Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(19):4125-4140. doi:10.7150/jca.21802 This issue Cite
Research Paper
Decreased KPNB1 Expression is Induced by PLK1 Inhibition and Leads to Apoptosis in Lung Adenocarcinoma
Noboru Sekimoto1 ![]() , Yutaka Suzuki2, Sumio Sugano1
, Yutaka Suzuki2, Sumio Sugano1 ![]()
1. Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Minato-ku, Tokyo 108-8639, Japan;
2. Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba 277-8562, Japan.
Received 2017-7-7; Accepted 2017-10-10; Published 2017-11-6
Abstract
Lung cancer is a major cause of death worldwide, with lung adenocarcinoma being the most frequently diagnosed subtype in Japan. Finding the target of an anticancer drug can improve lung adenocarcinoma treatments. Polo-like kinase 1 (PLK1) is an essential mitotic kinase in mitotic progression, and PLK1 inhibition induces cell cycle arrest and apoptosis in tumor cells. In addition, a variety of PLK1 inhibitors have been identified for cancer treatments. In this study, we looked for the target gene of the anticancer drug that has synergy with PLK1 inhibitors. We identified karyopherin beta 1 (KPNB1) as a possible target for lung adenocarcinoma treatment. We found that PLK1 inhibition decreased KPNB1 expression in lung adenocarcinoma cells and KPNB1 depletion inhibited cell proliferation via apoptosis. The same apoptosis signaling pathway may be activated because the expression of common apoptosis-related genes was decreased by PLK1 and KPNB1 silencing; however, the time course of cell growth inhibition was somewhat different. Cell cycle analysis showed that KPNB1 depletion increased the proportion of cells at the G0/G1 phase, although cells also accumulated at the G2/M phase in PLK1-depleted cells. Our findings suggest that decreased KPNB1 expression may be associated with the apoptosis induced by PLK1 inhibition.
Keywords: PLK1, KPNB1, Lung adenocarcinoma, Apoptosis.
Introduction
Lung cancer is the most frequently diagnosed cancer and a major cause of death worldwide (1). In the United States, as of 2016, lung cancer was the leading cause of death regardless of gender (2). Lung cancer is classified as non-small cell lung cancer (NSCLC) and small cell lung cancer. NSCLC is categorized as squamous cell carcinoma, adenocarcinoma, or large cell carcinoma and accounts for 85% of all lung cancers. In Japan, adenocarcinoma is the most common subtype and accounts for about 60% of resected lung cancers (3). Several useful anticancer drugs for lung adenocarcinoma patients have been discovered, such as Gefitinib and Erlotinib, which are tyrosine kinase inhibitors. However, these anticancer drugs are effective for only a fraction of lung adenocarcinomas and tumors often become resistant during treatment. New approaches to lung adenocarcinoma treatment are needed, including finding new targets for molecularly targeted therapies. To find a new target, a deep understanding of the molecular network in cancer cells is important. In particular, analysis of the signaling pathways driven by cancer-related genes may lead to the discovery of a new target for treatment.
Polo-like kinase 1 (PLK1) is an essential mitotic kinase in mitotic progression and spindle bipolarity (4, 5). PLK1 inhibition induces cell cycle arrest and apoptosis in tumor cells (6, 7). In addition, previous studies have shown that PLK1 is highly expressed in cancer cells, and its overexpression is related to poor prognosis (8-10). A previous study also showed that NSCLC patients with moderate PLK1 expression survived longer than patients with high PLK1 expression (11). To date, many PLK1 inhibitors have been developed, and some are currently in clinical trials for cancer treatment (12, 13). However, the side effects of PLK1 inhibitors are serious because PLK1 is an essential protein for cell-cycle progression (14). In addition, only limited knowledge exists regarding apoptosis induced by PLK1 inhibition, although the role of PLK1 in cell cycle progression has been extensively studied. We hypothesized that, by studying genes related to the cell death induced by PLK1 depletion, new targets for anticancer drugs may be revealed.
In this study, we comprehensively analyzed the RNA expression of the lung adenocarcinoma cell line H1299 using a next-generation sequencer. By comparing PLK1-depleted and non-depleted H1299 cells, we looked for apoptosis-related genes whose expression was changed by PLK1 depletion. These genes may be targets of anticancer drugs and may have synergy with PLK1 inhibitors. As a result of sequence data analysis, we identified karyopherin beta 1 (KPNB1) as a possible apoptosis-related gene that is down-regulated during the process of cell death induced by PLK1 depletion.
Materials and Methods
Cell lines
The human NSCLC adenocarcinoma cell lines H1299 and A549 and the human cervix adenocarcinoma cell line HeLa were obtained from the American Type Culture Collection (Manassas, VA, USA). H1299 cells were cultured in RPMI medium (Life Technologies, Palo Alto, CA, USA) supplemented with 10% v/v fetal bovine serum and 1% v/v antibiotic solution. A549 and HeLa cells were cultured in DMEM medium (Life Technologies) supplemented with 10% v/v fetal bovine serum and 1% v/v antibiotic solution. The culture medium used for the other cell lines has been described elsewhere (15). All cells were incubated at 37°C with 5% CO2.
Sequencing
Total RNA was extracted from the lung adenocarcinoma cell lines using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The mRNA library was prepared using the TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA, USA). mRNA sequence data were acquired using an HiSeq2500 system (Illumina). The detailed analytical method has been described elsewhere (16). All raw sequence data used in this study were deposited in the DNA Data Bank of Japan under the accession numbers DRA001846, DRA002311, and DRA005645.
RNA oligonucleotides and transfection
PLK1 and KPNB1 siRNAs were synthesized by Sigma-Aldrich Japan (Tokyo, Japan). AllStars Negative Control siRNA was purchased from Qiagen. siRNA transfection was conducted using lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer's manual. Briefly, siRNA was transfected into cells in six-well plates, and 7.5 μL of lipofectamine 3000 was diluted in 125 μL of Opti-MEM1 (Thermo Fisher Scientific) and mixed with 125 μL of siRNA (75 pmol) in Opti-MEM1. The mixed solutions were maintained at room temperature for 5 min and subsequently added to each well. When the cells were cultured in other plates, such as 96-well plates or 10 cm culture dishes, the volumes of siRNA and the other reagents used in the transfection were changed according to the size of the culture vessel. Plates were incubated at 37°C with 5% CO2 prior to use in further experiments.
Real-time PCR
Total RNA was extracted from cells using the miRNeasy Mini Kit according to the manufacturer's instructions. Primers were synthesized by Exigen (Tokyo, Japan). mRNA expression levels were detected using SYBR SELECT Master Mix (Applied Biosystems, Inc., Waltham, MA, USA). PCR was performed under the following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 s and 60°C for 1 min on a 7900HT Fast Real Time PCR System (Applied Biosystems, Inc.). The relative expression level of each gene was calculated and quantified using the ΔΔCt method after normalization to the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (17). The following specific primers were used for real-time PCR: PLK1, forward 5′-GTTCCCATCCCAACTCCTTGA-3′ and reverse 5′-TGCTCGCTCATGTAATTGCG-3′; KPNB1, forward 5′-GGCGGAGATCGAAGACTAACAA-3′ and reverse 5′-TTGTTGCCCATGTAGCAAGG-3′; and GAPDH, forward 5′-CCCTCAACGACCACTTTGTCA-3′ and reverse 5′-CCCTGTTGCTGTAGCCAAATT-3′.
Protein extraction and Western blotting analysis
To extract whole protein from cells, cells were harvested and lysed in 200~1000 μL of RIPA buffer (Wako Pure Chemical, Osaka, Japan) supplemented with 10% Complete Mini solution (Roche, Basel, Switzerland) and 1% Phosphatase Inhibitor Cocktail Solution (Wako Pure Chemical). To extract nuclear and cytoplasmic protein separately, NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) were used following the manufacturer's instructions. The extracted protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The detailed method has been explained in our previous study (16). Briefly, lysate aliquots containing equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred to Immun-Blot PVDF membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked in a solution containing 5% milk protein and 0.1% Tween-20 in Tris-buffered saline (TBS) and then incubated overnight with the appropriate primary antibody. The blots were then washed and incubated with an appropriate secondary antibody in TBS with 0.1% Tween-20 and 5% milk protein for 1 hour. Secondary antibody binding was visualized using a Las-3000 mini system (FujiFilm, Tokyo, Japan) and an ECL Prime Western Blotting Detection System (GE Healthcare, Buckinghamshire, UK).
The following primary antibodies were used for Western blotting: anti-alpha tubulin rabbit polyclonal antibody (abcam, Cambridge, UK; 1:1000 dilution), anti-GC1qR mouse monoclonal antibody (abcam; 1:5000 dilution), anti-NTF97/importin beta mouse monoclonal antibody (abcam; 1:5000 dilution), anti-PLK1 mouse monoclonal antibody (abcam; 1:10000 dilution), anti-lamin B1 rabbit polyclonal antibody (abcam; 1:1000 dilution), and anti-MCL1 rabbit monoclonal antibody (abcam; 1:1000 dilution). The following secondary antibodies were used, along with primary antibodies, for Western blotting: anti-Rabbit IgG, HRP-Linked Whole Ab Donkey (GE Healthcare; 1:100000), and anti-mouse IgG, HRP-linked Antibody (Cell Signaling Technology, Danvers, MA, USA).
Cell proliferation assay
Cells were counted using the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) following the manufacturer's manual. Briefly, 10 μL of CCK-8 solution was added to the cells in each well of a 96-well plate, followed by a 2-hour incubation at 37°C with 5% CO2. The solution absorbance was measured at 450 nm with an iMark Microplate Absorbance Reader (Bio-Rad).
Trypan blue stain
Cells were trypsinized and collected as 10 μL of cell suspension. Then, 10 μL of trypan blue (Bio-Rad) was added to the cell suspension and gently mixed. The numbers of total cells and stained cells were counted using a TC20 Automated Cell Counter (Bio-Rad).
Cell cycle analysis
Cells were transfected with PLK1 or KPNB1 siRNA. Following transfection, cells were incubated at 37°C with 5% CO2. Cells were stained using a Cell Cycle Phase Determination Kit (Cayman Chemical Company, Ann Arbor, MI, USA) following the manufacturer's protocol. The stained cells were analyzed in the FL2 channel of a flow cytometer BD FACSCalibur (BD Bioscience, Franklin Lakes, NJ, USA) with a 488 nm excitation laser. Data were analyzed using FlowJo (FlowJo, LLC, Ashland, OR, USA).
Immunoprecipitation
The cells were harvested and lysed in 1 mL of lysis buffer supplemented with 1% Nonidet P-40 (Nacalai Tesque), 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA (pH 8.0), 10% glycerol, 13% Complete Mini solution (Roche), and 1% Phosphatase Inhibitor Cocktail Solution (Wako Pure Chemical). SureBeads Protein A or G Magnetic Beads (Bio-Rad) were incubated with antibodies overnight at 4°C, followed by incubation with an equal amount of cell lysate overnight at 4°C. The immune complex was purified and analyzed by Western blotting analysis.
Immunofluorescence
Prior to cell seeding, coverslips were put on the six-well plates. Cells were cultured in the six-well plates and transfected with PLK1 or KPNB1 siRNA. Following transfection, cells were incubated at 37°C with 5% CO2. Cells were washed with phosphate-buffered saline (PBS) three times and fixed with 4% paraformaldehyde in PBS. Cells were rinsed with PBS three times, followed by incubation with 0.2% Triton X-100 in PBS for 5 min at room temperature. Cells were washed with PBS three times and blocked in PBS containing 1% BSA, followed by incubation with the appropriate primary antibody overnight at 4°C. Cells were washed with PBS five times, followed by incubation with the appropriate secondary antibody in the dark for 50 min at room temperature. Cells were then washed with PBS five times. The cells were mounted with Dapi-Fluoromount-G (Southern Biotechnology Associates Inc., Birmingham, AL, USA). Images of the cells were taken using an ECLIPSE Ti inverted research microscope (Nikon, Tokyo, Japan) and analyzed using ImageJ software (18).
ATP and caspase assay
H1299 cells were seeded in 96-well plates at a density of 1 × 103 per well (100 µL) one day prior to transfection.
The CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, Madison, WI, USA) was used for ATP assays following the manufacturer's instructions. Briefly, the plates were equilibrated for 30 min at room temperature. Then, 100 μL of Cell Titer-Glo® Reagent was added to each well, and the contents were mixed for 2 min on an orbital shaker. After a 10-min incubation at room temperature, the luminescent signal was detected using a FLUOstar OPTIMA system (BMG LABTECH, Ortenberg, Germany).
The caspase-3/7 activity was determined using the Caspase-Glo® 3/7 Assay System (Promega) following the manufacturer's instructions. Briefly, plates were equilibrated for 30 min at room temperature. Then, 100 μL of equilibrated Caspase-Glo 3/7 reagent was added to each well and the contents were mixed for 30 sec on an orbital shaker. After a 2-hour incubation at room temperature, the luminescent signal was detected using a FLUOstar OPTIMA system.
Annexin V and propidium iodide labeling
H1299 cells were seeded in six-well plates at a density of 3.8 × 104 per well (2 mL) one day prior to transfection. The cells were trypsinized and washed with PBS twice. The Annexin-V-FLUOS Staining Kit (Sigma-Aldrich, St. Louis, MO, USA) was used for Annexin V and propidium iodide (PI) labeling. The cells were suspended with 100 μL of incubation buffer. Then, Annexin V-FITC and PI were added to the cell suspension. After a 15 min incubation in the dark at room temperature, 400 μL of incubation buffer was added to the cell suspension and mixed. The cell suspension was analyzed using a FACS-Calibur system (BD Biosciences).
Results
Sequencing data showed that PLK1 expression levels were widely distributed (~nine times) in 25 human lung adenocarcinoma cell lines (Fig. 1a). PLK1 expression in the H1299 cell line was the highest among the lung adenocarcinoma cell lines. Therefore, we chose the H1299 cells for next-generation sequencing because we supposed that the effect of PLK1 knockdown in these cells may be remarkable compared with the other cell lines. We conducted the transfection of PLK1 siRNA using H1299 cells. As a result, we successfully decreased both PLK1 mRNA and protein expression levels to about one-third (Fig. 1b, c). To find the gene whose expression level changed following PLK1 depletion, RNA expression levels of H1299 cells transfected with PLK1 or negative control siRNA were comprehensively analyzed using a next-generation sequencer.
Transcriptome analysis of the lung adenocarcinoma cell line H1299. (a) PLK1 mRNA expression levels in lung adenocarcinoma cell lines (black bar) and small airway epithelial cells (SAEC) (white bar). (b) PLK1 mRNA expression levels of H1299 cells transfected with control or PLK1 siRNA. RNA was extracted 48 and 72 hours after the transfection. (c) Western blotting analysis of PLK1 protein expression levels of H1299 cells transfected with control or PLK1 siRNA. Protein was extracted 48 and 72 hours after the transfection. (d) The expression levels of PLK1 in H1299 cells (rpkm), which were obtained from the sequence data. (e) The correlation of PLK1 expression levels among various conditioned H1299 cells. This plot was composed of genes whose expression levels were >1 in the units of rpkm. (f) Venn diagram showing genes whose expression levels were decreased over 20% by PLK1 silencing. Rpkm is the abbreviation for “reads per kilobase per million.” Data represent mean values ± standard deviations (n = 6 replicates; *P < 0.01).
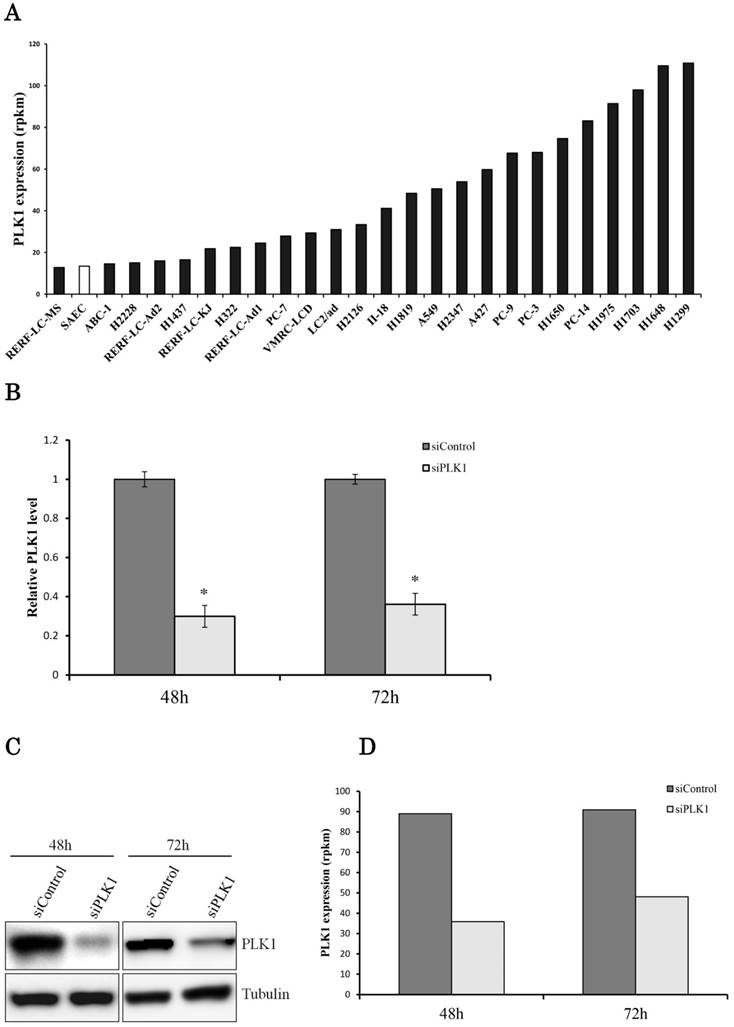
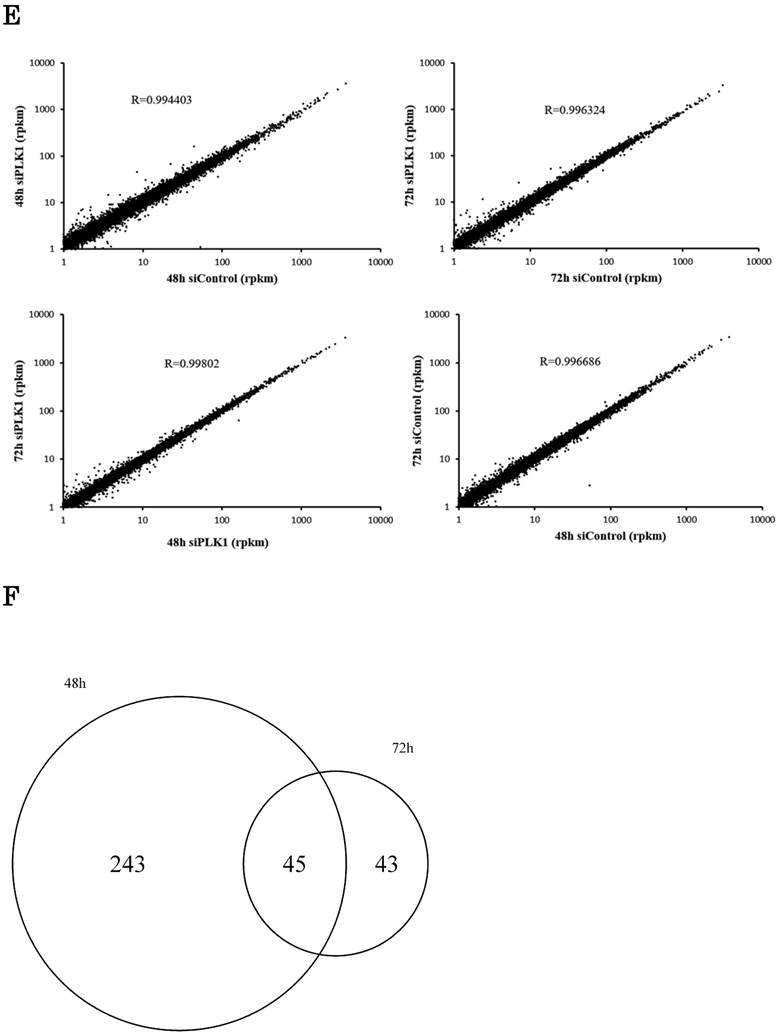
The sequence data showed that PLK1 expression levels decreased with the real-time PCR data (Fig. 1d). Although the expression of some genes was changed by PLK1 depletion, the strong correlation of gene expression levels between PLK1 and negative control siRNA-transfected cells implied that the majority of the gene expressions did not change drastically according to PLK1 depletion (Fig. 1e). The expression of more genes decreased at 48 hours than at 72 hours following PLK1 silencing; the majority of genes with decreased expression were the same at 48 and 72 hours (Fig. 1f). These data implied that the expression of most genes changed up to 48 hours, and many of these expression levels returned to normal before 72 hours.
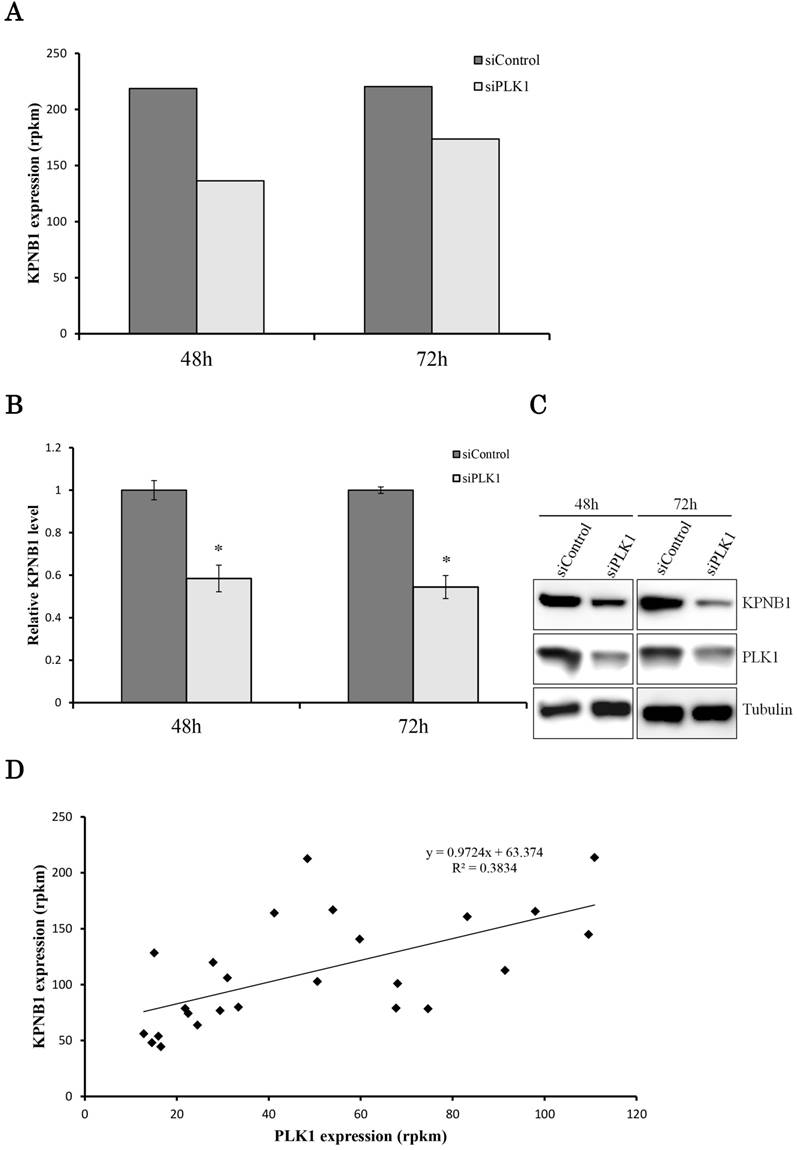
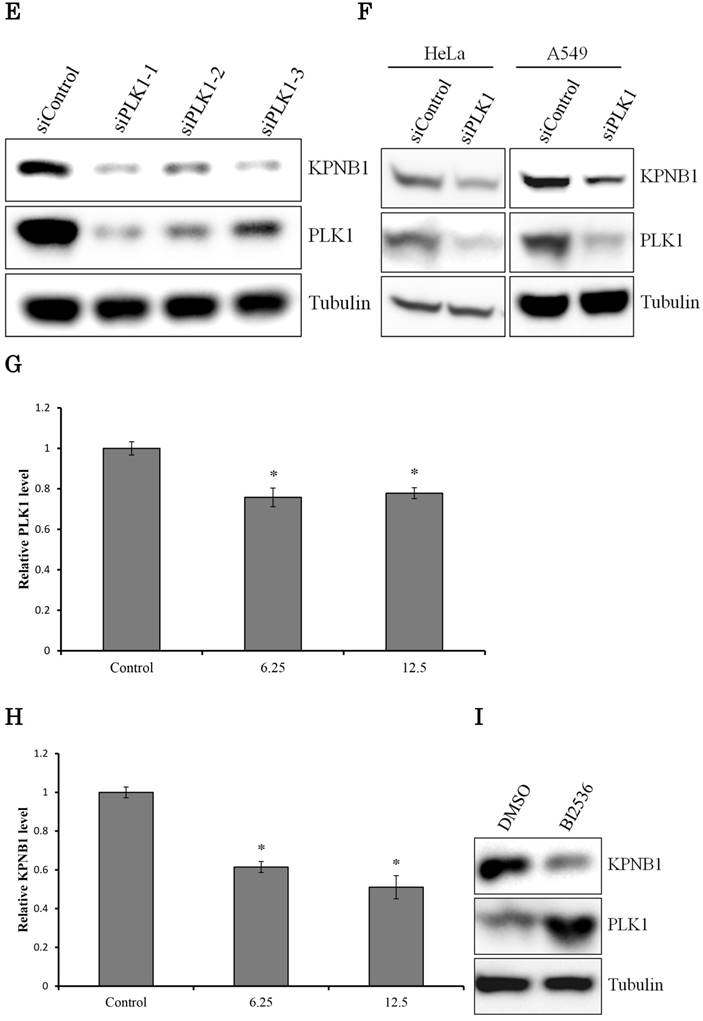
We hypothesized that the gene that contributed to cell apoptosis may be included among the genes down-regulated by PLK1 siRNA transfection. Indeed, we found that KPNB1 mRNA expression was significantly suppressed by silencing of PLK1 expression (Fig. 2a). KPNB1 is a member of the importin beta family, and a previous study indicated that it is important for tumor cell survival (19). The sequencing data were validated by real-time PCR and Western blotting in this study (Fig. 2b, c). KPNB1 expression levels were widely distributed and were positively correlated with PLK1 expression levels in the 25 lung adenocarcinoma cell lines (Supplementary Fig. 1, Fig. 2d). Furthermore, the RNA sequence data from The Cancer Genome Atlas (TCGA) showed that there was a slightly less but still positive correlation between PLK1 and KPNB1 expression levels in resected lung adenocarcinomas (Supplementary Fig. 2) (20-22). KPNB1 protein expression levels were also decreased by using other PLK1 siRNAs (Fig. 2e). KPNB1 protein expression levels were also analyzed using the lung adenocarcinoma cell line A549 and the human cervix adenocarcinoma cell line HeLa. We found that KPNB1 expression levels decreased in HeLa and A549 cell lines with the transfection of PLK1 siRNA (Fig. 2f). BI2536 is a selective inhibitor of PLK1 (23). The cellular proliferation of H1299 cells was inhibited by adding BI2536 (Supplementary Fig. 3). Real-time PCR analysis also revealed that BI2536 reduced both PLK1 and KPNB1 expression; in particular, KPNB1 expression was reduced by almost half (Fig. 2g, h). Western blotting data showed that the addition of BI2536 decreased KPNB1 protein expression levels along with mRNA expression (Fig. 2i). In contrast, PLK1 protein expression increased; this phenomenon has been reported in previous studies (24). From our data, we found that KPNB1 expression decreased during the apoptosis process induced by the depletion or inhibition of PLK1.
KPNB1 expression was decreased by PLK1 inhibition. (a) The expression levels of KPNB1 in H1299 cells (rpkm), which was obtained from the sequence data. (b) The relative KPNB1 levels of H1299 cells calculated with real-time PCR. RNA was extracted 48 and 72 hours after the transfection. (c) The protein expression levels of PLK1-silenced H1299 cells were detected by Western blotting. Protein was extracted 48 and 72 hours after the transfection of siRNA. (d) The correlation between PLK1 and KPNB1 expression levels of 25 lung adenocarcinoma cell lines. (e) The protein expression levels of PLK1-silenced H1299 cells were determined by Western blotting analysis. Protein was extracted 48 hours after transfection of siRNAs; each siRNA has a different sequence. (f) The protein expression levels of PLK1-silenced HeLa and A549 cells were examined by Western blotting analysis. Protein was extracted 48 hours after the transfection of siRNA. (g) The relative PLK1 levels of H1299 cells with the addition of BI2536 (6.25 or 12.5 nM) calculated with real-time PCR. RNA was extracted 48 hours after the addition of BI2536 to cells. (h) The relative KPNB1 levels of H1299 cells with the addition of BI2536 (6.25 or 12.5 nM) calculated with real-time PCR. RNA was extracted 48 hours after the addition of BI2536 to cells. (i) The protein expression levels of H1299 cells with the addition of BI2536 (10 nM) were determined by Western blotting analysis. Protein was extracted 48 hours after the addition of BI2536 to cells. Rpkm is the abbreviation for “reads per kilobase per million.” The data are given as means ± standard deviations (n = 6 replicates; *P < 0.01).
The effect of KPNB1 or PLK1 silencing on cellular growth was also tested using H1299 cells. The transfection of KPNB1 siRNA successfully decreased KPNB1 protein levels in H1299 cells (Fig. 3a). The number of living cells was counted 24, 48, and 72 hours after siRNA transfection. The number of PLK1-depleted cells hardly increased from the beginning (Fig. 3b). On the other hand, the number of KPNB1-depleted cells increased up to 48 hours and then decreased between 48 and 72 hours. The number of dead cells was also counted using trypan blue staining 24, 48, and 72 hours after siRNA transfection in H1299 cells. In these cells, the ratio of dead cells in control siRNA-transfected cells gradually decreased (Fig. 3c). This may be because some of the cells died from the toxicity of the lipofectamine reagent, and their ratio decreased as the number of surviving cells increased. However, the ratio of dead cells that were transfected with PLK1 or KPNB1 siRNA increased day by day. In particular, a number of cells died between 48 and 72 hours after transfection.
Cellular proliferation was inhibited by silencing PLK1 or KPNB1 expression in H1299 cells. (a) Western blotting analysis of the protein expression levels of KPNB1-silenced H1299 cells. Protein was extracted 48 and 72 hours after the transfection of siRNA. (b) The number of H1299 cells was counted 24, 48, and 72 hours after the transfection of siRNA. (c) The trypan blue staining of H1299 cells 24, 48, and 72 hours after the transfection of siRNA. (d) Cell cycle distributions of H1299 cells were detected by flow cytometry 48 hours after the transfection of siRNA. (e) The immunofluorescence analysis of H1299 cells 48 hours after the transfection of siRNA, staining for Ki67 (green) and DAPI (blue). Error bars represent standard deviations; * represents P < 0.01 versus control.
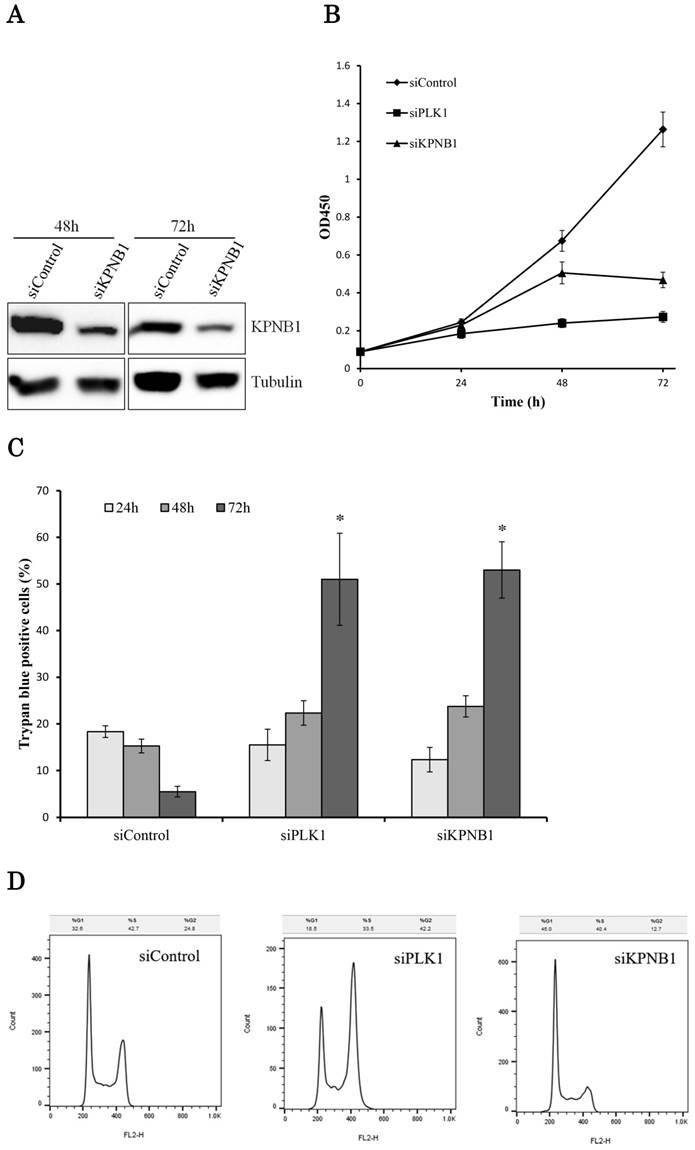
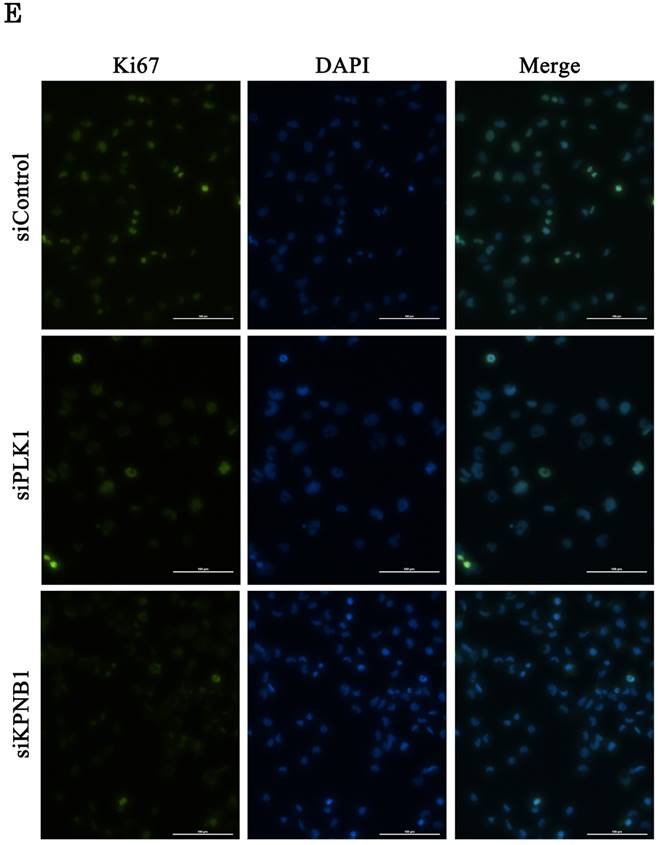
Cell cycle analysis was conducted using H1299 cells 48 hours after the transfection of control, PLK1, or KPNB1 siRNA. The ratio of cells at the G2/M phase in PLK1-silenced cells was higher compared with control cells (Fig. 3d). In contrast, the ratio of cells at the G0/G1 phase in KPNB1-silenced cells was higher compared with control cells. Expression of the cell proliferation marker Ki67 protein was then analyzed using immunofluorescence in H1299 cells. The ratio of stained cells was less in KPNB1-depleted cells compared with control cells (Fig. 3e). The ratio of Ki67-stained cells did not change greatly with PLK1 depletion. These data implied that cell cycle progression was inhibited at the G0 phase in KPNB1-depleted cells before cell death.
We further analyzed the cell death induced by KPNB1 depletion. Caspase -3 and -7 are the executioner caspases in apoptosis, and their activities were measured in H1299 cells transfected with control, PLK1, or KPNB1 siRNA. Although the caspase activities of cells transfected with PLK1 or KPNB1 siRNA gradually increased, the caspase activity of control cells barely changed (Fig. 4a). Next, we stained H1299 cells with Annexin V and PI in order to identify apoptotic cells. The number of Annexin V-positive cells was higher in PLK1- and KPNB1-depleted cells compared with control cells (Fig. 4b). The cellular ATP volume reflects the number of living cells; to examine the degree of apoptosis induced by PLK1 or KPNB1 depletion, we quantified the amount of ATP in H1299 cells transfected with control, PLK1, or KPNB1 siRNA. The ATP amount hardly changed from the beginning in PLK1-depleted cells (Fig. 4c). However, the ATP amount increased up to 48 hours and decreased between 48 and 72 hours in KPNB1-depleted cells. From these data, it was revealed that the depletion of PLK1 or KPNB1 induced apoptosis in H1299 cells.
KPNB1 depletion induced apoptosis with the silencing of PLK1. (a) Measuring caspase-3 and -7 activities in H1299 cells 24, 48, and 72 hours after the transfection of siRNA. (b) Flow cytometry analysis of Annexin V and PI labeling of H1299 cells. The cells were harvested 48 hours after the transfection of siRNA. (c) Quantification of ATP in H1299 cells 24, 48, and 72 hours after the transfection of siRNA. (d) C1qbp expression levels of H1299 cells (rpkm). The data were acquired using a next-generation sequencer. (e) The protein expression levels of H1299 cells were detected by Western blotting analysis. The protein was extracted 48 hours after the transfection of PLK1 or control siRNA. (f) The protein expression levels of H1299 cells were determined by Western blotting analysis. The protein was extracted 48 hours after the transfection of KPNB1 or control siRNA. Rpkm is the abbreviation for “reads per kilobase per million.” Error bars represent standard deviations.
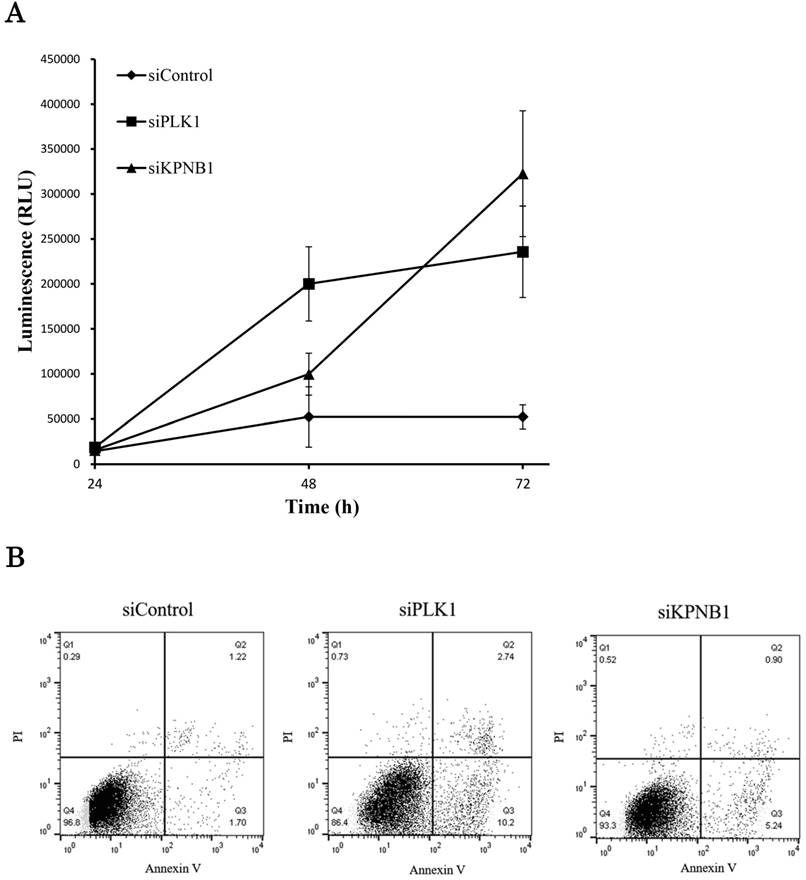
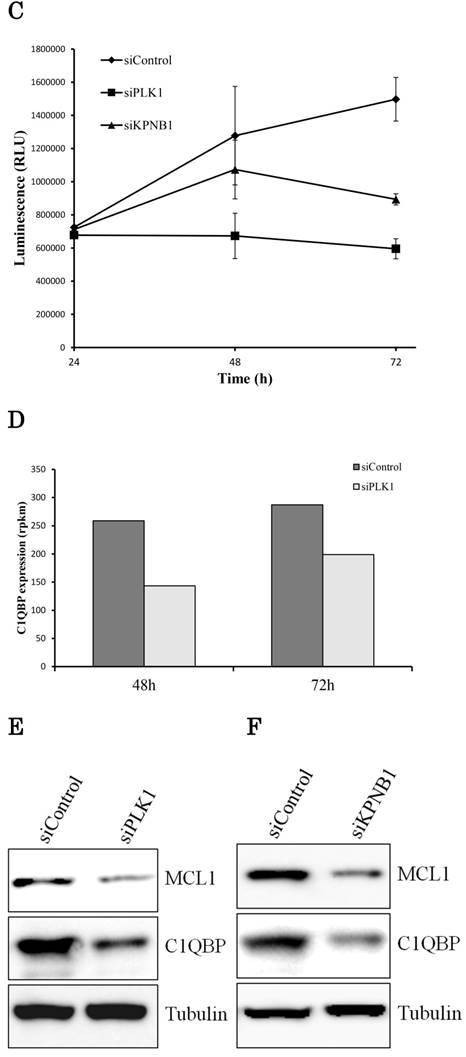
If the expression of the same apoptosis-related gene was changed by PLK1 or KPNB1 depletion, there should be a high possibility that the same apoptosis signaling pathway was activated. Therefore, we investigated the expression of apoptosis-related genes in PLK1- or KPNB1-depleted H1299 cells. First, we analyzed C1qbp expression because a previous study showed that C1qbp promotes cellular proliferation and resistance to cell death in breast cancer cell lines (25). Second, we analyzed Mcl-1 expression because a previous study reported that Mcl-1 suppression was related to apoptosis caused by PLK1 inhibition in sarcomas (26). In the sequencing data, C1qbp mRNA expression decreased depending on PLK1 knockdown (Fig. 4d). The results of Western blotting analysis revealed that both Mcl-1 and C1qbp expressions decreased with PLK1 knockdown (Fig. 4e). Furthermore, these protein expression levels were also decreased by KPNB1 siRNA transfection (Fig. 4f). From the above findings, it appeared there was a high possibility that the same apoptosis signaling pathway was activated when PLK1 or KPNB1 expression was silenced.
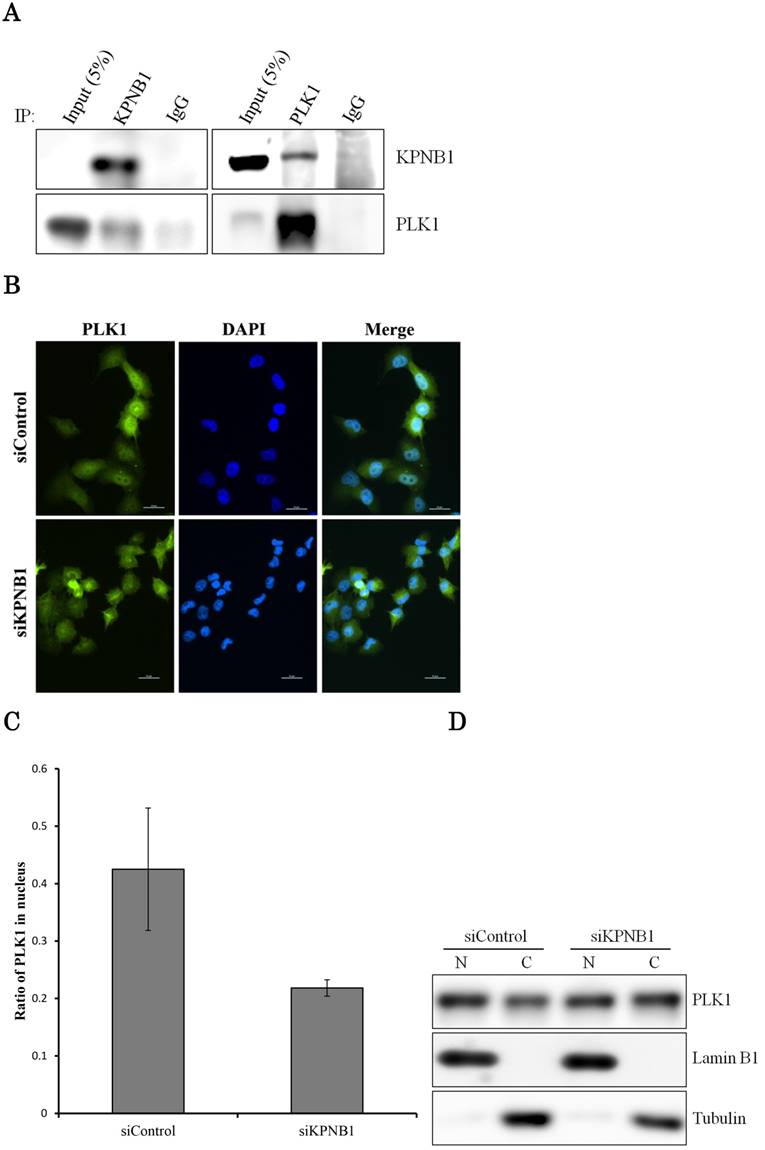
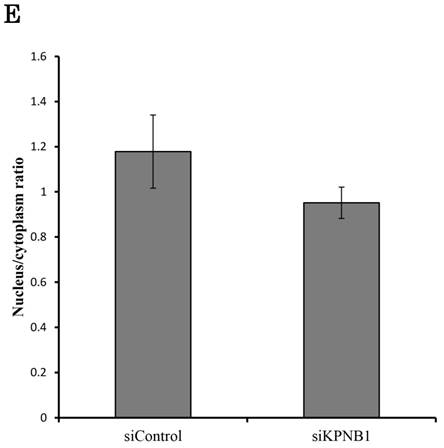
PLK1 primarily works as a protein kinase in the nucleus; thus, PLK1 may need a transport protein to allow its importation into the nucleus. KPNB1 is a member of the importin beta family that is involved in importing cargo proteins into the nucleus. We considered the possibility that PLK1 may regulate KPNB1 expression to achieve importation of PLK1 into the nucleus. First, immunoprecipitation and Western blotting analysis were conducted to determine whether PLK1 and KPNB1 form a complex. The analysis showed that PLK1 and KPNB1 antibodies precipitated both PLK1 and KPNB1 proteins (Fig. 5a). Next, to determine whether KPNB1 is an important protein for the nuclear importation of the PLK1 protein, the intracellular location of the PLK1 protein was detected in KPNB1-depleted H1299 cells by immunofluorescence. The assay showed that the intracellular location of PLK1 was changed slightly by KPNB1 siRNA transfection (Fig. 5b, c). Next, protein was extracted from the nucleus and cytoplasm separately in KPNB1-silenced H1299 cells, and the extracted proteins were analyzed by Western blotting analysis. The analysis showed that the ratio of PLK1 in the nucleus was not significantly changed by KPNB1 depletion (Fig. 5d, e), indicating that KPNB1 depletion was insignificant for the importation of PLK1 into the nucleus, although KPNB1 may play a part in the importation of PLK1 into the nucleus.
There is a possibility that KPNB1 functions in the importation of PLK1 into the nucleus. (a) Immunoprecipitation with KPNB1 and PLK1 antibodies using whole lysate extracted from H1299 cells. The lysate and immunoprecipitates were detected by Western blotting analysis with anti-KPNB1 and -PLK1 antibodies. (b) Immunofluorescence staining of PLK1 (green) and DAPI (blue) using H1299 cells. The assay was performed 48 hours after control or PLK1 siRNA transfection (scale bar, 25 μm). (c) The nucleus/cytoplasm ratios of PLK1 fluorescence were calculated from the immunofluorescent images of H1299 cells. The data represent the average of the ratios from three independent images. (d) The expression levels of proteins in the nucleus and cytoplasm were determined by Western blotting. The protein was separately extracted from H1299 cells 48 hours after KPNB1 siRNA transfection. (e) The nucleus/cytoplasm ratios for the PLK1 protein were calculated from the Western blotting images of H1299 cells. The data represent the average of the ratios from three independent images. Error bars represent standard deviations.
Discussion
To identify an apoptosis-related gene that potentially could be used as a target of an anticancer drug, we looked for a gene whose expression level changed depending on the silencing of PLK1. We assumed that the expression of some apoptosis-related genes would be changed in the process of cell death induced by PLK1 depletion. That kind of gene might contribute to the apoptosis induced by PLK1 depletion, and its inhibitor would have synergy with the PLK1 inhibitor. A previous study showed that PLK1 is highly expressed in a variety of human tumors (27). From our sequence data, the PLK1 expression of human small airway epithelial cells (SAECs) was low compared with the lung adenocarcinoma cell lines. It was interesting that PLK1 expression differed greatly among the lung adenocarcinoma cell lines. Indeed, a previous study demonstrated that PLK1 expression levels of tissues differed more than ten times among NSCLC patients (11). Considering the function of PLK1 in cancer cells, it is possible that cancer cells with high PLK1 expression have a high grade of malignancy.
Our sequence data demonstrated that KPNB1 mRNA expression levels decreased along with PLK1 knockdown. Moreover, there was a positive correlation between PLK1 and KPNB1 expression levels in lung adenocarcinoma cell lines. Therefore, we examined the possibility that KPNB1 expression was changed downstream of PLK1. Our Western blotting analysis indicated that the protein expression levels of KPNB1 were also decreased, along with mRNA expression levels, by knockdown of PLK1 in three different cell lines: H1299, A549, and HeLa. This analysis indicated that decreased KPNB1 expression was not induced only in a specific cell line. We also confirmed that KPNB1 expression was decreased by the addition of the PLK1 inhibitor BI2536. This implied that PLK1 function (protein phosphorylation) is important for the regulation of KPNB1 expression.
In this study, the knockdown of KPNB1 induced cell apoptosis in H1299 cells. These data are consistent with previous studies that have shown that the inhibition of KPNB1 expression induces apoptosis and inhibits cellular proliferation in cervical cancer cells and hepatocellular carcinoma (28, 29). PLK1 knockdown induced apoptosis a bit earlier than KPNB1 knockdown in H1299 cells. Therefore, a factor other than KPNB1 may relate to the apoptosis induced by PLK1 depletion. However, the expression levels of the apoptosis-related genes C1qbp and Mcl-1 decreased similarly following PLK1 and KPNB1 depletion. Therefore, KPNB1 depletion may play a part in the apoptosis induced by PLK1 depletion.
The time-course of cell growth inhibition was somewhat different between PLK1- and KPNB1-silenced H1299 cells. The PLK1-silenced cells showed hardly any growth from the beginning. On the other hand, the number of KPNB1-silenced cells increased up to 48 hours after transfection. As discussed, this phenomenon may exist partly because PLK1 depletion induced apoptosis earlier than KPNB1 depletion. Furthermore, the different mechanism of inducing cell cycle arrest might affect the speed of cellular proliferation. Our cell cycle analysis revealed that the cell cycle was arrested at the G2/M phase in PLK1-silenced cells. On the other hand, the proportion of cells at the G0/G1 phase was increased by KPNB1 silencing. The larger number of cells at the G0/G1 phase may be derived from the accumulation of cells at the G0 phase because Ki67-stained cells increased with KPNB1 silencing. This is consistent with previous research demonstrating that KPNB1 is one of the key regulators for mitotic exit in human cells (30). Therefore, the different time-course of cell growth inhibition between PLK1- and KPNB1-depleted cells may result from the different mode of cell cycle arrest. Taken together, KPNB1 might not be involved in the cell cycle arrest caused by PLK1 depletion, although KPNB1 may play a part in the apoptosis induced by PLK1 silencing.
The KPNB1 protein is a member of the importin beta family and imports cargo proteins or RNAs into the nucleus in cooperation with importin alpha family members. We hypothesized that KPNB1 may take part in the importation of PLK1 into the nucleus because we speculated that apoptosis might be induced owing to the stagnation of PLK1 importation into the nucleus when the KPNB1 protein was depleted. Immunoprecipitation indicated that PLK1 and KPNB1 form a complex; however, KPNB1 depletion hardly changed the PLK1 intracellular location. There are a wide variety of proteins that take part in the importation of cargo proteins into the nucleus. Therefore, it is reasonable to believe that another importin family protein imports PLK1 into the nucleus, even if KPNB1 is depleted.
In summary, we showed that KPNB1 expression was decreased by PLK1 inhibition. It is possible that KPNB1 depletion plays a part in the apoptosis observed in PLK1-depleted cells. PLK1 has been extensively studied, and a wide variety of drugs targeting PLK1 have been discovered, since PLK1 is considered a potent target of anticancer drugs. However, the drug dosage is limited considering its side effects, as PLK1 is essential for the cell cycle progression of normal cells. Previous research has shown that the effect of the PLK1 inhibitor BI2536 was modest for NSCLC patients (31). A KPNB1 inhibitor may be used as an anticancer drug that may enhance the therapeutic effects of PLK1 inhibitors in the future.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research in the Priority Area “Genome Science” [221S0002] from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65(2):87-108
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7-30
3. Asamura H, Goya T, Koshiishi Y, Sohara Y, Eguchi K, Mori M. et al. A Japanese Lung Cancer Registry study: prognosis of 13,010 resected lung cancers. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2008;3(1):46-52
4. Qian YW, Erikson E, Li C, Maller JL. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Molecular and cellular biology. 1998;18(7):4262-71
5. Sunkel CE, Glover DM. Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. Journal of cell science. 1988;89(Pt 1):25-38
6. Bu Y, Yang Z, Li Q, Song F. Silencing of polo-like kinase (Plk) 1 via siRNA causes inhibition of growth and induction of apoptosis in human esophageal cancer cells. Oncology. 2008;74(3-4):198-206
7. de Oliveira JC, Brassesco MS, Pezuk JA, Morales AG, Valera ET, Montaldi AP. et al. In vitro PLK1 inhibition by BI 2536 decreases proliferation and induces cell-cycle arrest in melanoma cells. Journal of drugs in dermatology: JDD. 2012;11(5):587-92
8. Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. Journal of cutaneous pathology. 2002;29(6):354-8
9. Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K. et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer science. 2003;94(2):148-52
10. Tut TG, Lim SH, Dissanayake IU, Descallar J, Chua W, Ng W. et al. Upregulated polo-like kinase 1 expression correlates with inferior survival outcomes in rectal cancer. PloS one. 2015;10(6):e0129313
11. Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ. et al. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14(5):543-9
12. Van den Bossche J, Lardon F, Deschoolmeester V, De Pauw I, Vermorken JB, Specenier P. et al. Spotlight on volasertib: preclinical and clinical evaluation of a promising Plk1 inhibitor. Medicinal research reviews. 2016;36(4):749-86
13. Liu X. Targeting polo-like kinases: a promising therapeutic approach for cancer treatment. Translational oncology. 2015;8(3):185-95
14. Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R. et al. Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Current oncology (Toronto, Ont). 2012;19(1):e28-35
15. Suzuki A, Makinoshima H, Wakaguri H, Esumi H, Sugano S, Kohno T. et al. Aberrant transcriptional regulations in cancers: genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucleic acids research. 2014;42(22):13557-72
16. Sekimoto N, Suzuki A, Suzuki Y, Sugano S. Expression of miR26a exhibits a negative correlation with HMGA1 and regulates cancer progression by targeting HMGA1 in lung adenocarcinoma cells. Molecular medicine reports. 2017;15(2):534-42
17. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402-8
18. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9(7):671-5
19. Martens-de Kemp SR, Nagel R, Stigter-van Walsum M, van der Meulen IH, van Beusechem VW, Braakhuis BJ. et al. Functional genetic screens identify genes essential for tumor cell survival in head and neck and lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(8):1994-2003
20. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-4
21. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1
22. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543-50
23. Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M. et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Current biology: CB. 2007;17(4):316-22
24. Choi M, Kim W, Cheon MG, Lee CW, Kim JE. Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe following activation of the spindle assembly checkpoint in non-small cell lung cancer cells. Cancer letters. 2015;357(2):591-601
25. McGee AM, Douglas DL, Liang Y, Hyder SM, Baines CP. The mitochondrial protein C1qbp promotes cell proliferation, migration and resistance to cell death. Cell cycle (Georgetown, Tex). 2011;10(23):4119-27
26. Nair JS, Schwartz GK. Inhibition of polo like kinase 1 in sarcomas induces apoptosis that is dependent on Mcl-1 suppression. Cell cycle (Georgetown, Tex). 2015;14(19):3101-11
27. Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nature reviews cancer. 2006;6(4):321-30
28. Angus L, van der Watt PJ, Leaner VD. Inhibition of the nuclear transporter, Kpnbeta1, results in prolonged mitotic arrest and activation of the intrinsic apoptotic pathway in cervical cancer cells. Carcinogenesis. 2014;35(5):1121-31
29. Yang L, Hu B, Zhang Y, Qiang S, Cai J, Huang W. et al. Suppression of the nuclear transporter-KPNbeta1 expression inhibits tumor proliferation in hepatocellular carcinoma. Medical oncology (Northwood, London, England). 2015;32(4):128
30. Schmitz MH, Held M, Janssens V, Hutchins JR, Hudecz O, Ivanova E. et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nature cell biology. 2010;12(9):886-93
31. Sebastian M, Reck M, Waller CF, Kortsik C, Frickhofen N, Schuler M. et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2010;5(7):1060-7
Author contact
![]() Corresponding author: Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 4-6-1 Sirokanedai, Minato-ku, Tokyo 108-8639, Japan E-mail: nsekimotok.u-tokyo.ac.jp (Noboru Sekimoto), ssuganok.u-tokyo.ac.jp (Sumio Sugano).
Corresponding author: Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 4-6-1 Sirokanedai, Minato-ku, Tokyo 108-8639, Japan E-mail: nsekimotok.u-tokyo.ac.jp (Noboru Sekimoto), ssuganok.u-tokyo.ac.jp (Sumio Sugano).