. Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511-24
21. Murphy G. The ADAMs: significant scissors in the tumor microenvironment. Nat Rev Cancer. 2008;8:929-41
22. Lendeckel U, Kohl J, Arndt M. et al. Increased expression of ADAM family members in human breast and breast cancer lines. J Cancer Res Clin Oncol. 2005;131:41-8
23. Zheng X, Jiang F, Katakowski M. et al. Inhibition of ADAM17 reduces hypoxia-induced brain tumor cell invasiveness. Cancer Sci. 2007;98:674-84
24. Blanchot-Jossic F, Jarry A, Masson D. et al. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156-63
25. Roemer A, Schwettmann L, Yung M. et al. Increased mRNA expression of ADAMs in renal cell carcinoma and their association with clinical outcome. Oncol Rep. 2004;11:529-36
26. Zhou BB, Peyton M, He B. et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39-50
27. Tanaka Y, Miyamoto S, Suzuki SO. et al. Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin Cancer Res. 2005;11:4783-92
28. Karan D, Lin FC, Bryan M. et al. Expression of ADAM (a disintegrin and metalloproteases) and TIMP-3 (tissue inhibitor of metalloproteinase-3) in human prostate adenocarcinomas. Int J Oncol. 2003;23:1365-71
29. McGowan PM, McKiernan E Bolster F. et al. ADAM-17 predict adverse outcome in patients with breast cancer. Ann Oncol. 2008;19:1075-81
30. Gavert N, Conacci-Sorrell M, Gast D. et al. L1, a novel target of betta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168:633-42
31. Yoshimura T, Tomita T, Dixon MF. et al. ADAMs (a disintegrin and metalloprotease) messenger RNA expression in Helicobacter pylori-infected, normal and neoplastic gastric mucosa. J Infect Dis. 2002;185:332-40
32. Wu E, Croucher PI, McKie N. Expression of members of the novel membrane linked metalloproteinase family ADAM in cells derived from a range of haematological malignancies. Biochem Biophys Res Commun. 1997;235:437-42
33. McCulloch DR, Aki P, Samaratunga H. et al. Expression of the disintegrin metalloprotease, ADAM-10, in prostate cancer and its regulation by dihydrotestosterone, insulin-like growth factor 1, and epidermal growth factor in the prostate cancer cell model LNCaP. Clin Cancer Res. 2004;10:314-23
34. Fogel M, Gutwein P, Mechtersheimer S. et al. L1 expression as a predictor of progression and survival in patients with uterine and ovarian carcinomas. Lancet. 2003;362:869-75
35. Miller MA, Barkal L, Jeng K. et al. Proteolytic activity matrix analysis (PrAMA) for simultaneous determination of multiple protease activities. Integr Biol (Camb). 2010;3:422-38
36. Moss ML, Rasmussen FH, Nudelman R. et al. Fluorescent substrates useful as high throughput screening tools for ADAM9. Comb Chem High Throughput Screen. 2010;13:358-65
37. Lambert MH, Blackburn RK, Seaton TD. et al. Substrate specificity and novel selective inhibitors of TNF-alpha converting enzyme (TACE) from two-dimensional substrate mapping. Comb Chem High Throughput Screen. 2005;8:327-39
38. Moss ML, Rasmussen FH. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem. 2007;366:144-8
39. Alabi RO, Glomski K, Haxaire C. et al. ADAM10-dependent signaling through Notch1 and Notch4 controls development of organ-specific vascular beds. Circ Res. 2016;119:519-31
40. Gelling RW, Yan W, Al-Noori S. et al. Deficiency of TNF alpha converting enzyme (TACE/ADAM17) causes a lean, hypermetabolic phenotype in mice. Endocrinology. 2008;149:6053-64
41. Paudel S, Kim YH, Huh MI. et al. ADAM10 mediates N-cadherin ectodomain shedding during retinal ganglion cell differentiation in primary cultured retinal cells from the developing chick retina. J Cell Biochem. 2013;114:942-54
Author contact
![]() Corresponding author: Nikola L. Vujanovic, University of Pittsburgh Cancer Institute, Hillman Cancer Center, Research Pavilion, 51117 Centre Avenue, Pittsburgh PA, 15213; phone number, 412-623-3211; fax number, 412-623-1119; e-mail address, vujanovicnledu
Corresponding author: Nikola L. Vujanovic, University of Pittsburgh Cancer Institute, Hillman Cancer Center, Research Pavilion, 51117 Centre Avenue, Pittsburgh PA, 15213; phone number, 412-623-3211; fax number, 412-623-1119; e-mail address, vujanovicnledu