Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(1):41-53. doi:10.7150/jca.21520 This issue Cite
Research Paper
Modulated electro-hyperthermia induced loco-regional and systemic tumor destruction in colorectal cancer allografts
Tamas Vancsik1, Csaba Kovago2, Eva Kiss1, Edina Papp3, Gertrud Forika1, Zoltan Benyo4, Nora Meggyeshazi1*, Tibor Krenacs1* ![]()
1. 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary;
2. Department of Pharmacology and Toxicology, Faculty of Veterinary Science, St. Istvan University, Budapest, Hungary;
3. Faculty of Bionics, Pazmany Peter Catholic University, Budapest, Hungary;
4. Institute of Clinical Experimental Research, Semmelweis University, Budapest, Hungary.
* These authors equally contributed to this paper
Received 2017-6-19; Accepted 2017-9-26; Published 2018-1-1
Abstract
Background: Modulated electro-hyperthermia (mEHT), a non-invasive intervention using 13.56 MHz radiofrequency, can selectively target cancers due to their elevated glycolysis (Warburg-effect), extracellular ion concentration and conductivity compared to normal tissues. We showed earlier that mEHT alone can provoke apoptosis and damage associated molecular pattern (DAMP) signals in human HT29 colorectal cancer xenografts of immunocompromised mice.
Materials: Here we tested the mEHT induced stress and immune responses in C26 colorectal cancer allografts of immunocompetent (BALB/c) mice between 12-72 h post-treatment. The right side of the symmetrical tumors grown in both femoral regions of mice were treated for 30 minutes, while the left side tumors served for untreated controls.
Results: Loco-regional mEHT treatment induced an ongoing and significant tumor damage with the blockade of cell cycle progression indicated by the loss of nuclear Ki67 protein. Nuclear shrinkage, apoptotic bodies and DNA fragmentation detected using TUNEL assay confirmed apoptosis. Cleaved/activated-caspase-8 and -caspase-3 upregulation along with mitochondrial translocation of bax protein and release of cytochrome-c were consistent with the activation of both the extrinsic and intrinsic caspase-dependent programmed cell death pathways. The prominent release of stress-associated Hsp70, calreticulin and HMGB1 proteins, relevant to DAMP signaling, was accompanied by the significant tumor infiltration by S100 positive antigen presenting dendritic cells and CD3 positive T-cells with only scant FoxP3 positive regulatory T-cells. In addition, mEHT combined with a chlorogenic acid rich T-cell promoting agent induced significant cell death both in the treated and the untreated contralateral tumors indicating a systemic anti-tumor effect.
Conclusions: mEHT induced caspase-dependent programmed cell death and the release of stress associated DAMP proteins in colorectal cancer allografts can provoke major immune cell infiltration. Accumulating antigen presenting dendritic cells and T-cells are likely to contribute to the ongoing tumor destruction by an immunogenic cell death mechanism both locally and through systemic effect at distant tumor sites.
Keywords: modulated electro-hyperthermia, colorectal cancer, apoptosis, damage signaling, immunogenic cell death, systemic effect.
Introduction
Colorectal cancer is one of the most common fatal malignancies worldwide affecting more than 1.2 million new patients every year [1, 2]. At least 50% of the cases relapse after surgical removal of the tumor without distant metastasis and 20% of colorectal cancers have distant metastasis at the time of diagnosis [3]. Though the 5 year survival of localized cancer is over 90%, it is sharply reduced to <12% in patients with distant tumor spread [4]. Since traditional treatment options of colorectal cancer are limited, boosting of the host's immune response offers a promising addition as shown by clinical trials utilizing immune modulation therapies [2, 5]. We earlier revealed that modulated electro-hyperthermia (mEHT) treatment can effectively damage colorectal cancer allografts in immunocompromised mice and provoke the release of damage associated molecular pattern (DAMP) signals, which can potentially promote immunogenic tumor cell death [6, 7]. In this study we investigated the mEHT related tumor stress and immune response in cancer allografts of immunocompetent mice both locally and at distant tumor sites after combining mEHT with a T-cell promoting agent.
Immunogenic cell death (ICD) can be stimulated by the spatiotemporal occurrence of DAMP signals during programmed cell death as a result of diverse cancer treatments [8]. DAMP signals support the activation of NK-cells and the functional maturation of professional antigen presenting cells (APC) in combination with tumor antigens and trigger cell-mediated antitumor immune responses [9]. Several oncological treatments can induce DAMP signals with slightly diverse patterns including chemotherapeutic agents, such as anthracyclines, oxaliplatin or cyclophosphamide; γ-irradiation, or anti-epidermal growth factor receptor (EGFR) specific target therapy [8, 10].
Modulated EHT treatment is a non-invasive complementary to chemo- and radiation tumor therapy using electromagnetic field generated by amplitude modulated 13.56 MHz radiofrequency. The electromagnetic field, which can instantly penetrate into cancer lumps, can induce heat shock response at controlled 42oC, besides interfering with cell membrane lipid rafts gathering receptor molecules [11]. Tumor selectivity of mEHT is linked to the elevated glycolysis (also known as the Warburg effect), ion concentration and permittivity of tumors compared to adjacent normal tissues [12]. In HT29 human colorectal cancer grown in immunodeficient BALB/c nu/nu mice we previously confirmed a significant, dominantly caspase-independent, AIF (apoptosis inducing factor) mediated tumor destruction by mEHT [7]. The programmed cell death response was accompanied by the progressive release of DAMP signals and we observed the accumulation immature immune cells suggestive of an ICD reaction [6]. In line with this, mEHT pre-treatment has recently been described to augment tumor antigen primed dendritic cell immunotherapy through supporting T-cell, macrophage and eosinophil leukocyte invasion of CT26 murine colorectal cancer allografts [13].
Extracts from Marsdenia tenacissima (MTE) herb has been used as a complementary to chemo- and targeted receptor tyrosine kinase (RTK) inhibitory therapy (e.g. with gefitinib) of lung, gastric and liver cancers [14]. In non-small cell lung cancer MTE provoked apoptosis in combination with cisplatin and also suppressed EGFR signaling [15, 16]. Also, MTE treatment in esophageal cancer contributed to the temporary inhibition of MAPK pathway through G0/G1 arrest [14]. High pressure liquid chromatography-mass spectrometry (HPLC-MS) analysis of MTE extracts detected massive chlorogenic acid levels [17]. Chlorogenic acid has been demonstrated to support the efficacy of 5-fluorouracil treatment in hepatocellular carcinoma through inhibiting ERK1/2 signaling [18], and tumor infiltration by T-cells [19].
In this study, we investigated the mEHT induced tumor cell death in C26 colon cancer allografts using immunocompetent BALB/c mice, with particular focus on stress responses, DAMP signaling and anti-tumor immune response. We also tested the effect of mEHT after MTE administration to see how combined therapy affects local and distant tumor sites.
Materials and Methods
Tumor model
C26 murine colorectal adenocarcinoma cell line (CLS Cell Lines Service GmbH, Eppelheim, Germany, #400156) was grown in RPMI 1640 + GlutaMax, medium including 10% heat inactivated fetal calf serum (FCS) and 5% streptomycin-penicillin (5000 units penicillin and 5 mg streptomycin/ml). Cells were released from a sub-confluent monolayer using 0.25% trypsin and 0.22 mg/ml ethylenediaminetetraacetic acid (EDTA) for 5 min and suspended in a serum free medium to reach the required 107 cell/ml concentration. All reagents were from GIBCO (Invitrogen, Carlsbad, CA).
BALB/c mice (females) were maintained in sterile environment, kept on sterilized food and water ad libitum under 12 h dark/12 h light cycles. The C26 cell line was subcutaneously injected with 1 million cells/100 µl into both femoral regions of 6 week old BALB/c mice, which were kept for 14 days until the diameter of symmetrical tumor implants reached ~1.5 cm diameter.
Laboratory animals were kept and treated in compliance with the Hungarian Laws No. XXVIII/1998 and LXVII/2002 on the protection and welfare of animals, and the animal welfare regulations of the European Union. The study was approved by the Governmental Ethical Committee under No. SzIE ÁOTK MÁB 233/2012 and No. SzIE ÁOTK MÁB 26/2013.
Treatments
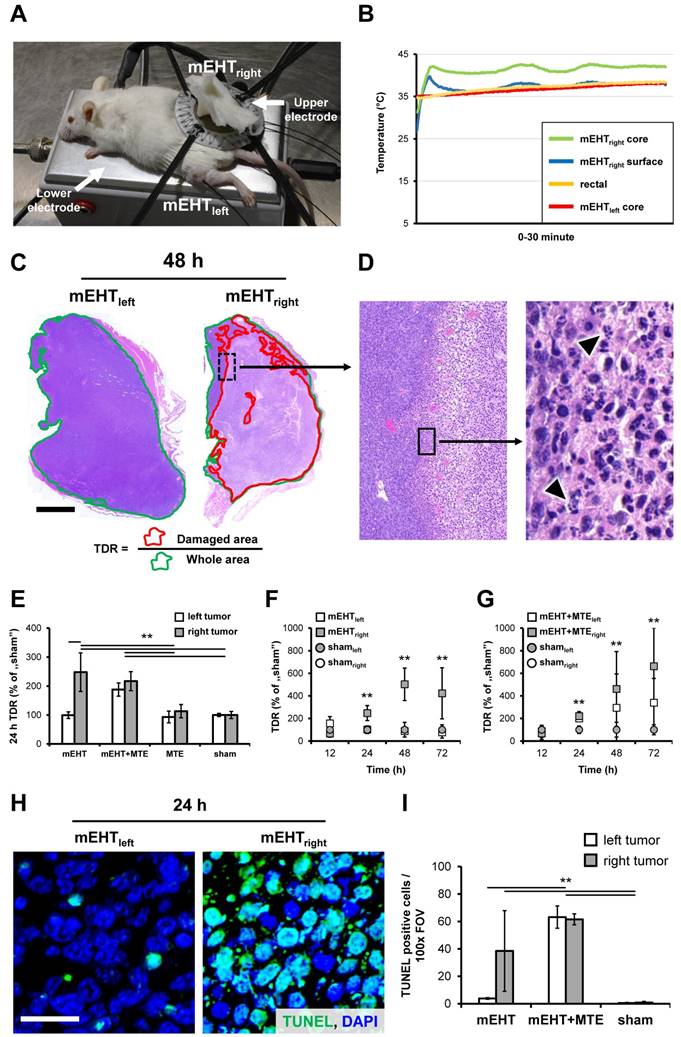
Only the right leg tumors were treated with mEHT (mEHTright) of the symmetrical C26 implants using plan-parallel electric condensers embracing the tumors (Figure 1A). The left-leg tumors served both for untreated internal controls (mEHTleft) and for monitoring the potential systemic effect of the treatment options tested. A rectangular grounded (lower) aluminum electrode of 72.0 cm2 (kept at 37 °C) was below the animals and a 2.5 cm2 round copper-silver-tin coated flexible textile electrode was overlaid on the tumors, which were cooled under control using a wet pad. Electromagnetic heating was generated by capacitive coupled, amplitude modulated 13.56 MHz radiofrequency (LabEHY, Oncotherm kft, Budaors, Hungary). Animals were treated under 100 mg/kg ketamine and 10 mg/kg xylazine anesthesia, with a single shot of mEHT for 30 min using 1-3W average power. Intratumoral temperature was measured with optical sensors (Luxtron FOT Lab Kit, LumaSense Technologies, Inc. CA) and kept at ~42°C (+/- 0.5°C) (Figure 1B). The subcutaneous temperature under the electrode was kept at ~40 °C and the rectal temperature at ~37°C.
mEHT induced tumor destruction and DNA fragmentation. Experimental model of mEHT treatment (A). Temperature curves during treatment (B). Tumor destruction ratio (TDR) calculated as the damaged per whole tumor area in hematoxylin-eosin stained sections was significantly elevated after mEHT treatment of C26 colorectal cancer allografts (C). Chromatin condensation and apoptotic bodies (arrowheads) are typical signs of apoptosis (D). Elevated TDR both in the mEHT treated right-leg tumors compared to the untreated left sides or sham controls in monotherapy, and in both tumor sides after combing mEHT and MTE (mEHT+MTE) compared to the sham controls, 24 h after treatment (E). Progressive tumor damage both after single (F) and combined mEHT+MTE treatments (G). Significantly elevated numbers of TUNEL positive cell nuclei (green) in the mEHT-treated (right) tumor (H), and in both sides of the mEHT+MTE combination group compared to sham controls 24 h post-treatment (I). p**<0.001. Scale bar shows 2 mm on C, 100 µm (left) and 10 µm (right) on D; and 20 µm (H-I).
Marsdenia tenacissima ingredients extracted using warm water followed by alcohol precipitation (MTE, Xiao-Ai-Ping, Sanhome Pharmaceutical Co., Nanjing, China). Mice were injected with 7.5 ml/kg extract intraperitoneally (i.p.). In the combined treatment group, MTE administration was followed 30 min later by mEHT treatment of the right-leg tumors (mEHT+MTEright), as described above. In sham treated animals the experimental conditions were the same as in mEHT groups except that the electric circuit was off. Tumor samples were collected from at least 3 animals at each time point from each group 12, 24, 48 and 72 h after treatment following the sacrificing of mice by cervical dislocation. Excised tumors were fixed in 10% formalin, dehydrated and embedded into paraffin wax.
Measuring mEHT related tumor destruction
Serial cross sections of 4 µm thickness were cut from whole tumor blocks were used both for hematoxylin and eosin (H&E) and immunostaining. H&E stained slides were digitalized using Pannoramic Scan and analyzed with the HistoQuant module of Pannoramic Viewer software (all from 3DHISTECH, Budapest, Hungary) based on image color and intensity segmentation. The whole tumor area (W) was correlated with the damaged (paler) tumor tissue (D) for calculating the tumor destruction ratio (TDR=W/D) (Figure 1C).
Immunohistochemistry
Both whole cross sections and tissue microarrays (TMA) were used for immunohistochemistry. Three representative cores of 2 mm diameter taken from standard areas of each tumor samples were collected into TMA blocks. Two cores from the border between degraded and intact tumor regions and 1 from the degraded tumor center based on H&E stained slides using the computer-driven TMA Master (3DHISTECH). For antigen retrieval dewaxed, rehydrated and endogenous peroxidase-blocked (20 min in 1:60; H2O2:methanol) slides were heated at ~100oC for 40 min using microwave oven (Whirlpool, Benton Harbor, MI) either in citrate buffer pH 6.0 (0.01 M sodium citrate-citric acid) or in Tris-EDTA (TE) buffer pH 9.0 (0.1 M Trisbase and 0.01 M EDTA) followed by a blocking in 0.1 M Tris-buffered saline (TBS, pH7.4) containing 5% bovine serum albumin (BSA) and 0,1 % sodium-azide (all from Sigma-Aldrich, St Luis, MO) for 20 min. Then, sections were incubated in a humidity chamber at room temperature overnight (16 h) using the antibodies and conditions listed in Table 1. For immunofluorescence Alexa Fluor 546 (orange-red) coupled anti-rabbit Ig (1:200) was used for 90 min. Cell membranes were highlighted with a wheat germ agglutinin (WGA) alexa fluor 488 conjugate (1:200) and cell nuclei were stained blue with 4', 6-diamidino-2-phenylindole (DAPI). All fluorescence probes were from Invitrogen/Molecular Probes (Carlsbad, CA, USA). For immunoperoxidase staining the labelled polymer-peroxidase anti-rabbit Ig detection system (Dako, Glostrup, Denmark) was applied for 30 min and the reactions were developed using DAB/hydrogen peroxide kit (Leica-NovoCastra, Newcastle Up-on-Tyne, UK). Between incubation steps the slides were washed 3x3 minutes in TBS (pH 7.4) buffer. Stained slides were digitalized and the immunofluorescence reactions were evaluated using image (color, intensity and size) segmentation based software tools HistoQuant and CellQuant (3DHISTECH). Relative mask areas (rMA) representing positive immunoreactions were calculated by dividing stained area with the whole annotation area for cleaved-caspase-3, HMGB1, CD3 and S100 staining. For calreticulin and Hsp70 staining the number of biomarker positive cells was counted in 5 representative microscopic fields (FOV) at x40 objective magnification in each sample at each time point and case. The number of cytoplasmic cytochrome c positive and nuclear FoxP3 positive cells was counted in 3 FOV and 5 FOV/sample at x50 objective magnification, respectively.
Antibodies and conditions used for immunohistochemistry and immunofluorescence. (T-E tris-EDTA)
| Antigen | Type | Reference no. | Dilution | Antigen retrieval | Vendor |
|---|---|---|---|---|---|
| AIF | Rabbit, pAb | #4642 | 1:25 | T-E | Cell Signaling |
| Bax | Rabbit, pAb | #HPA027878 | 1:50 | T-E | Sigma |
| Calreticulin | Rabbit, pAb | #12238 | 1:200 | T-E | Cell Signaling |
| CD3 | Rabbit, pAb | #IS503 | 1:2 | T-E | DAKO |
| Cleaved caspase-3 | Rabbit, pAb | #9664 | 1:100 | Citrate | Cell Signaling |
| Cleaved caspase-8 | Rabbit, pAb | #8592 | 1:100 | T-E | Cell Signaling |
| Cytochrome c | Rabbit, pAb | #4280 | 1:50 | T-E | Cell Signaling |
| FoxP3 | Rat, pAb | #14-5773 | 1:600 | T-E | eBioscience |
| HMGB1 | Rabbit, pAb | #6893 | 1:200 | Citrate | Cell Signaling |
| Hsp70 | Rabbit, pAb | #4872 | 1:50 | T-E | Cell Signaling |
| S100 | Rabbit, pAb | #RB-9018 | 1:500 | T-E | Thermo |
Vendor specification: Cell Signaling (Danvers, MA, USA), Thermo (Waltham, MA, USA), Dako (Glostrup, Denmark), eBioscience (San Diego, CA, USA), Sigma (St. Louis, MO, USA); T-E: Tris-EDTA, pH 9.0.
TUNEL assay
Terminal deoxynucleotidyl transferase nick end labeling (TUNEL, Alexa Fluor 488 Assay, Invitrogen) assay was used according to manufacturer's instructions. This assay is based on the detection and linking of DNA nick ends by terminal deoxynucleotidyl transferase (TdT) with fluorochrome labeled deoxyuridine triphosphate (dUTP) nucleotides. Labeling of cell nuclei is proportional with the amount of fragmented DNA as a result of programmed cell death. Briefly, dewaxed and rehydrated slides were heated in a citrate based pH 6.0 antigen unmasking solution (H-3300, Vector Lab, Burlingame, CA) for 45 minutes using microwave oven (Whirlpool, as above). Then slides were incubated at 37°C for 60 min with a cocktail of alkyne substituted dUTP and TdT followed by the fluorochrome for 30 min at room temperature which coupled to dUTP under copper (I) catalysis. Slides were digitalized using Pannoramic Scan after nuclear DNA was stained with DAPI. The TUNEL positive cells were counted at x100 objective magnification in 5 FOV in each sample.
Statistical analysis
The SPSS v.20 package was used for statistics (IBM, New York, NY). Since our data did not show normal distribution the non-parametric Kruskal-Wallis test was used for analyzing significance among groups, followed by Dunn's post hoc test with Bonferroni correction to see pairwise statistical significance between groups. Significance was declared at p<0.05, or less after correction.
Results
mEHT induced programmed tumor cell death
Tumor destruction ratio (TDR) calculated by dividing the damaged areas with the whole tumor areas (Figure 1C-D), measured in H&E stained digital slides, was significantly higher in mEHT treated (mEHTright) than untreated sides (mEHTleft) or sham control tumors between 24-72 h post-treatment (Figure 1E-F). Combination therapy (mEHT+MTE) led to tumor damage not only in the treated right tumors (mEHT+MTEright) but also in the untreated left tumors (mEHT+MTEleft) within the same time-frame (Figure 1G). Kruskal-Wallis test proved highly significant differences (p<0.001) among groups 24 h (χ2(5)=26.081), 48 h (χ2(5)=20.911) and 72 h χ2(5)=28.297) post-mEHT treatment. The effect of MTE administration alone was not significant.
Nuclear chromatin condensation and widespread apoptotic bodies indicated mEHT induced programmed cell death response (Figure 1D), which was confirmed by elevated DNA fragmentation from 24 h post-treatment (χ2(5)=72.415, p<0.001) (Figure 1H). Post hoc test revealed significantly elevated TUNEL positivity in: mEHTright vs. shamright and shamleft; in the mEHT+MTEright or mEHT+MTEleft vs. sham controls and mEHTleft (Figure 1I).
Mechanism of mEHT induced programmed tumor cell death
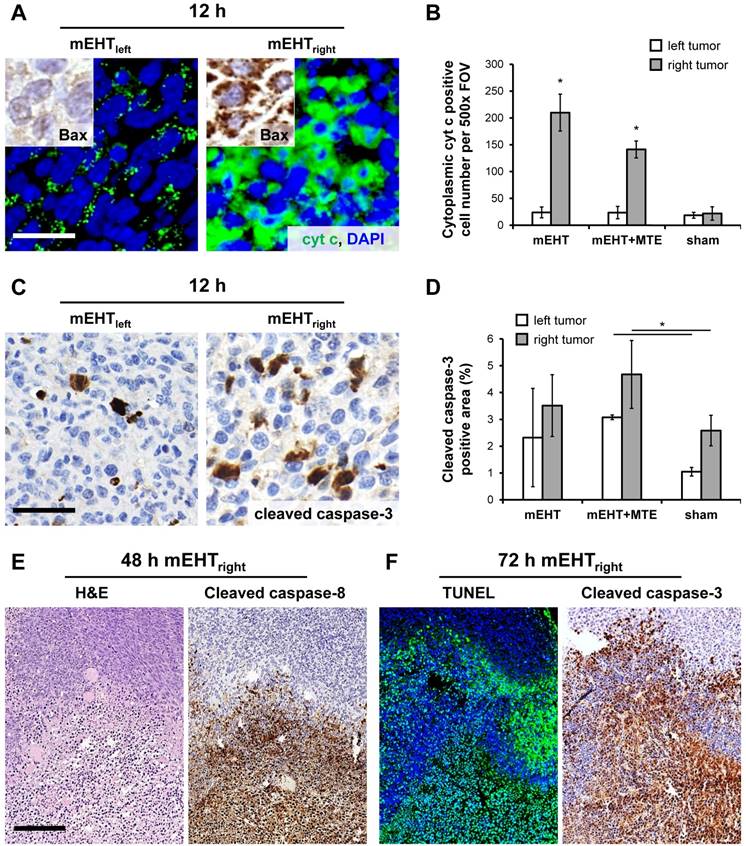
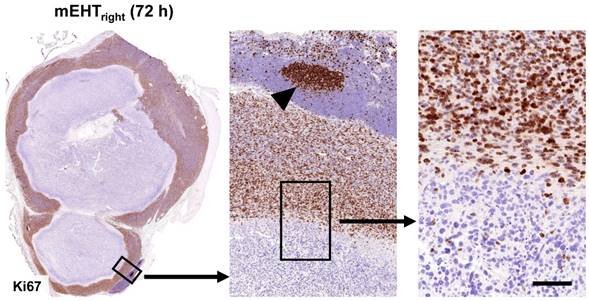
mEHT treatment induced the significant mitochondrial translocation of bax protein and the cytoplasmic release of cytochrome c between 12-24 h (χ2(5)=11,989, χ2(5)=14,497, p<0.05) in the treated (mEHTright) tumors either after single or combined mEHT treatment (Figure 2A-B). Cytochrome c became significantly delocalized (χ2(5)=11,943, p<0.05) also in the mEHT untreated (left-leg) tumors of the combined mEHT+MTE group between 48-72 h (not shown). Major elevation of cleaved/activated caspase-3 positive cell numbers were seen from 12 h post-treatment compared to sham controls (χ2(5)=9,842, p<0.05) (Figure 2C-D). Overlapping cleaved caspase-8, -caspase-3 and TUNEL positive cell fractions were significantly elevated in the apoptotic tumor regions including the intact-damaged marginal zone, particularly at advanced stages, 48-72 h post-mEHT treatment (Figure 2E-F). These findings are consistent with the activation of caspase-dependent extrinsic and intrinsic apoptotic pathways. After treatment the nuclear Ki67 protein expression disappeared completely from tumor cells which showed minor morphological signs of apoptosis (Figure 3).
mEHT induced pro-apoptotic factors. Mitochondrial cytochrome c in untreated tumors show significant cytoplasmic (green, immunofluorescence after mEHT treatment (A) in parallel with the mitochondrial translocation of bax protein (insets in A, brown immunoperoxidase) in 12h samples (p*<0.05) (B). At the same time, cleaved caspase-3 positive cells (brown, immunoperoxidase) are also significantly more common at both tumor sides after combined (mEHT+MTE) treatment (D). Overlapping cleaved caspase-8 positive (brown, immunoperoxidase) (E), TUNEL (green fluorescence) and cleaved caspase-3 (brown) positive cell fractions (F) concentrated in damaged tumor regions proved both extrinsic and intrinsic caspase dependent apoptosis, 48 h and 72 h after treatment, respectively. Scale bars, A and C: 20 µm; E and F: 100 µm.
mEHT induced blocking of cell cycle progression. Nuclear Ki67 positivity indicating tumor cell proliferation is ceased in the mEHT damaged central tumor regions (left), even in cells showing only early signs of apoptosis. Gradually higher magnifications (middle and right). The lymphoid germinal center (middle, arrowhead) serves for Ki67 positive control. Scale bar shows 2 mm on left, 200 µm on middle and 60 µm on right images.
There was no statistical difference in nuclear apoptosis inducing factor (AIF) levels between the treated and untreated tumors (not shown).
mEHT induced damage associated molecular pattern (DAMP) signals
Massive cytoplasmic to cell-membrane relocalization of calreticulin was observed from 12 h post-treatment (χ2(5)=44.720, p<0.001). Pairwise statistical analysis confirmed significant cell membrane concentration in mEHTright (mEHT treated) vs. mEHTleft and sham control tumors; and in mEHT+MTEright or mEHT+MTEleft vs. mEHTleft and sham control tumors (Figure 4A-B).
mEHT induced damage signaling. Release of calreticulin (red) protein from the mitochondria and colocalization with the cell membrane marker WGA (green) confirms its membrane accumulation (yellow) after mEHT treatment (A). Significantly increased proportion of tumor cells with overlapping WGA and calreticulin reactions (arrowheads) in both treatment groups (p**<0.001) compared to sham controls (B). Elevated intracytoplasmic Hsp70 protein (red) in overlap with the cell membrane WGA (green) 48h after treatment. The number of Hsp70 positive cells was significantly increased (p**<0.001) in the mEHTright and in both tumors of mEHT+MTE groups compared to sham controls (D). Early nuclear to cytoplasmic release of HMGB1 (red) group 48 h after treatment (E). The proportion of HMGB1 negative areas indicating extracellular release were significantly lower (p*<0.05) in the mEHTright and in both tumor sides of mEHT+MTE groups compared to sham controls (F). Scale bar in large images shows 20 µm, and 10 µm in the insets.
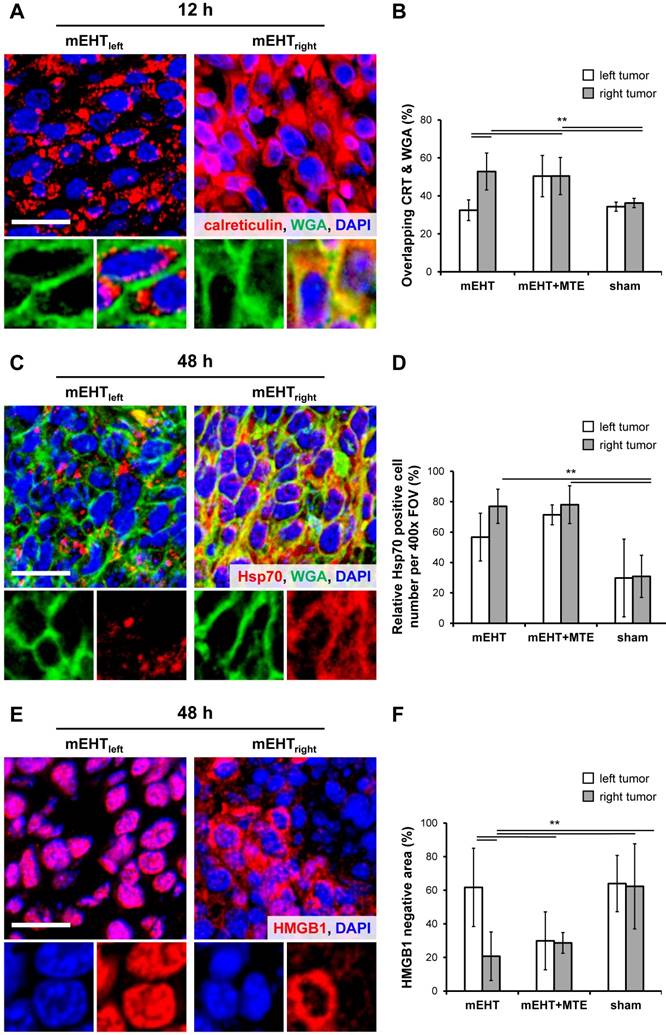
This was followed by the significant cell-membrane accumulation of Hsp70 protein in the mEHT-treated groups peaking at 48 h post-treatment (χ2(5)=54.912, p<0.001). Pairwise significance was seen in mEHTright vs. mEHTleft and sham control tumors; and in mEHT+MTEright or mEHT+MTEleft vs. mEHTleft and sham control tumors (Figure 4C-D).
Also, significant nuclear to cytoplasmic release or complete loss in the damaged areas of HMGB1 protein was seen at 48 h post-mEHT treatment (χ2(5)=12.969, p=0.024). Post hoc test confirmed significant disappearance of HMGB1 in mEHTright vs. sham controls; and in mEHT+MTEleft vs. mEHTleft tumors (Figure 4E-F).
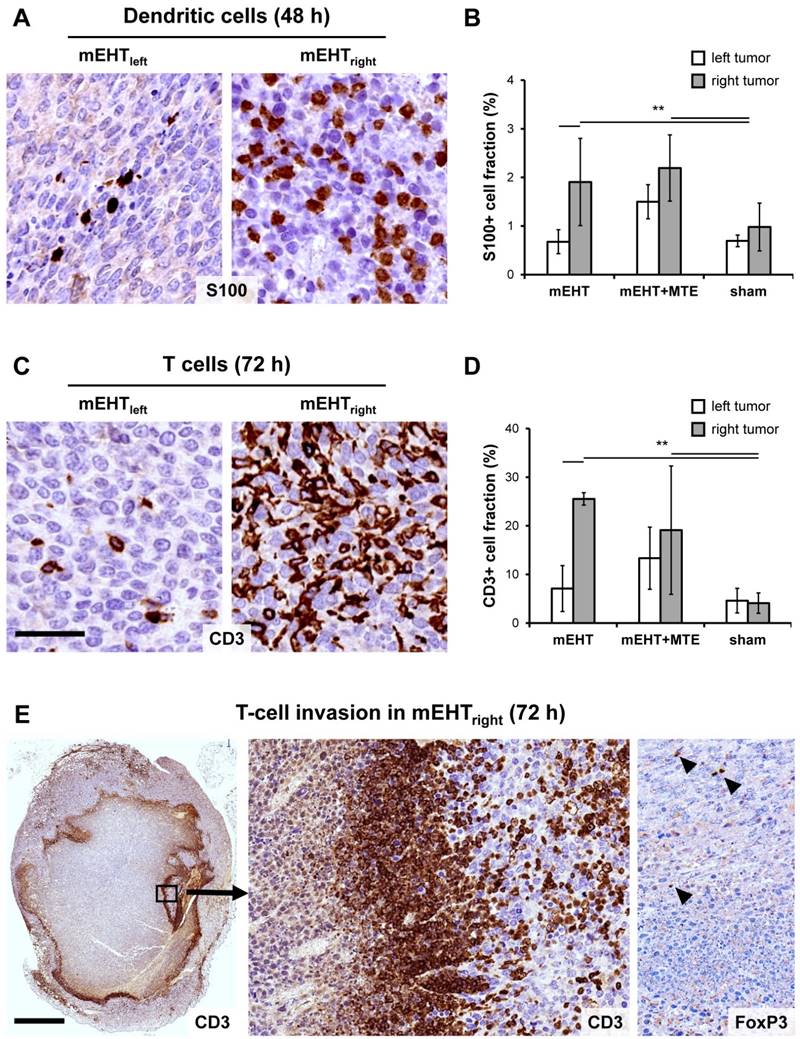
mEHT induced tumor infiltration by antigen presenting cells and T-cells
DAMP signal sequence was accompanied by the massive tumor infiltration by S100 positive APC from 48 h post-treatment (χ2(5)=12.483, p=0.029). Post hoc test revealed significant numbers of APC in mEHTright vs. mEHTleft and shamright and shamleft tumors; and in mEHT+MTEright vs. sham tumors (Figure 5A-B). Also, significant numbers of CD3 positive T-cells infiltrated the treated tumors with a peak at 72 h post-mEHT treatment (χ2(5)=14.156, p=0.015) (Figure 5C). Pairwise testing confirmed significantly more T-cells in mEHTright vs. mEHTleft tumors; and in mEHT+MTEright vs. shamleft and shamright tumors (Figure 5C-D). APC and T-cell invasion also showed a nearly significant trend between mEHT+MTEleft and sham control tumors. A dense ring of T-cell invasion was seen in the margin between the damaged and “intact”-looking tumor regions, with the latter also infiltrated heavily (Figure 5E). FoxP3 positive T-cells were rare and their number did not differ between treated and untreated tumors.
mEHT induced tumor infiltration by immune cells. Significantly elevated numbers of S100 positive (brown) antigen presenting dendritic cells (APC) in the mEHTright tumors, and in both tumor sides of the mEHT+MTE combined group 48 h after treatment (A), as summarized in a graph (B). Increase in T-cell numbers also proved to be significant (p*<0.05) in the mEHTright tumors, and in both tumor sides of the mEHT+MTE combined group 72 h after treatment (C), shown also in a graph (D). Massive T-cell infiltration rim separates damaged central tumor regions from the intact periphery (E), which latter is also heavily infiltrated as seen at higher magnification (middle), with only scant FoxP3+ T cells (right). Scale bars in A-B and D-E: 40 µm. Scale bar in C-left: 2 mm, middle and right: 100 µm.
Discussion
Previously we showed that the mEHT can trigger a programmed cell death response in HT29 human colorectal cancer xenografts of immunocompromised mice, which was dominantly mediated by caspase independent AIF activation [7]. This was accompanied by the translocation and release of DAMP signal elements relevant to inducing ICD and the tumor infiltration by immature immune cells [6]. In the current study, C26 colorectal cancer allografts grown in immunocompetent mice were used to test mEHT induced cell stress, programmed cell death and anti-tumor immune responses. Here also, we detected apoptosis mediated tumor damage, which however, followed a caspase-dependent subroutine. It seems that the preferred programmed cell death pathway after mEHT treatment is rather depend on the inherent genetic and epigenetic makeup of the given tumor cell line, than the treatment itself. This view is supported by detecting the same death pathways as after mEHT treatment in small damaged areas, probably induced by hypoxia, in the untreated sham tumors both in the HT29 and C26 models. In this study, the mEHT induced DAMP signal sequence was followed by significant APC and T-cell invasion of the treated tumors, which was also seen in the untreated contralateral neoplasms when mEHT was delivered together with MTE, a T-cell promoting agent (summarized in Figure 6). These data suggest that besides its loco-regional effect mEHT can also contribute to tumor damage distant from the directly targeted regions.
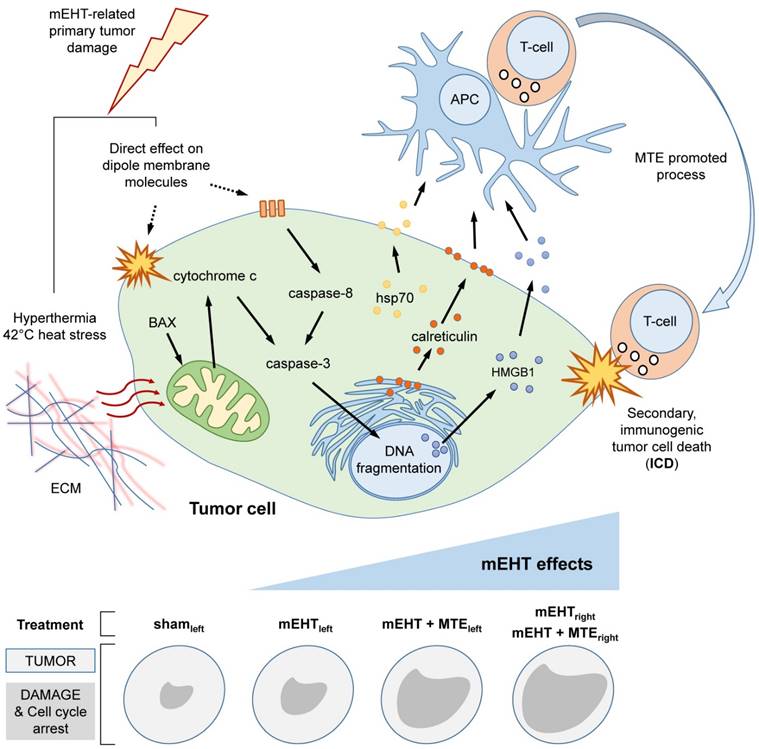
Summary drawing on mEHT induced anti-tumor damage mechanisms. Upper panel. Heat stress at 42oC and direct effect of electric field on dipole molecules both in the extracellular matrix (ECM) and the tumor cell membrane induce caspase-dependent apoptosis resulting in DNA fragmentation. The concomitant release of DAMP signals (Hsp70, calreticulin and HMGB1) support the maturation of antigen presenting cells (APC), which activate cytotoxic T-cells for mediating a secondary, immunogenic cell death (ICD) response. Systemic mEHT effect is possibly facilitated by MTE promoted T-cell migration/activation. Lower panel sums up semiquantitatively how mEHT induced pathways, tumor destruction effect and cell cycle arrest are manifested after using different treatment modules.
It has already been revealed that mEHT treatment can more effectively destroy tumor cells than conventional hyperthermia at the same (42°C) intratumoral temperature [20]. This and all of our earlier works suggest that modulated electric field can instantly penetrate deep into tumors and provoke tumor damage spreading inside-out from tumor centers [6, 7, 20]. It can efficiently mediate an irreversible cell stress by inducing heat shock and affecting charged molecules particularly in tumor cell membranes [11]. In human cancer therapy, mEHT (oncothermia; and traditional hyperthermia too) is used in sequentially repeated treatment modules, usually alternating either with radio- or chemotherapy. Experimentally, so far we only characterized the effects of single mEHT treatment. Therefore, in our present model, for standardizing single treatment, we precisely measured and kept intratumoral temperature at 42oC. We demonstrated that mEHT treatment caused significant destruction of C26 colorectal cancer cells by inducing apoptosis confirmed by chromatin condensation and nuclear DNA fragmentation. C26 cell line form aggressive tumors in vivo, which show almost 100% proliferation index by detecting Ki67 protein a general cell cycle marker expressed from G1- to the end of M-phase [21]. Furthermore, we showed that early apoptotic commitment induced by mEHT can result in the complete blockade of the cell cycle regulation machinery indicated by the loss nuclear Ki67 expression.
The progressive accumulation of DNA fragmentation and apoptotic bodies between 12-72 h post mEHT treatment was consistent with a programmed cell death response without obvious necrosis. The significant tumor damage caused by mEHT treatment proved to be caspase-dependent indicated by the massive expansion of the cleaved/activated caspase-3 positive apoptotic cell fractions. The upregulation of cleaved/activated caspase-8 protein besides the mitochondrial translocation of bax and the mitochondrial release of cytochrome c confirmed the activation of both the extrinsic and intrinsic caspase dependent pathways [22]. Elevated apoptotic signaling detected initially, between 12-24 h, only in the mEHT treated tumors became bilateral by 48 h post-treatment in mEHT+MTE treated animals. This finding suggests that apoptosis can be provoked by mEHT both directly and later on through mediators which recruit the immune system (see Figure 6). The latter may involve factors discharged by damaged tumor cells and those of MTE extract leading to a systemic effect on distant tumor sites.
Tumor treatment can contribute to the elimination of cancer cells through interactions between immune-independent (initial apoptosis) and immune-dependent (activation of immune response) pathways [23]. Immunogenic tumor cell death response can extend and maintain the treatment induced primary apoptotic tumor damage. ICD can be triggered by DAMPs including extracellular release of HMGB1 and ATP which act similar to cytokines [24]. It is also supported by the cell membrane localization and access by antigen presenting and immune cells of the chaperons Hsp70 and calreticulin [6]. Conventional hyperthermia can induce only partial DAMPs in tumors mainly through Hsp70 release which alone is less efficient in supporting APC maturation as well as NK and cytotoxic T-cell functions [25, 26, 27]. Complex DAMP signals can activate APC by promoting the uptake and presentation of tumor antigens through Toll-like-, purinergic- and pattern recognition receptors for enhancing tumor-specific T-cell response [23]. In line with our results in immunocompromised tumor model [6], mEHT in this study also induced complex DAMP signals including calreticulin, HSP70 and HMGB1 release. The potential immune stimulatory effect of these signals was supported by our finding of progressive tumor infiltration by S100 positive APC and CD3 positive T-cells in immunocompetent mice with only negligible number of FoxP3+ immune inhibitory T-cells. Though elevated tumor damage, DAMP signaling and immune cell infiltration was frequently seen in the contralateral (left) tumors after single mEHT treatment, this became significant only after combined mEHT+MTE treatment in the mEHT untreated left tumors.
Similar to our results, local mEHT treatment, in combination with antigen-primed syngeneic APC injection, significantly inhibited the growth of CT26 tumors (similar to C26 but from different vendor) and protected animals from developing new tumors after CT26 re-challenge [13]. This intervention primed IFN-γ secretion and supported the recruitment of both the innate immunity and the adaptive cytotoxic T-cell mediated immunity. A similar mEHT primed APC treatment of squamous cell carcinoma allografts also induced elevated numbers of S100 positive APC and CD8 positive cytotoxic T-cells in line with the inhibition of tumor growth both in the treated and distant tumor sites [28]. In agreement with our results, mEHT (when combined with APC therapy) did not influence the number of the immune-inhibitory FoxP3 positive regulatory T-cells [29]. Thus, massive CD3 positive T-cell infiltration with insignificant number of FoxP3 positive regulatory T-cells, can mediate an effective anti-tumor immune response [26, 29]. This was supported by the progressive nature and positive correlation between tumor damage and immune cell infiltration in our present study.
MTE treatment has been demonstrated to support the cellular immune response. MTE administration synergized with the growth inhibitory and apoptosis inducing effect of cisplatin in Lewis lung cancer xenografts and increased the number of tumor infiltrating CD8 positive T-cells and the expression of interferon-γ, perforin-1, and granzyme-B [16]. It also contributed to the suppression of growth of EGFR inhibitor resistant, K-ras and EGFR-mutant non-small cell lung cancer cells through hampering downstream RTK signaling pathways (Akt and MAPK) [15]. This effect was primarily attributed to chlorogenic acid identified in MTE by enhancing IFN-γ, IL-2, IL-12, MCP-1 levels and promoting the recruitment of TH1 lymphocytes, as well as stimulating T-lymphocyte, NK-cell and macrophage functions [19, 30]. The systemic effect of loco-regional cancer therapy, also known as abscopal effect, which was confirmed e.g. after chemotherapy or γ-irradiation, is most probably mediated by the immune system [31]. Therefore, both published reports and our data suggest that MTE can contribute to APC and T-cell recruitment and tumor infiltration in synergy with DAMP signals and tumor antigen release induced by mEHT. However, the accurate mechanism of action and the messengers involved need to be further clarified in molecular and functional studies.
In conclusion, mEHT treatment can cause irreversible cell stress and the blockade of tumor cell cycle leading to caspase-dependent programmed cells death in C26 colorectal cancer allografts of immunocompetent mice. DAMP signals released from mEHT damaged cells are associated with tumor infiltration by APC and T-cells, which are likely to contribute to the ongoing tumor damage. Local mEHT treatment can also facilitate a systemic antitumor effect in combination with a T-cell promoting agent. The contribution of MTE derived chlorogenic acid in the immune response need to be further elucidated.
Abbreviations
AIF: apoptosis inducing factor; APC: antigen presenting cells; ATP: adenosine triphosphate; bax: Bcl-2-associated X protein; BSA: bovine serum albumin; CD3: cluster of differentiation 3; DAB: 3,3'-Diaminobenzidine; DAPI: 4',6-diamidino-2-phenylindole; DNA: deoxyribonucleic acid; dUTP: deoxyuridine triphosphate; EDTA: ethylenediaminetetraacetic acid; EGFR: epidermal growth factor receptor; FCS: fetal calf serum; FOV: field of view; FoxP3: forkhead box P3; H&E: hematoxylin and eosin; HMGB1: high mobility group box 1; HPLC-MS: high pressure liquid chromatography-mass spectrometry; hsp70: 70 kilodalton heat shock protein; ICD: immunogenic cell death; IFN-γ: interferon gamma; IL-2 and -12: interleukin-2 and -12; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemoattractant protein-1; mEHT: modulated electro-hyperthermia; MTE: Marsdenia tenacissima; NK: natural killer cell; rMA: relative mask area; RTK: receptor tyrosine kinase; TBS: Tris-buffered saline; TDR: Tumor destruction ratio; TdT: terminal deoxynucleotidyl transferase; TE: Tris-EDTA; TH1: T helper 1 cell; TMA: tissue microarrays; TUNEL: terminal deoxynucleotidyl transferase nick end labeling; WGA: wheat germ agglutinin.
Acknowledgements
We are grateful to LifeRay Inc. for some reagents, to Edit Parsch and Renata Popp for skillful technical assistance and to Zsofia Csupor for contributing to the evaluation of some immunoreactions on digital slides.
Funding
This work was supported by the Hungarian National Research, Development and Innovation Office (NVKP_16-1-2016-0042) grant.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383(9927):1490-502
2. Boland PM, Ma WW. Immunotherapy for Colorectal Cancer. Cancers (Basel). 2017:9 (5)
3. Koido S, Ohkusa T, Homma S, Namiki Y, Takakura K, Saito K. et al. Immunotherapy for colorectal cancer. World J Gastroenterol. 2013;19(46):8531-42
4. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T. et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220-41
5. Lynch D, Murphy A. The emerging role of immunotherapy in colorectal cancer. Ann Transl Med. 2016;4(16):305
6. Andocs G, Meggyeshazi N, Balogh L, Spisak S, Maros ME, Balla P. et al. Upregulation of heat shock proteins and the promotion of damage-associated molecular pattern signals in a colorectal cancer model by modulated electrohyperthermia. Cell Stress Chaperones. 2015;20(1):37-46
7. Meggyeshazi N, Andocs G, Balogh L, Balla P, Kiszner G, Teleki I. et al. DNA fragmentation and caspase-independent programmed cell death by modulated electrohyperthermia. Strahlentherapie und Onkologie: Organ der Deutschen Rontgengesellschaft [et al]. 2014;190(9):815-22
8. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51-72
9. Liu Y, Zeng G. Cancer and innate immune system interactions: translational potentials for cancer immunotherapy. J Immunother. 2012;35(4):299-308
10. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014;21(1):26-38
11. Gabor Andocs MUR, Qing-Li Zhao, Edina Papp, Takashi Kondo Andras Szasz. Nanoheating without Artificial Nanoparticles Part II. Experimental Support of the Nanoheating Concept of the Modulated Electro-Hyperthermia Method, Using U937 Cell Suspension Model. Biology and Medicine. 2015;7(4):9
12. Szasz A, Szasz N, Szasz O. Oncothermia: Principles and Practices. Springer Netherlands. 2010:36-8
13. Tsang YW, Huang CC, Yang KL, Chi MS, Chiang HC, Wang YS. et al. Improving immunological tumor microenvironment using electro-hyperthermia followed by dendritic cell immunotherapy. BMC Cancer. 2015;15:708
14. Fan W, Sun L, Zhou JQ, Zhang C, Qin S, Tang Y. et al. Marsdenia tenacissima extract induces G0/G1 cell cycle arrest in human esophageal carcinoma cells by inhibiting mitogen-activated protein kinase (MAPK) signaling pathway. Chin J Nat Med. 2015;13(6):428-37
15. Han SY, Zhao W, Sun H, Zhou N, Zhou F, An G. et al. Marsdenia tenacissima extract enhances gefitinib efficacy in non-small cell lung cancer xenografts. Phytomedicine. 2015;22(5):560-7
16. Li W, Yang Y, Ouyang Z, Zhang Q, Wang L, Tao F. et al. Xiao-Ai-Ping, a TCM Injection, Enhances the Antigrowth Effects of Cisplatin on Lewis Lung Cancer Cells through Promoting the Infiltration and Function of CD8(+) T Lymphocytes. Evid Based Complement Alternat Med. 2013;2013:879512
17. Zhao L, Xiang B, Tan X. Fingerprinting of Marsdenia tenacissima by capillary electrophoresis compared to HPLC. J Chromatogr Sci. 2010;48(5):417-20
18. Yan Y, Li J, Han J, Hou N, Song Y, Dong L. Chlorogenic acid enhances the effects of 5-fluorouracil in human hepatocellular carcinoma cells through the inhibition of extracellular signal-regulated kinases. Anticancer Drugs. 2015;26(5):540-6
19. Kang TY, Yang HR, Zhang J, Li D, Lin J, Wang L. et al. The studies of chlorogenic Acid antitumor mechanism by gene chip detection: the immune pathway gene expression. J Anal Methods Chem. 2013;2013:617243
20. Andocs G, Renner H, Balogh L, Fonyad L, Jakab C, Szasz A. Strong synergy of heat and modulated electromagnetic field in tumor cell killing. Strahlentherapie und Onkologie: Organ der Deutschen Rontgengesellschaft [et al]. 2009;185(2):120-6
21. Brown DC, Gatter KC. Ki67 protein: the immaculate deception? Histopathology. 2002;40(1):2-11
22. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516
23. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P. et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3(9):e955691
24. Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19(15):4046-57
25. Mukhopadhaya A, Mendecki J, Dong X, Liu L, Kalnicki S, Garg M. et al. Localized hyperthermia combined with intratumoral dendritic cells induces systemic antitumor immunity. Cancer Res. 2007;67(16):7798-806
26. Skitzki JJ, Repasky EA, Evans SS. Hyperthermia as an immunotherapy strategy for cancer. Curr Opin Investig Drugs. 2009;10(6):550-8
27. Repasky EA, Evans SS, Dewhirst MW. Temperature matters! And why it should matter to tumor immunologists. Cancer Immunol Res. 2013;1(4):210-6
28. Qin W, Akutsu Y, Andocs G, Suganami A, Hu X, Yusup G. et al. Modulated electro-hyperthermia enhances dendritic cell therapy through an abscopal effect in mice. Oncol Rep. 2014;32(6):2373-9
29. Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD. et al. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun. 2007;7:7
30. Lee CH, Yoon SJ, Lee SM. Chlorogenic acid attenuates high mobility group box 1 (HMGB1) and enhances host defense mechanisms in murine sepsis. Mol Med. 2013;18:1437-48
31. Derer A, Deloch L, Rubner Y, Fietkau R, Frey B, Gaipl US. Radio-immunotherapy-induced immunogenic cancer cells as basis for induction of systemic anti-tumor immune responses - pre-clinical evidence and ongoing clinical applications. Frontiers in Immunology. 2015:6
Author contact
![]() Corresponding author: Dr. Tibor Krenacs PhD, DSc, 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary Tel: +36 20 825 9700; E-mail: krenacstcom
Corresponding author: Dr. Tibor Krenacs PhD, DSc, 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary Tel: +36 20 825 9700; E-mail: krenacstcom