Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(19):4027-4039. doi:10.7150/jca.21256 This issue Cite
Research Paper
UHRF1 is an Independent Prognostic Factor and a Potential Therapeutic Target of Esophageal Squamous Cell Carcinoma
Jiecheng Ye1, 2*, Yong Zhang3*, Weiye Liang1, 2, Jianxian Huang1, 2, Lihui Wang1, 2 ![]() , Xueyun Zhong1, 2
, Xueyun Zhong1, 2 ![]()
1. Department of Pathology, Medical College, Jinan University, Guangzhou 510632, China;
2. Guangdong Province Key Laboratory of Molecular Immunology and Antibody Engineering, Jinan University, Guangzhou 510632, China;
3. Institute of Pathology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
*Contributed to this work equally
Received 2017-5-30; Accepted 2017-10-10; Published 2017-10-23
Abstract
Purpose: Ubiquitin-like with plant homeodomain and ring-finger domains 1 (UHRF1) plays an essential role in DNA methylation, and the overexpression of UHRF1 is associated with poor prognosis in various cancers. Esophageal squamous cell carcinoma (ESCC) accounts for approximately 90% of esophageal cancer cases in China, but the five-year survival rate for patients is less than 10% due to limited clinical approaches for early diagnosis and treatment. The present research aimed to investigate the expression of UHRF1 in ESCC and its biological role in ESCC development.
Methods: UHRF1 expression in ESCC and normal esophageal tissues was examined using immunohistochemical staining, followed by analysis of the correlation between UHRF1 expression and clinical features. In addition, the effects of lentivirus-mediated RNA interference of UHRF1 on global DNA methylation, cell proliferation, cell cycle progression and apoptosis and were investigated in ESCC cells.
Results: UHRF1 was overexpressed in ESCC tissues and was an independent prognostic factor for ESCC patients. In ESCC cells, knockdown of UHRF1 caused global DNA hypomethylation, inhibited cell proliferation and induced apoptosis. Furthermore, UHRF1 depletion induced cell cycle arrest at the G2/M phase, accompanied by activation of Wee1 and DNA damage response pathway.
Conclusions: Our findings identify UHRF1 as a promising prognostic marker for ESCC and suggest that UHRF1 may be a potential therapy target for ESCC patients with elevated UHRF1 expression.
Keywords: DNA methylation, esophageal squamous cell carcinoma, prognosis, UHRF1, Wee1
Introduction
Esophageal cancer is the eighth most common cancer and the sixth leading cause of cancer-related death worldwide [1]. China accounts for approximately 50% of esophageal cancer cases, and 90% of these cases are esophageal squamous cell carcinoma (ESCC) [1, 2]. Although the diagnostic approaches and treatments for ESCC have improved in recent years, the over 5-year survival rate for patients is still less than 10% [3]. Therefore, understanding the molecular mechanisms responsible for ESCC pathogenesis will help identify biomarkers for early diagnosis, prognostic assessment and therapeutic targeting.
Epigenetic alterations such as DNA methylation, histone modification, and loss of genome imprinting play a crucial role in ESCC tumorigenesis, along with genetic changes [4, 5]. Aberrant DNA methylation is correlated with ESCC pathogenesis through gene promoter hypermethylation or global DNA hypomethylation [4, 6]. DNA hypermethylation within the gene promoter region of tumor suppressor genes (TSGs) usually leads to transcriptional repression. Many TSGs, such as CDKN2A, CDKN2B, RASSF1 and RARB, have been reported to be frequently silenced by promoter hypermethylation in ESCC [6, 7]. DNA hypomethylation of long interspersed transposable element-1 (LINE-1) may predispose cells to chromosomal defects and rearrangements that result in genetic instability. Global DNA hypomethylation increases chromosomal instability and leads to initiation and development of ESCC [5, 8].
Ubiquitin-like with plant homeodomain and ring-finger domains 1 (UHRF1) was identified as a multidomain nuclear protein, which plays a major role in DNA methylation and histone methylation [9]. UHRF1 recognizes hemimethylated CpG sites on newly synthesized DNA strands and recruits DNA methyltransferase 1 (DNMT1) to catalyze maintenance of DNA methylation [10-13]. UHRF1 is related to various physiological and pathological phenomena, including embryogenesis, cell migration and cancer development and progression [14]. Many studies have reported that UHRF1 is overexpressed in various cancer types, such as bladder cancer, lung cancer and colorectal cancer, and confirmed UHRF1 as a valuable diagnostic and prognostic biomarker for timely cancer detection, progression and therapeutic response monitoring[15-19]. Furthermore, UHRF1 depletion inhibits cell proliferation and migration, and induces cell cycle arrest and apoptosis through DNA demethylation, suggesting that UHRF1 can be a useful therapy target of cancers [17, 20-22]. Thus many strategies were proposed to target UHRF1, including use of small molecular compounds [19, 22].
Previous studies have demonstrated that UHRF1 is involved in the initiation and progression of cancer and can be a valuable biomarker for cancer diagnosis and prognosis, as well as a therapeutic target [15-19, 22]. However, the prognostic value and potential biological role of UHRF1 in ESCC remain unclear. In the present study, we aimed to investigate the expression and prognostic value of UHRF1 in ESCC and explore its molecular role in DNA maintenance, cell proliferation, cell cycle progression and apoptosis.
Materials and Methods
Patients and tissue samples
Paraffin-embedded samples of ESCC, which were diagnosed clinically and pathologically, were collected from 84 patients between 2004 and 2008 in Meizhou People's Hospital, China. None of the patients received radiotherapy or chemotherapy before surgery, and none of them had multiple cancers in other organs. Prior informed consent was obtained from all patients, and this study was approved by the research Ethics Committee of Meizhou People's Hospital. The following clinicopathological parameters were collected from the medical records: age, sex, histological grade, depth of invasion, and clinical stage. The histopathological diagnosis was based on the World Health Organization criteria. Tumor staging was determined according to the 6th edition of the tumor-node-metastasis (TNM) classification of the International Union Against Cancer.
Immunohistochemical staining
The paraffin-embedded samples of ESCC and normal tissues were cut into 5-µm-thick sections and placed on pathological slides for immunohistochemical staining. Tissue sections were heated at 100 °C in citrate buffer solution (pH=6.0) for 10 min to facilitate antigen retrieval. Then, the sections were incubated with rabbit antibody against human UHRF1 (1:400, Novus, USA) for 3 h followed by incubation with secondary antibody (Dako REAL EnVision, USA). Immunoreacted cells were visualized using diaminobenzidine, and nuclei were counterstained with hematoxylin. Phosphate-buffered saline (PBS) was substituted for the primary antibody as a negative control. Sections were independently evaluated microscopically by two pathologists (Jiecheng Ye and Yong Zhang) without knowledge of the clinicopathological features. UHRF1 staining was considered positive if ≥5% of cells presented strong nuclear staining.
Cell culture and transfection
The ESCC cell lines ECA109 and TE-1 were purchased from Shanghai Institute of Cell Biology (Shanghai, China) and were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were maintained in an atmosphere containing 5% CO2 at 37 °C. Lentiviruses carrying shRNA targeting human UHRF1 or non-targeting control lentiviral vectors (GV248) were constructed by GeneChem (Shanghai, China). TE-1 and ECA109 cells were transfected with the viruses according to the manufacturer's instructions. After 48 h, medium containing puromycin was used to select stable clones. The clones with stable knockdown of UHRF1 were identified and verified using qRT-PCR and Western blotting.
CCK-8 assay
Cell proliferation was detected using Cell Counting Kit-8 (CCK-8) assays according to the manufacturer's instructions. Briefly, the cells were seeded into a 96-well plate at a density of 3000 cells/well. After incubation, 10 µl of CCK-8 reagent (Dojindo, Japan) was added to each well, and the cells were cultured for another 2 h. At the end of the incubation, the optical density at 450 nm was analyzed with a microplate reader (BioTek, USA).
Colony formation assay
A total of 120 cells were placed in a well of a 6-well plate and maintained in media containing 10% FBS; the medium was replaced every 4 days. After 14 days, the cells were fixed with 4% formaldehyde and stained with 0.1% crystal violet (Beyotime, China). Visible colonies were counted manually, and each group included triplicate wells.
Flow cytometry assay
Briefly, 1×105 cells were seeded in a well of a 6-well plate and maintained in medium containing 10% FBS. After 48 h, the cells were harvested and then resuspended with cold PBS. For cell cycle analysis, cells were fixed in 70% ethanol overnight at 4 °C. Fixed cells were washed with PBS, incubated with 50 µg/ml propidium iodide (BD Pharmingen, USA) and 100 µg/ml RNaseA in the dark for 30 min. Cell cycle phase distribution was assessed using a FACScan flow cytometer (BD Biosciences, USA), and data were analyzed using Becton-Dickinson Cell Quest software. For apoptosis analysis, the cells were resuspended in binding buffer and incubated with Annexin V-APC/PI (eBioscience, USA) at room temperature for 15 min. After incubation, the percentage of apoptotic cells was measured with a FACSCalibur flow cytometer (BD Biosciences, USA).
RNA extraction and quantitative RT-PCR
Total RNA was isolated using TRIzol (Invitrogen, USA) following the protocol supplied by the manufacturer. Reverse transcription PCR was performed using a PrimeScript RT Reagent kit (TaKaRa, Japan). The qPCR analysis was performed in a CFX96 Real-Time PCR Detection System (Bio-Rad, USA) using a SYBR Green Real-Time PCR kit (TaKaRa, Japan). The PCR reaction conditions were 30 s at 95 °C followed by 40 cycles of 5 s at 95 °C and 30 s at 60 °C. Relative mRNA values were normalized to the expression of the GAPDH gene using the 2-∆∆Ct method. The primer sequences used were as follows: UHRF1, (forward) 5'-AAGGTGGAGCCCTACAGTCTC-3' and (reverse) 5'-CACTTTACTCAGGAACAACTGGAAC-3' [23]; human GAPDH, (forward) 5'-ATCAATGGAAATCCCATCACCA-3' and (reverse) 5'-GACTCCACGACGTACTCAGCG-3' [24].
Western blotting
Protein expression was detected using Western blotting. In brief, cells were lysed in radio immunoprecipitation assay buffer containing protease inhibitors. The obtained protein samples were subjected to10% SDS-PAGE and transferred to PVDF membranes (Millipore, USA). After being blocked with 5% fat-free milk in Tris-buffered saline containing 0.2% Tween-20, the membranes were incubated overnight at 4 °C with primary antibodies. Following incubation with horseradish peroxidase-conjugated secondary antibodies, immunoreactive bands were visualized using enhanced chemiluminescence reagent (Millipore, USA). Densities of the immunoreactive bands were determined by ImageJ (National Institutes of Health, USA).
Immunofluorescence
Cells were cultured on cover slips for 48 h. Then, the cells were fixed in 4% formaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for 10min. After blocking in 5% bovine serum albumin (BSA) for 1h, cells were incubated with rabbit anti-human monoclonal phosphorylated H2AX (1:400; Cell Signaling Technology, USA) overnight at 4°C. Cell slides were then incubated with Alexa Fluor 647-conjugated anti-rabbit IgG (1:1000; Cell Signaling Technology, USA) for 1 h and counterstained with DAPI for 15 min at room temperature. Finally, the slides were imaged at a magnification of ×400 under a fluorescence microscope (Leica, Germany) and analyzed with Image-Pro Plus softwafe(Media Cybernetics, USA).
DNA extraction and bisulfite modification
Genomic DNA was extracted from transfected cells using a TaKaRa MiniBEST Universal Genomic DNA Extraction Kit (TaKaRa, Japan) according to the manufacturer's protocol. DNA concentration and purity were determined using a Nano-1000 spectrophotometer (Thermo, USA). After DNA quantification, bisulfite modification was performed using an EZ DNA Methylation-Gold Kit (Zymo Research, Germany) following the manufacturer's instructions. This treatment converted unmethylated cytosine residues into uracil, while the methylated cytosine residues remained unchanged.
Measurement of LINE-1 methylation by pyrosequencing
PCR and pyrosequencing for LINE-1 were performed as previously reported[25] using TaKaRa Taq™ Hot Start Version (Takara, Japan). A total volume of 40 µl of PCR amplification reagent consisted of the forward and reverse primers (each 0.1 µmol/l), 0.2 mmol/l dNTPs, 10× PCR buffers, 2.5 U of Takara Hot Start Taq polymerase, and 2 µl of bisulfited template DNA. PCR conditions were as follows: initial denaturing at 95 °C for 3 min; 45 cycles of 95 °C for 15 s, 50 °C for 20 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min. The primers used in the amplification were as follows: LINE-1, (forward) 5'-TTTTTTGAGTTAGGTGTGGGATA-3' and (reverse) 5'-AAAAATCAAAAAATTCCCTTTCC-3'. The PCR products were purified and sequenced with pyrosequencing PyroMark ID (Qiagen, USA) according to the manufacturer's instructions. The number of C residues divided by the sum of the C and T residues at each CpG site was calculated as a percentage. The average of the relative number of C residues in the 3 CpG sites was considered the overall LINE-1 methylation level in the specimen.
Statistical analysis
All data were analyzed with SPSS 19.0 software (IBM, USA). The association between UHRF1 expression and clinicopathological variables was assessed using a chi-square test. The Kaplan-Meier method and log-rank tests were used to compare the overall survival. Multivariate analyses of variables were conducted using a Cox proportional hazards regression model. The numerical data are reported as the mean ± standard deviation (SD), and mean values among multiple groups were compared using one-way ANOVA. Two-way ANOVA was used to compare the LINE-1 methylation levels among different groups. All analyses were considered statistically significant when P<0.05 or P<0.01 was obtained.
Results
UHRF1 expression was elevated and found to be an independent prognostic factor in ESCC
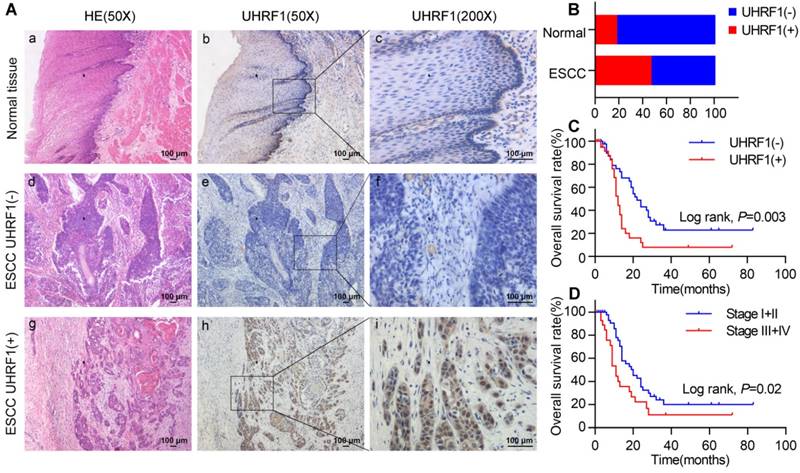
We evaluated the expression of UHRF1 in 84 ESCC tissues and 40 adjacent normal esophageal tissues using immunohistochemistry (IHC). Representative tissues with IHC staining are shown in Fig 1A. The expression of UHRF1 presented mainly in the nucleus, and thus, brown nuclear immunoreactivity for UHRF1 was identified as positive staining. We defined the expression of UHRF1 in ESCC as positive when the percentage of positive-staining tumor cells was more than 5. Of the 84 patients studied, 39 cases (46.4%) showed positive expression, while 7 of the 40 corresponding normal tissues (17.5%) showed positive expression. The results revealed that the UHRF1 expression level in ESCC was significantly higher than in normal tissue (P=0.002, Fig 1B).
UHRF1 expression is elevated and correlated with poor prognosis in ESCC. A. HE staining and UHRF1 immunostaining in ESCC and normal esophageal tissue (magnification, 50× and 200×). (a, d, g) Representative HE staining of ESCC and normal esophageal tissue; (b, c) Negative expression of UHRF1 in nuclei of cells in normal esophageal tissue; (e, f) Negative expression of UHRF1 in nuclei of ESCC cells; (h, i) Positive expression of UHRF1 in nuclei of ESCC cells. B. The histogram illustrates the percentage of UHRF1 immunostaining in ESCC and normal esophageal tissue. ESCC tissues showed significantly higher UHRF1 expression levels than normal esophageal tissue (chi-square test, P=0.002). C. Kaplan-Meier curves according to UHRF1 expression status in ESCC. Patients with positive UHRF1 expression have shorter overall survival time than patients with negative UHRF1 expression (log-rank test, P=0.003). D. Kaplan-Meier curves according to TNM stage in ESCC. Patients at stage III+IV have shorter overall survival time than patients at stage I+II (log-rank test, P=0.02).
The correlations between UHRF1 expression and clinicopathological variables are shown in Table 1. Positive expression of UHRF1 was not related to clinicopathological characteristics, including gender, age, histological grade, depth of invasion, lymph node metastasis and tumor stage (P>0.05).
Relationships between UHRF1 expression and clinical pathological parameters in ESCC patients
| Variables | N | UHRF1 Expression | P | |
|---|---|---|---|---|
| Negative | Positive | |||
| Sex | 0.115 | |||
| Male | 67 | 33 | 34 | |
| Female | 17 | 12 | 5 | |
| Age | 0.274 | |||
| ≤55 | 42 | 20 | 22 | |
| >55 | 42 | 25 | 17 | |
| Tumor Size | 0.491 | |||
| ≤5.0 | 44 | 22 | 22 | |
| >5.0 | 40 | 23 | 17 | |
| Histological Grade | 0.108 | |||
| Ⅰ | 12 | 9 | 3 | |
| Ⅱ-Ⅲ | 72 | 36 | 36 | |
| T stage | 0.945 | |||
| T1-T2 | 24 | 13 | 11 | |
| T3-T4 | 60 | 32 | 28 | |
| N stage | 0.726 | |||
| N0 | 50 | 26 | 24 | |
| N1 | 34 | 19 | 15 | |
| Tumor Stage | 0.344 | |||
| Ⅰ-Ⅱ | 54 | 31 | 23 | |
| Ⅲ-Ⅳ | 30 | 14 | 16 | |
In a univariate analysis, Kaplan-Meier curves showed that patients with negative UHRF1 expression survived significantly longer than patients with positive UHRF1 expression (P=0.003, Fig 1C and Table S1). The mean survival time of patients with negative UHRF1 expression was 33.2±4.9 months, but it decreased to 17.0±3.4 months in patients with positive UHRF1 expression. The Kaplan-Meier analysis also demonstrated that tumor stage was a prominent prognostic factor (P=0.02, Fig 1D). Multivariate analysis revealed that positive UHRF1 expression (HR=2.161, 95%CI=1.219-3.833, P=0.008, Table 2) was independent prognostic factors for poor prognosis in ESCC along with tumor stage (HR=1.772, 95%CI=1.004-3.128, P=0.049).
Multivariate analysis for overall survival of ESCC patients
| Variables | Hazard Radio | 95% confidence interval | P Value |
|---|---|---|---|
| Tumor Stage* | 1.772 | 1.004-3.128 | 0.049 |
| UHRF1 Expression** | 2.161 | 1.219-3.833 | 0.008 |
*Stages Ⅲ-Ⅳ versus stages Ⅰ-Ⅱ; **Positive expression versus negative expression.
Knockdown of UHRF1 using lenti-shRNA induced global DNA hypomethylation in ESCC cells
To investigate the role of UHRF1 in ESCC cells, UHRF1 protein expression levels were first detected in ESCC cell lines. All of the ESCC cell lines examined showed positive UHRF1 expression (Fig 2A). Then, the ECA109 and TE-1 cell lines were chosen for transfection with lenti-shUHRF1 or shNC containing GFP because of their relatively high UHRF1 expression. The efficiency of lenti-shRNA infection in ECA109 and TE-1 cells was measured by observing the GFP expression using fluorescence microscopy and was confirmed to be over 80 percent (Fig 2B). To confirm the knockdown efficiency, both UHRF1 mRNA and protein levels were analyzed. The results showed that the mRNA and protein levels of UHRF1 were significantly downregulated in lenti-shUHRF1 transfected cells (shUHRF1 group) compared with lenti-shNC transfected cells (shNC group) and untransfected cells (Blank group) (Fig 2C and 2D).
Knockdown of UHRF1 using lenti-shRNA caused DNA hypomethylation in ESCC cell lines. A. UHRF1 protein expression in ESCC cell lines. B. The transfection efficiency was determined by visualizing the GFP label with a fluorescence microscope (magnification, 200×). C. The knockdown efficiency of UHRF1 shRNA was confirmed by qRT-PCR. D. The knockdown efficiency of UHRF1 shRNA was confirmed by immunoblotting in ECA109 and TE-1 cells. E. UHRF1 knockdown caused LINE-1 hypomethylation in ESCC cells. (Two-way ANOVA, P<0.01). The average of the relative methylation levels in the 3 CpG sites was considered the overall LINE-1 methylation level.
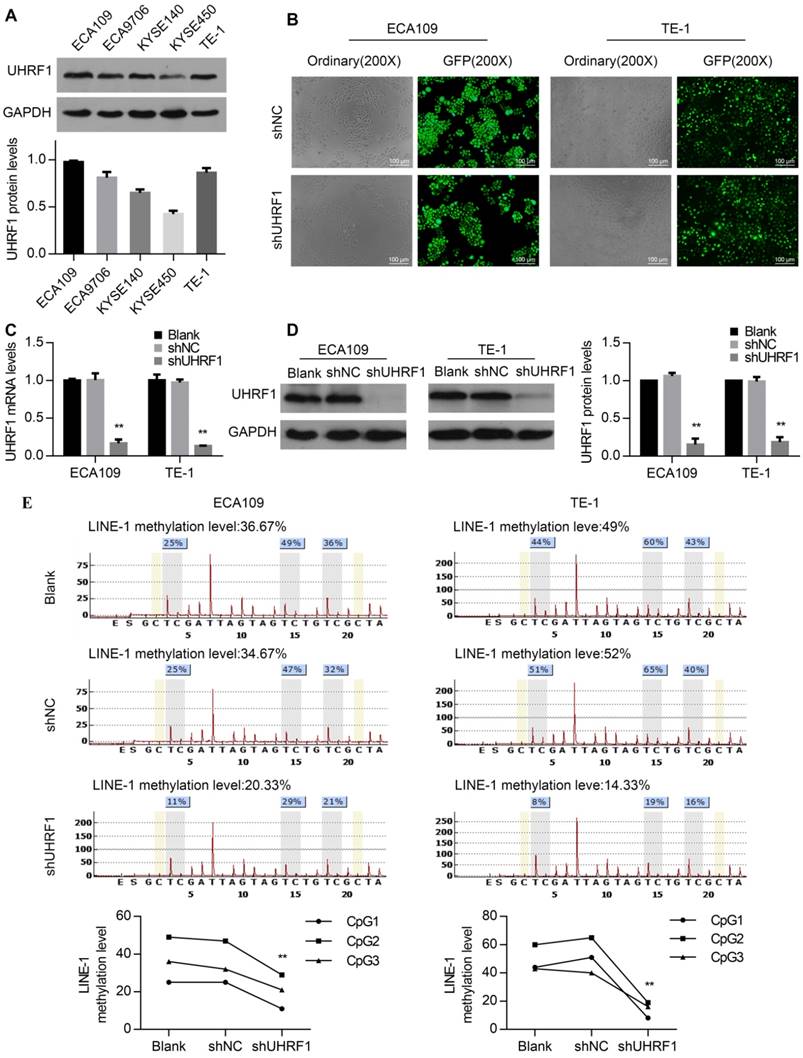
UHRF1 plays an important role in maintaining DNA methylation, UHRF1 alteration can affect the global DNA methylation levels in human, mouse and zebrafish cells [26, 27], but the role in ESCC cells remains unclear. Because LINE-1 constitutes approximately 17% of the entire human genome, the methylation of LINE-1 has been regarded as a surrogate for global DNA methylation status [28]. To investigate whether UHRF1 knockdown downregulated global DNA methylation levels, we detected the LINE-1 methylation levels using pyrosequencing. The results showed that the DNA methylation levels of LINE-1 decreased in UHRF1-silenced cells (P<0.01, Fig 2E). This suggested that UHRF1 regulated the global DNA methylation level in ESCC cells.
Knockdown of UHRF1 inhibited the proliferation of ESCC cells
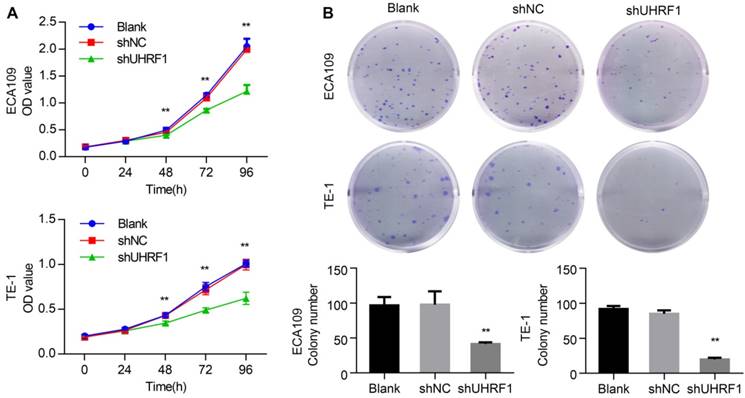
Previous studies have shown that the proliferation of cancer cells was inhibited by UHRF1 knockdown [17, 29]. In this study, we investigated the effect of UHRF1 knockdown in ESCC cell proliferation using CCK-8 assays. The results showed that growth of ECA109 and TE-1 cells was inhibited after knockdown of UHRF1 with shUHRF1 (P<0.01, Fig 3A). In addition, the effect of UHRF1 inhibition was also investigated with colony formation assays. Similarly, the results revealed that the number of colonies decreased significantly following UHRF1 inhibition (P<0.01, Fig 3B). These results suggested that UHRF1 knockdown inhibited the proliferative ability of ESCC cells.
Knockdown of UHRF1 inhibited the growth of ESCC cell lines. A. CCK-8 assays were performed at different time points to detect the effect of knockdown of UHRF1 on the growth of ECA109 and TE-1 cells. B. Colony-forming assays were performed on the same cell lines; the histograms illustrate the number of colonies. Data are presented as the mean ± SD of three independent experiments. **P<0.01.
Depletion of UHRF1 induced apoptosis in ESCC cells
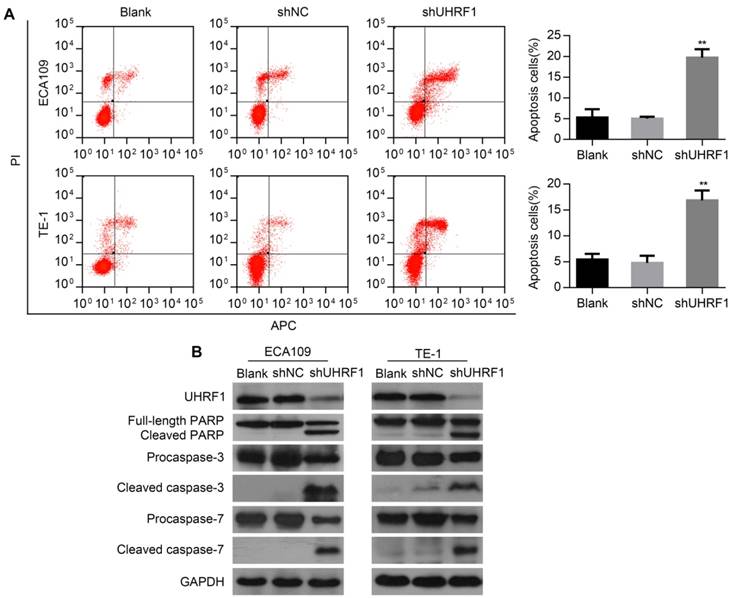
To determine whether depletion of UHRF1 induced apoptosis in ESCC cells, flow cytometry (FCM) was used to detect the apoptotic changes in transfected cells. The results showed that the percentage of apoptosis cells markedly increased both in ECA109 and TE-1 cells depleted of UHRF1 (P<0.01, Fig 4A). Then, Western blotting results showed that depletion of UHRF1 significantly increased the activity of caspase-3 and caspase-7 in ESCC cells (Fig 4B). Additionally, proteolytic cleavage of poly (ADP-ribose) polymerase (PARP) by activated caspase-3 and caspase-7, serving as a marker of apoptosis [30], was also detected in cells depleted of UHRF1 (Fig 4B). These data demonstrated the knockdown of UHRF1 induced apoptosis in ESCC cells.
Knockdown of UHRF1 induced apoptosis in ESCC cells. A. Apoptotic changes in cells after knockdown of UHRF1 were examined with FCM. B. Western blot analysis of apoptosis-related markers in ESCC cells after knockdown of UHRF1. Data are presented as the mean ± SD of three independent experiments. **P<0.01.
The knockdown of UHRF1 induced cell cycle arrest at the G2/M transition in association with activation of Wee1
To characterize the cell cycle distribution of ESCC cells depleted of UHRF1, transfected cells were harvested for FCM. The results showed that the proportion of cells in G2/M phase increased after UHRF1 knockdown in ECA109 and TE-1 cells (P<0.01, Fig 5A), while there was no significant change in the number of cells in G1 phase. In addition, the percentage of cells in S phase decreased after knockdown of UHRF1 in ECA109 cells but not TE-1 cells. The G2/M phase transition is primarily controlled by activation of the Cyclin B1/CDC2 complex [31, 32]. To explore the potential mechanism of cell cycle arrest in G2/M phase, proteins regulating G2/M phase transposition were examined. Western blotting showed that phosphorylation of CDC2Tyr15, the inactivated state of CDC2, increased in cells depleted of UHRF1, while expression of Cyclin B1 decreased (Fig 5B). In addition, phosphorylation of histone H3Ser10 was reduced in cells depleted of UHRF1, indicating a block in mitotic entry. These results demonstrated that UHRF1 depletion induced a cell cycle block at the G2/M transition.
Knockdown of UHRF1 induced cell cycle arrest at G2/M stage in association with activation of Wee1. A. Changes in cell cycle after knockdown of UHRF1 were detected with FCM in ECA109 and TE-1 cells. B. The key regulators of the G2/M transition were analyzed using Western blotting. C. Western blot analysis of key proteins in the G2/M transition after Wee1 inhibition in UHRF1-depleted cells. Data are presented as the mean ± SD of three independent experiments. **P<0.01.
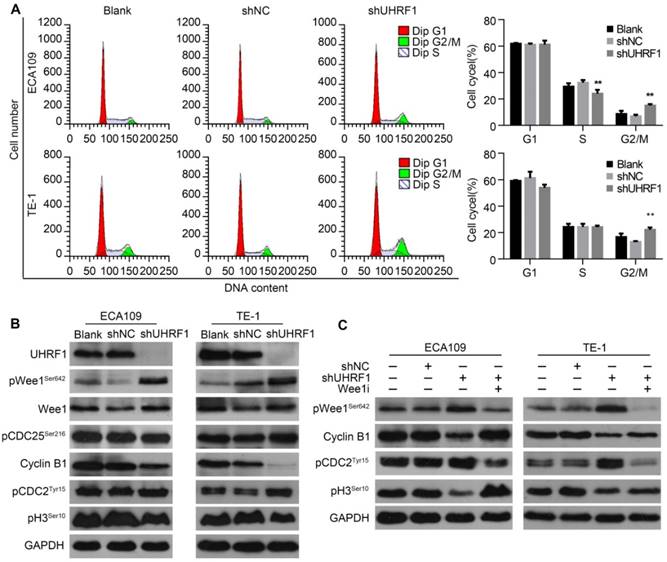
As cells approach the G2/M boundary, the inhibitory phosphorylation of CDC2 at Tyr15 is primarily catalyzed by Wee1 [33]. The critical regulatory step in activating CDC2 during progression into mitosis appears to be dephosphorylation of CDC2 at Tyr15 by CDC25 phosphatase [34]. As shown in Fig 5B, inhibitory phosphorylation of CDC25 at Ser216 remained constant in UHRF1-depleted cells. Surprisingly, phosphorylation of Wee1 at Ser642, the activated state of Wee1, was enhanced in cells depleted of UHRF1, while total Wee1 remained constant. To further confirm the role of Wee1 in cell cycle arrest in cells depleted of UHRF1, we inhibited Wee1 in UHRF1-depleted cells with MK1775, an inhibitor of Wee1, and measured the expression of downstream proteins. The results showed that inhibition of Wee1 induced activation of the Cyclin B1/CDC2 complex and subsequently catalyzed mitosis, indicated by enhanced phosphorylation of histone H3Ser10 (Fig 5C). These results suggested that UHRF1 depletion induced cell cycle arrest at the G2/M transition was correlated with activation of Wee1.
UHRF1 depletion activated the DNA damage response pathway
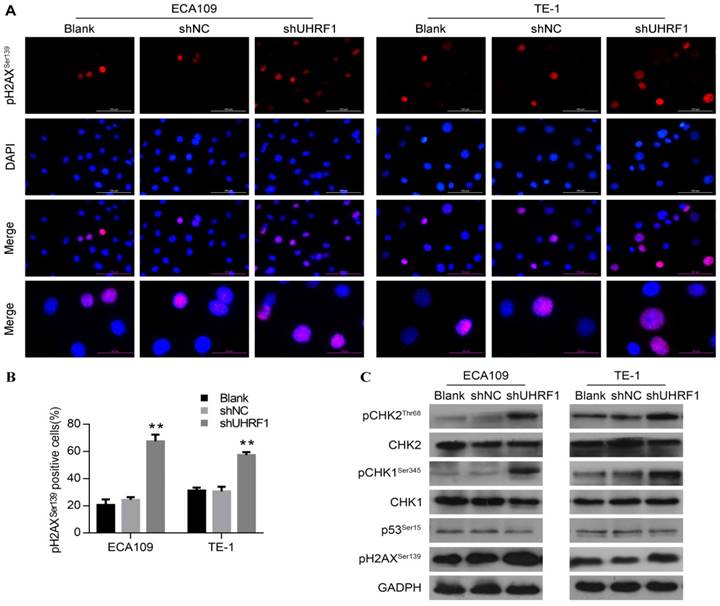
Various studies have demonstrated that the G2/M phase checkpoint can be activated by DNA damage, leading to cell cycle arrest and even apoptosis [31, 35, 36]. In response to DNA damage, Wee1 can be phosphorylated by pCHK1Ser345, resulting in maintenance of phosphorylation of CDC2 at Tyr15 and hence G2 delay [37]. Thus, we hypothesized that the DNA damage response was involved in the G2/M phase arrest and apoptosis observed in UHRF1-knockdown cells. Phosphorylation of H2AX at serine 139 (pH2AXSer139) is a rapid and sensitive cellular response to the presence of DNA damage [38]. To understand whether knockdown of UHRF1 induced DNA damage, we measured pH2AXSer139 content in ESCC cells by immunofluorescence firstly. As shown in Fig 6A and Fig 6B, cells depleted of UHRF1 showed a higher percentage of cells positive for pH2AXSer139 compared to control cells (P<0.01). Subsequently, we detected DNA damage response-related proteins by Western blotting. Consistent with immunofluorescence results, Western blotting showed that pH2AXSer139 increased in cells depleted of UHRF1 (Fig 6C). In addition, phosphorylation of CHK1Ser345 and CHK2Thr68 in both ECA109 and TE-1 cells was increased, while total CHK1 and CHK2 remained unchanged (Fig 6C). However, phosphorylation of p53 at Ser15 remained constant in cells depleted of UHRF1, indicating that p53 may not be involved in this response. Taken together, the results indicated that the DNA damage response pathway was activated in response to UHRF1 knockdown.
Knockdown of UHRF1 activated the DNA damage response in ESCC cells. A. Cells were analyzed with immunostaining for pH2AXSer139, and counterstained with DAPI (magnification, 400×). B. The histogram illustrates the percentage of cells expressing pH2AXSer139 in ESCC cells. Data are presented as the mean ± SD of three independent experiments. **P<0.01. C. Western blot analysis of DNA damage response factors in ESCC cells after knockdown of UHRF1.
Discussion
In the present study, we determined that positive expression of UHRF1 was significantly related to poor prognosis in ESCC, although there were no correlations between UHRF1 expression and clinicopathological features. In ESCC cell lines, knockdown of UHRF1 caused global DNA hypomethylation, inhibited cell proliferation and induced apoptosis. Furthermore, we showed that UHRF1 depletion induced cell cycle arrest at G2/M phase by activation of the DNA damage response pathway.
Like other cancers, in our present study, UHRF1 was overexpressed in ESCC tissues compared with normal esophageal mucosa. In addition, positive expression of UHRF1 was significantly associated with poor prognosis in ESCC. This was consistent with a recent study that reported that UHRF1 overexpression was correlated with poor prognosis in ESCC patients [39]. Moreover, the multivariate survival analysis showed, for the first time, that positive UHRF1 expression was an independent prognostic factor for ESCC patients. However, there were no correlations between UHRF1 expression and clinicopathological features in this study, different with a previous study which reported that UHRF1 overexpression correlated significantly with advanced T-stage, positive lymph node metastasis and poor differentiation in ESCC [40]. Taken together, these findings demonstrated that UHRF1 is a valuable diagnostic and prognostic marker for ESCC patients.
UHRF1 plays an essential role in DNA maintenance and affects genome-wide DNA demethylation [27, 41]. Previous studies have found that both UHRF1 overexpression and inhibition can cause DNA hypomethylation in zebrafish hepatocytes [21, 29, 42]. Another study showed that loss of UHRF1 resulted in a strong loss of DNA methylation in plant and animal genomes, suggesting that UHRF1 plays a conserved and global role in the maintenance of DNA methylation [27]. This study revealed that UHRF1 knockdown led to global DNA hypomethylation in ESCC cell lines, indicated by LINE-1 hypomethylation. It is interesting that global DNA hypomethylation can result from both UHRF1 overexpression and depletion, but the responsible mechanism remains to be clarified. Based on this study and the previous studies, we propose that the mechanisms of UHRF1 depletion and overexpression responsible for DNA hypomethylation are different. 1) Because UHRF1 plays an important role in maintaining DNA methylation by recognizing hemimethylated CpG sites during DNA replication and subsequently recruiting DNMT1, depletion of UHRF1 could directly cause a failure to write the DNA methylome [14, 27, 43]. Therefore, loss of UHRF1 results in a strong decrease in DNA methylation in both gene bodies and transposons of plant and animal genomes [21, 27, 43, 44]. 2) In contrast, UHRF1 overexpression can ubiquitinate and degrade DNMT1 through E3 ubiquitin ligase activity [45, 46]. In addition, UHRF1 overexpression delocalizes DNMT1 to intranuclear structures and blocks the appropriate function of DNMT1, leading to global DNA hypomethylation [21, 47].
UHRF1 is involved in cancer development and progression by regulating cell cycle, proliferation and migration ability [14]. In this study, to explore the biological role of UHRF1 in ESCC, the effect of UHRF1 knockdown on proliferation and apoptosis were detected in ESCC cells. We showed that the proliferation of ESCC cells was clearly inhibited after knockdown of UHRF1. Furthermore, depleting ESCC cells of UHRF1 caused apoptosis in a caspase-dependent manner. This finding was similar to a previous study which reported that inhibition of UHRF1 expression could increase the spontaneous apoptosis and radiosensitivity in ESCC cells [40]. Thus, UHRF1 plays an important role in proliferation and survival in ESCC cells. We proposed that UHRF1 may be a potential ESCC therapeutic target due to its significant tumor promoting effects.
Many studies regarding the role of UHRF1 in cell cycle progression have been performed, but the results have varied. A previous study reported that transfection of colorectal cancer cells with lenti-shUHRF1 induced cell cycle arrest in G0/G1 phase [18]. In contrast, it has also been proposed that UHRF1 depletion causes G2/M phase arrest [20, 48]. In the present study, knockdown of UHRF1 in ESCC cells induced cell cycle arrest at G2/M phase, accompanied by inactivation of the Cyclin B1/CDC2 complex. In addition, the decrease of pH3Ser10 indicated a block to mitotic entry. Moreover, phosphorylation of Wee1, which is the activated state, was also elevated in cells depleted of UHRF1. Wee1 is the major kinase responsible for the inhibitory phosphorylation of CDC2 on Tyr15 [33]. Thus, Wee1 plays an essential role in the proper timing of entry into mitosis by regulating the activity of CDC2 [33, 49]. In this study, when Wee1 was inhibited, the cell cycle block in the G2/M transition was abolished, as indicated by reactivation of pH3Ser10. Altogether, these results support the finding that UHRF1 depletion caused cell cycle arrest at the G2/M transition through Wee1 activation in ESCC cells. Notably, UHRF1 knockdown has been shown to cause different types of cell cycle blocks in various studies. This may occur because the role of UHRF1 in cell cycle control varies in different cancer cells.
The G2/M checkpoint prevents cells from entering mitosis in response to DNA damage, providing an opportunity for repair [31]. The DNA damage response is primarily accomplished by phosphorylation of CHK1 and CHK2, leading to their activation and further transmission of the checkpoint signal [50]. In our study, both immunofluorescence and western blotting showed that UHRF1 knockdown caused phosphorylation of H2AX at Ser139, suggesting that DNA damage occurred. In addition, UHRF1 depletion induced the DNA damage response through phosphorylation of CHK1 and CHK2 and subsequent activation of Wee1. Activation of CHK1 and CHK2 induced by DNA damage can also promote cell cycle arrest and apoptosis via p53 [36]. However, p53Ser15 remained constant in cells depleted of UHRF1, indicating that the cell cycle arrest and apoptosis induced by UHRF1 knockdown in ESCC cells may not be p53 dependent. This apoptosis may be triggered by another mechanism, such as the death-receptor-driven mechanism or the endogenous mitochondrial damage pathway, both ending with activation of caspase-3[36]. These results suggest that depletion of UHRF1 caused DNA damage and activated the DNA damage response, followed by cell cycle arrest. This effect was similar, in part, to a previous study that reported that UHRF1 depletion caused DNA damage and activated CHK2-CDC25 [20].
However, what is the mechanism by which UHRF1 depletion leads to activation of the DNA damage response? Our results showed that UHRF1 knockdown induced global DNA hypomethylation, accompanied by activation of the DNA damage response system and subsequent cell cycle arrest. We propose that activation of the DNA damage response was induced by global DNA hypomethylation in cells depleted of UHRF1. In support of this, recent studies have reported that without maintenance of DNA methylation, both perinatal intestinal progenitor cells and postnatal hepatocytes displayed severe DNA damage, activation of the DNA damage response, cycle arrest and apoptosis [51, 52]. In colorectal cancer cells, loss of DNA methylation was also observed to result in genomic instability, DNA damage, activation of the DNA damage checkpoint and subsequent G2 phase arrest [53]. However, more comprehensive studies need to perform to clearly understand the relationship between DNA hypomethylation and the DNA damage response.
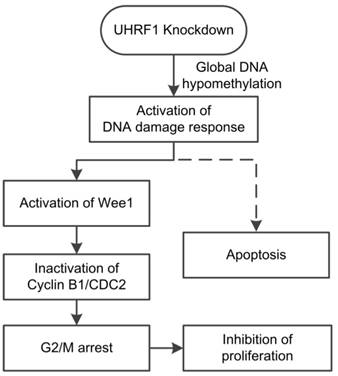
In conclusion, our study showed that positive UHRF1 expression was an independent prognostic factor of ESCC patients. UHRF1 depletion caused global DNA hypomethylation and activated the DNA damage response system. Activation of the DNA damage response pathway induced cell cycle arrest at G2/M phase and subsequent apoptosis (Fig 7). Thus, we propose that UHRF1 plays an essential role in ESCC progression, and it may be used as a potential prognostic marker and therapy target for ESCC.
Model of the effects of UHRF1 knockdown in ESCC cells. The solid lines indicate the correlations demonstrated in this study, whereas the dashed line indicates a tentative relationship.
Abbreviations
UHRF1: Ubiquitin-like with plant homeodomain and ring-finger domains 1; ESCC: Esophageal squamous cell carcinoma; TSGs: Tumor suppressor genes; LINE-1: long interspersed transposable element-1; DNMT1: DNA methyltransferase 1; TNM: tumor-node-metastasis; PBS: Phosphate-buffered saline; CCK-8: Cell Counting Kit-8; FBS: fetal bovine serum; SD: standard deviation; IHC: immunohistochemistry; FCM: flow cytometry; PARP: proteolytic cleavage of poly (ADP-ribose) polymerase.
Supplementary Material
Supplementary figure S1 and table S1.
Acknowledgements
This work was supported by the National High Technology Research and Development Program of China (863 Program), No.2012AA02A503, People's Republic of China; Science and Technology Program of Guangzhou, No.2014J4100103, People's Republic of China; Medical Research Program of Guangdong High Level University Construction, No.88016013042, People's Republic of China.
Ethical approval
This study was carried out in strict accordance with the recommendations in the Guide for the Chinese Ethics Review Committees and with the 1964 Helsinki declaration and its later amendments. This study was approved by the research Ethics Committee of Meizhou People's Hospital.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
2. Tran GD, Sun XD, Abnet CC, Fan JH, Dawsey SM, Dong ZW. et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian General Population Trial cohort in China. International Journal of Cancer. 2005;113:456-63
3. Song Y, Li L, Ou Y, Gao Z, Li E, Li X. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91-5
4. Lima SC, Hernandez-Vargas H, Simao T, Durand G, Kruel CD, Le Calvez-Kelm F. et al. Identification of a DNA methylome signature of esophageal squamous cell carcinoma and potential epigenetic biomarkers. Epigenetics. 2011;6:1217-27
5. Baba Y, Watanabe M, Baba H. Review of the alterations in DNA methylation in esophageal squamous cell carcinoma. Surgery today. 2013;43:1355-64
6. Hoshimoto S, Takeuchi H, Ono S, Sim MS, Huynh JL, Huang SK. et al. Genome-Wide Hypomethylation and Specific Tumor-Related Gene Hypermethylation are Associated with Esophageal Squamous Cell Carcinoma Outcome. J Thorac Oncol. 2015;10:509-17
7. Kaz AM, Grady WM. Epigenetic biomarkers in esophageal cancer. Cancer letters. 2014;342:193-9
8. Kawano H, Saeki H, Kitao H, Tsuda Y, Otsu H, Ando K. et al. Chromosomal instability associated with global DNA hypomethylation is associated with the initiation and progression of esophageal squamous cell carcinoma. Annals of surgical oncology. 2014;21(Suppl 4):S696-702
9. Bronner C, Fuhrmann G, Chedin FL, Macaluso M, Dhe-Paganon S. UHRF1 Links the Histone code and DNA Methylation to ensure Faithful Epigenetic Memory Inheritance. Genetics & epigenetics. 2010;2009:29-36
10. Bostick M, Kim JK, Esteve P-O, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760-4
11. Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818-U12
12. Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C. et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822-U13
13. Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826-U14
14. Bronner C, Krifa M, Mousli M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochemical pharmacology. 2013;86:1643-9
15. Unoki M, Kelly JD, Neal DE, Ponder BA, Nakamura Y, Hamamoto R. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. British journal of cancer. 2009;101:98-105
16. Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. British journal of cancer. 2010;103:217-22
17. Sabatino L, Fucci A, Pancione M, Carafa V, Nebbioso A, Pistore C. et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene. 2012;31:5061-72
18. Wang F, Yang YZ, Shi CZ, Zhang P, Moyer MP, Zhang HZ. et al. UHRF1 promotes cell growth and metastasis through repression of p16(ink(4)a) in colorectal cancer. Annals of surgical oncology. 2012;19:2753-62
19. Ashraf W, Ibrahim A, Alhosin M, Zaayter L, Ouararhni K, Papin C. et al. The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget. 2017
20. Tien AL, Senbanerjee S, Kulkarni A, Mudbhary R, Goudreau B, Ganesan S. et al. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. The Biochemical journal. 2011;435:175-85
21. Jacob V, Chernyavskaya Y, Chen X, Tan PS, Kent B, Hoshida Y. et al. DNA hypomethylation induces a DNA replication-associated cell cycle arrest to block hepatic outgrowth in uhrf1 mutant zebrafish embryos. Development. 2015;142:510-21
22. Unoki M, Brunet J, Mousli M. Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochemical pharmacology. 2009;78:1279-88
23. Geng Y, Gao Y, Ju H, Yan F. Diagnostic and prognostic value of plasma and tissue ubiquitin-like, containing PHD and RING finger domains 1 in breast cancer patients. Cancer science. 2013;104:194-9
24. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
25. Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ. et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. International journal of cancer Journal international du cancer. 2008;122:2767-73
26. Krifa M, Alhosin M, Muller CD, Gies JP, Chekir-Ghedira L, Ghedira K. et al. Limoniastrum guyonianum aqueous gall extract induces apoptosis in human cervical cancer cells involving p16 INK4A re-expression related to UHRF1 and DNMT1 down-regulation. Journal of experimental & clinical cancer research: CR. 2013;32:30
27. Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG. et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010;107:8689-94
28. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nature reviews Genetics. 2009;10:691-703
29. Babbio F, Pistore C, Curti L, Castiglioni I, Kunderfranco P, Brino L. et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene. 2012;31:4878-87
30. Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. The Journal of biological chemistry. 1998;273:33533-9
31. Stark GR, Taylor WR. Control of the G2/M transition. Molecular biotechnology. 2006;32:227-48
32. Smits VA, Medema RH. Checking out the G(2)/M transition. Biochimica et biophysica acta. 2001;1519:1-12
33. Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955-7
34. Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng CY, Piwnica-Worms H. Cell cycle regulation of the p34cdc2 inhibitory kinases. Mol Biol Cell. 1994;5:989-1001
35. Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 1994;31:1-13
36. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends in molecular medicine. 2006;12:440-50
37. O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. The EMBO journal. 1997;16:545-54
38. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone phosphorylation on serine 139. The Journal of biological chemistry. 1998;273:5858-68
39. Nakamura K, Baba Y, Kosumi K, Harada K, Shigaki H, Miyake K. et al. UHRF1 regulates global DNA hypomethylation and is associated with poor prognosis in esophageal squamous cell carcinoma. Oncotarget. 2016
40. Yang C, Wang Y, Zhang F, Sun G, Li C, Jing S. et al. Inhibiting UHRF1 expression enhances radiosensitivity in human esophageal squamous cell carcinoma. Molecular biology reports. 2013;40:5225-35
41. Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. The EMBO journal. 2013;32:340-53
42. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer cell. 2014;25:196-209
43. Kent B, Magnani E, Walsh MJ, Sadler KC. UHRF1 regulation of Dnmt1 is required for pre-gastrula zebrafish development. Developmental biology. 2016;412:99-113
44. Ramesh V, Bayam E, Cernilogar FM, Bonapace IM, Schulze M, Riemenschneider MJ. et al. Loss of Uhrf1 in neural stem cells leads to activation of retroviral elements and delayed neurodegeneration. Genes Dev. 2016;30:2199-212
45. Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S. et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Science signaling. 2010;3:ra80
46. Qin W, Leonhardt H, Spada F. Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. Journal of cellular biochemistry. 2011;112:439-44
47. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA. et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908-12
48. Liu X, Ou H, Xiang L, Li X, Huang Y, Yang D. Elevated UHRF1 expression contributes to poor prognosis by promoting cell proliferation and metastasis in hepatocellular carcinoma. Oncotarget. 2017
49. Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463-74
50. Medema RH, Macurek L. Checkpoint control and cancer. Oncogene. 2012;31:2601-13
51. Elliott EN, Sheaffer KL, Schug J, Stappenbeck TS, Kaestner KH. Dnmt1 is essential to maintain progenitors in the perinatal intestinal epithelium. Development. 2015;142:2163-72
52. Kaji K, Factor VM, Andersen JB, Durkin ME, Tomokuni A, Marquardt JU. et al. DNMT1 is a required genomic regulator for murine liver histogenesis and regeneration. Hepatology. 2016;64:582-98
53. Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B. et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nature genetics. 2007;39:391-6
Author contact
![]() Corresponding authors: Xueyun Zhong, E-mail: tzxyedu.cn, Tel: +86-20-85228363; Lihui Wang, E-mail: maillihuicom, Tel: +86-20-85220252.
Corresponding authors: Xueyun Zhong, E-mail: tzxyedu.cn, Tel: +86-20-85228363; Lihui Wang, E-mail: maillihuicom, Tel: +86-20-85220252.