Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(1):117-128. doi:10.7150/jca.21965 This issue Cite
Research Paper
Antitumor Activity and Mechanism of a Reverse Transcriptase Inhibitor, Dapivirine, in Glioblastoma
Weiwen Liu1*, Xian-lu Song2*, Shan-chao Zhao3, Minyi He4, Hai Wang5 6, Ziyang Chen5,6, Wei Xiang5,6, Guozhong Yi5,6, Songtao Qi5,6 ![]() , Yawei Liu5,6
, Yawei Liu5,6 ![]()
1. First College of Clinical Medicine, Southern Medical University, Guangzhou 510515, China;
2. Department of Radiotherapy, Guangzhou Medical University Cancer Institute and Hospital, Guangzhou 510095, China;
3. Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China;
4. Center for Clinical Medical Education, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China;
5. Department of Neurosurgery, Nanfang hospital, Southern Medical University, Guangzhou 510515, China;
6. Laboratory for Precision Neurosurgery, Nanfang hospital, Southern Medical University, Guangzhou 510515, China.
* Weiwen Liu and Xianlu Song contribute equally.
Received 2017-7-17; Accepted 2017-10-18; Published 2018-1-1
Abstract
Ethnopharmacological relevance: Dapivirine is one of reverse transcriptase inhibitors (RTIs). It is the prototype of diarylpyrimidines (DAPY), formerly known as TMC120 or DAPY R147681 (IUPAC name: 4- [[4-(2, 4, 6-trimethylphenyl) amino]-2-pyrimidinyl] amino]-benzonitrile; CAS no.244767-67-7).
Aim: The purpose of this study is to investigate the antitumor activity of dapivirine, one of the RTIs, on U87 glioblastoma (GBM) cells in vitro and in vivo.
Materials and Methods: U87 GBM cells were cultured and treated with or without dapivirine. Cell viability was evaluated by CCK-8 (Cell Counting Kit 8, CCK-8) assay; apoptosis was analyzed by flow cytometry; cell migration was evaluated by Boyden Chamber assay; Western blotting was performed to detect proteins related to apoptosis, epithelial-to-mesenchymal transition and autophagy. PathScan intracellular signaling array kit was used to detect important and well-characterized signaling molecules. Tumor xenograft model in nude mice was used to evaluate the antitumorigenic effect in vivo.
Results: Dapivirine weakened proliferation of glioma cells and induced the apoptosis of U87 glioblastoma cells. Furthermore, dapivirine regulated autophagy and induced Akt, Bad and SAPK/JNK activations. Moreover, the inhibition of glioma cell growth by dapivirine was also observed in nude mice in vivo.
Conclusion: In summary, in our study dapivirine exposure induces stress, resulting in JNK and PI3K/Akt pathway activation through diminished inhibition of the apoptosis and autophagy cascade in U87 GBM cells, which inhibits cell growth in vitro and in vivo.
Keywords: Dapivirine, Antitumor activity, Drug metabolism, Glioblastoma, Autophagy
Introduction
Glioblastoma (WHO IV) is the most commonly diagnosed malignant adult primary brain tumor. Median survival for GBM is 12 to 15 months [1]. Current treatment of GBM includes surgical resection, diagnostic biopsy, adjuvant radiation and chemotherapy [2]. Temozolomide (TMZ) is the standard-of-care chemotherapy for GBM as the first line treatment with radiotherapy or as a single agent for maintenance therapy. This approach doubles the 2-year survival rate to 27%, but overall prognosis remains poor [3]. Therefore, new drugs for treating GBM are urgently needed.
Reverse transcriptase inhibitors (RTIs) may have a potential therapeutic effect against cancer cells. Azydothymidine (AZT), a retrovirus reverse transcriptase inhibitor, is one of the most studied RTIs. AZT can inhibit proliferation of cancer cells by suppressing telomerase activity [4-10]. In addition, zidovudine, which is an inhibitor of a viral enzyme called reverse transcriptase, has been recommended as the standard first-line chemotherapeutic drug for adult T-cell leukemia (ATL) [11]. Moreover, tenofovir could decrease both accumulation of cytoplasmic DNA and pro-inflammatory signaling caused by Fanconi Anemia (FA), which is a perspective of RTIs in the prevention of adverse effects of chronic inflammation in tumorigenesis [12]. Recently, ritonavir, another RTI, has been demonstrated to be as effective for treating glioblastoma and lung adenocarcinoma [13, 14]. Choy. W et al. [14] retrospectively reviewed 17 HIV-positive patients with glioblastoma (histologically-proven World Health Organization grade IV astrocytoma) and reported that highly active antiretroviral therapy (HAART) is associated with improved survival in patients with HIV-associated glioblastoma, although the precise mechanisms underlying this improvement remain unclear. In HAART, RTIs play a pivotal role, and it is likely to function as a kind of new chemo-therapeutic drug in glioblastoma.
Dapivirine, a RTI now undergoing late stage clinical testing, is widely known as the active ingredient in antiretroviral agents, for example, vaginal rings [15]. Dapivirine is the prototype of diarylpyrimidines (DAPY), formerly known as TMC120 or DAPY R147681 (IUPAC name: 4-[[4-(2, 4, 6-trimethylphenyl) amino]-2-pyrimidinyl]amino]-benzonitrile; CAS no. 244767-67-7) [16]. The chemical structure and relevant physicochemical properties of dapivirine are presented in Fig. 1A. Although clinical trials of dapivirine have emerged, it has not been used for cancer treatment. In this study, with the respect to GBM, we demonstrated that dapivirine has the antitumor activity on U87 GBM cells in vitro and in vivo by regulating cell proliferation-, apoptosis- and autophagy-related signaling pathways.
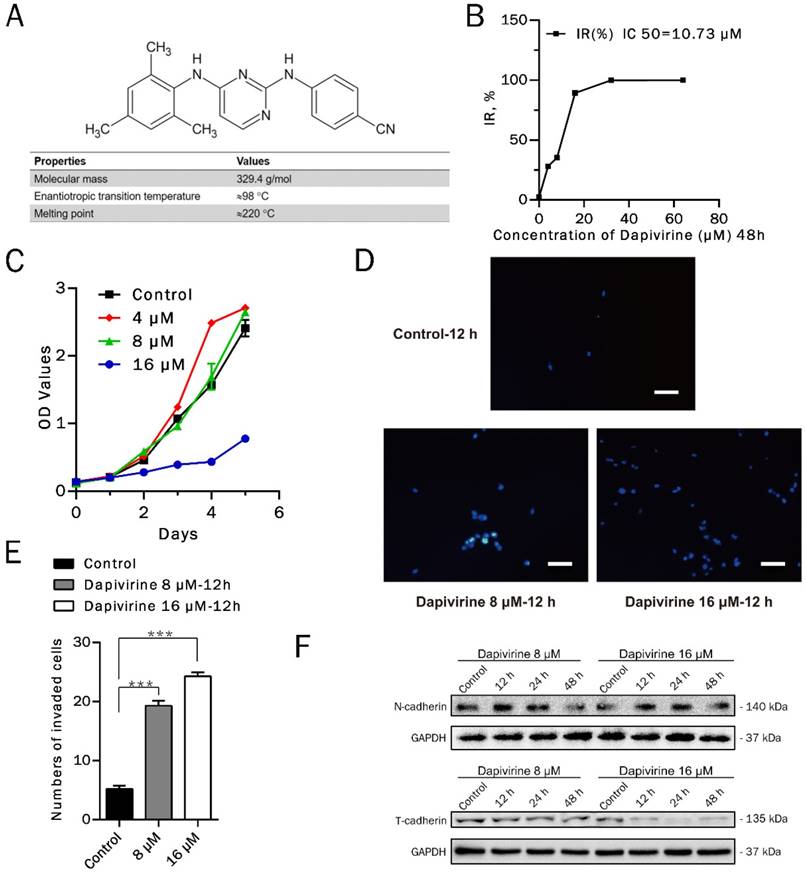
Inhibition of glioma cell growth by Dapivirine. (A) Chemical structure and relevant physicochemical properties of dapivirine. (B) glioblastoma cells were treated with Dapivirine at various concentrations (0, 4, 8, 16, 32, 64 μM) for 48h. Cell viability was measured by Cell Counting Kit assays. IC 50 is 10.73 μM. (C) Cell Counting Kit assays on proliferating U87 glioblastoma cells in control conditions or treated with 4, 8, 16 μM Dapivirine for 24, 48, 72, 96 and 120 hours. (D) The invasion ability of U87 cells was measured following treatment with dapivirine (16 μM and 8 μM) for 12 h. (E) Cells penetrating the artificial basement membrane were counted under a light microscope in 5 random visual fields (200X). The error bars represent the standard deviation of the mean (n = 3; #, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001). (F) Expression of invasion-related proteins, N-cadherin and T-cadherin, in U87 cells treated with dapivirine (8 μM and 16 μM) for 12 h, 24 h and 48 h. All of the results were confirmed in three independent experiments.
Methods
Reagents
Dapivirine (4-[[4-(2, 4, 6-trimethylphenyl) amino]-2-pyrimidinyl]amino]-benzonitrile) was purchased from Selleck (CAS No. 244767-67-7) and was dissolved in DMSO (Sigma, D2650). The dissolved dapivirine was then added to complete culture medium at final concentrations of 4, 8 and 16 μM.
Cell lines and culture conditions
U87 cells was purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, containing 4.5 g/L glucose; Gibco, 11995-065) with 10% fetal bovine serum (Gibco, 16140-071), 100 U/mL penicillin, and 100 mg/mL streptomycin (Gibco, 15140-122). Cells were cultured at 37 ℃ in a humidified atmosphere of 5% CO2.
Determination of IC50
The cells were seeded in 96-well plates (5×104 cells/ml) with 100 μL cell suspension in each well with either the indicated concentrations of dapivirine (4, 8, 16, 32, 64 μM) or controls (DMSO). To determine IC50 concentrations, cells were treated for 48 h, and the viability of cells was determined according to Cell Counting Kit-8 manufactures (Dojindo Laboratories, Japan). Cells were cultured at 37 ℃ in 5% for 3 h, after which the optical density was measured at 450 nm. The inhibition rate (IR) = [OD450 (control) - OD450 (dapivirine)] / [OD450 (control) - OD450 (blank)]. Each experiment was repeated 3 times.
Cell proliferation assay
The cells were seeded in 96-well plates (2×104 cells/ml) with 100 μL cell suspension in each well with either the indicated concentrations of dapivirine (4, 8, 16 μM) or control (DMSO), and were incubated for 5 days respectively. The viability of cells was determined according to Cell Counting Kit-8 manufactures (Dojindo Laboratories, Japan). Cells were cultured at 37ºC in 5% for 3 h, after which the optical density was measured at 450 nm. Each experiment was repeated 3 times.
Cell migration assay
For cell migration assays, 5×104 cells in 200 μL DMEM without fetal bovine serum were seeded on fibronectin-coated polycarbonate membrane inserts in a transwell apparatus (Corning, 3422) with either the indicated concentrations of dapivirine (8, 16 μM) or control (DMSO). In the lower chamber, 600 μL of conditioned medium was added as a chemoattractant. After the cells were incubated for 12 h at 37 ℃ in a 5% CO2 atmosphere, the inserts were washed with PBS, and cells on the top surface of the inserts were removed with cotton swabs. Cells adhering to the lower surface were fixed with methanol, stained with DAPI, and counted under a fluorescence microscope in 6 high-resolution (200x). Each experiment was repeated 3 times.
Flow Cytometry-based Annexin-V Assay
Cells were plated between 60% and 70% confluency into 6-well plates. In each well, cells were treated with the indicated concentrations of dapivirine (8, 16 μM) or control (DMSO) for 12 h, 24 h and 48 h. After incubation for different hours, these cells were washed 3 times with PBS (pH 7.4) then resuspended in 500 μL of annexin binding buffer. 100 μL of this suspension (1×105 cells) was removed to a new 1.5-mL tube. 6 μL of Annexin-Vallophycocyanin (APC) (BD Pharmingen) was added to each tube and gently mixed. Tubes were incubated at room temperature for 15 min. After incubation, 400 μL of annexin binding buffer (containing 1 ng/ml propidium iodide (PI)) was added to each tube. Samples were gently mixed, placed on ice, and analyzed by flow cytometry within 60 mins.
Antibodies and western blotting
Cells were lysed in RIPA Buffer (Cell Signaling Technology, 9800), and protein concentrations were determined by using BCA assays (Cell Signaling Technology, 7780). Total protein (40 mg) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 12% gel and electro transferred to polyvinylidene fluoride membranes. Membranes were then blocked with 5% nonfat dry milk in Tris-buffered saline (Sigma, T7777, pH 7.6) and were incubated overnight at 4 ℃ with rabbit monoclonal anti-N-cadherin, anti-LC3B, anti-Beclin-1, anti-ATG7 antibodies (Cell Signaling Technology, EMT Antibody Sampler Kit 9782 and Autophagy Antibody Sampler Kit 4445) and rabbit polyclonal antibody anti-T-cadherin (Millipore, ABT121). Alternatively, membranes were incubated with mouse monoclonal anti-GAPDH (Abclonal, AC002), apoptosis antibody sampler kit (Cell Signaling Technology 9915), anti-phospho SAPK/JNK (Cell Signaling Technology 81E11) and anti-SAPK/JNK (Cell Signaling Technology 9252), anti-Akt (Cell Signaling Technology C67E7) and anti-phospho Akt antibodies (Cell Signaling Technology D9E). HRP-conjugated anti-rabbit IgG and anti-mouse IgG antibodies were obtained from Cell Signaling Technology (7074).
Intracellular signaling array
Thaw 1X Cell Lysis Buffer (Cell Signaling Technology 7018) and mix thoroughly. Supplement Cell Lysis Buffer with phenylmethylsulfonyl fluoride (PMSF) (Solarbio, Beijing P0100) to a final concentration of 1 mM with a protein phosphatase inhibitor (Solarbio, Beijing P1260). Keep lysis buffer on ice. Remove media and wash cells once with ice-cold 1X PBS (Gibco, 20012027). Remove PBS and add ice-cold Cell Lysis Buffer. For adherent cells, use 0.5 ml cell lysis buffer for each plate (10 cm in diameter). Incubate on ice for 2 minutes. Tilt the plate and collect the lysate into a clean micro tube. Optional step: microcentrifuge the lysate at maximum speed for 3 minutes at 4°C and transfer the supernatant to a new tube. Intracellular signaling molecules were detected using a PathScan® intracellular signaling array kit (Cell Signaling Technology 7323) according to the manufacturer's procedure.
Tumor xenograft assays in nude mice
The protocol for the mouse xenograft model was approved by the Experimental Animal Center of Southern Medical University. In brief, GBM cells were subcutaneously injected in the right flank of nude mice (4-6week-old). Weights of mice were measured every 3 days for 12 days. Tumors were measured with calipers to estimate volume at the end of study, 28 days post-U87 cell implantation. The study was conducted in accordance with the internationally accepted principles for laboratory animal use and care.
Immunohistochemistry
Tumor specimens from nude mice were embedded by paraffin as routine methods. The tissue slices were obtained for immunohistochemistry according to manufacturer's protocol. Anti-human Ki67 (ZSGB-BIO ZM-0167) was used.
TUNEL assay
Tissue sections were dewaxed and rehydrated according to the standard protocols of in situ cell death detection kit, POD (Roche 11684817910). TUNEL reaction mixture were added on samples, lids were added and sections were incubated for 60 min at 37 °C in a humidified atmosphere in the dark. Slides were rinsed 3 times with PBS. Then Converter-POD were added on samples, slides were incubated in a humidified chamber for 30min at 37 °C; DAB Substrate were added, slides were incubated for a few minutes at 15-25 °C Samples were counterstained with hematoxylin and preserved with resinene prior to analysis by light microscope. Each sample was taken pictures by the microscope and the ratio of apoptotic cells (TUNEL-positive cells) were counted.
Statistical analyses
Results are shown as means; error bars represent the standard error of the mean (SEM), Analysis of variance (ANOVA) was used to determine the significance of difference in CCK-8, the annexin-V assay and migration assays. Paired, 2-tailed t tests were used to determine the significance of differences in tumor volumes in the xenograft study. Differences were considered significant when P < 0.05.
Results
Effect of dapivirine on the human glioblastoma cells in vitro
Dapivirine inhibited proliferation of glioma cells
To investigate the anticancer activity of dapivirine in vitro, U87 GBM cells were cultured in absence (DMSO as control) or presence of increasing concentrations of dapivirine for 48h. The viability of cells was determined according to Cell Counting Kit-8 manufactures and the IC50 value of dapivirine was 10.73 μM (Fig. 1B).
Proliferation inhibiting effects of dapivirine on U87 GBM cells were analyzed in long-term cell proliferation assays. As shown in Fig. 1C, when the concentration of dapivirine is 16 μM, it was able to retard cell growth.
Dapivirine induced apoptosis of U87 glioblastoma cells
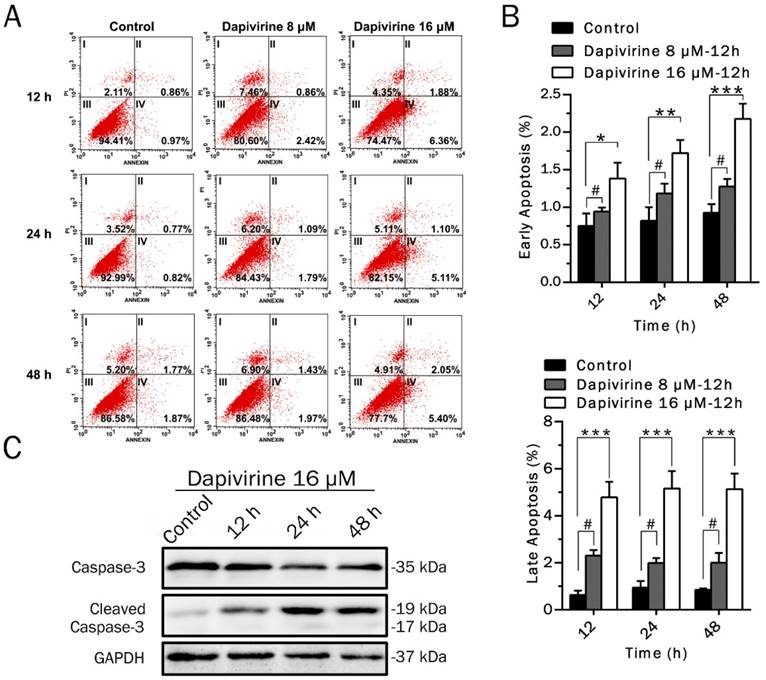
Flow cytometry was used to measure early and late apoptosis and necrosis of U87 GBM cells induced by dapivirine (Fig. 2A). As shown in Fig. 2B, the number of early and late apoptotic cells were significantly increased with treatment of dapivirine (16 μM, administered for 12h, 24h and 48h) as compared to controls. However, there were no significant changes in apoptosis or necrosis to U87 GBM with dapivirine (8 μM). To further confirm the presence of apoptosis, a central regulator of apoptosis, caspase-3 was examined. Western blotting analysis showed that expression of cleaved caspase-3 was accumulated in U87 cells treated with dapivirine, in addition with decreased caspase-3 (Fig. 2C).
Dapivirine moderately induces the apoptosis of U87 glioblastoma cells. (A) After Annexin V and propidium iodide staining, the levels of apoptosis in U87 glioblastoma cells were quantified by flow cytometry. (B) Statistical analysis of the early apoptosis and late apoptosis. (C) Western blotting was used to detect the expression of apoptosis-related proteins in U87 cells treated with dapivirine (16 μM) for 12 h, 24 h and 48 h. All of the results were confirmed in three independent experiments. The error bars represent the standard deviation of the mean (n = 3; #, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Dapivirine enhanced invasion of glioma cells
Results of cell invasion assays indicated that U87 cell invasion was stimulated by the treatment of dapivirine (16 μM & 8 μM, 12 h) (Fig. 1D). The number of cells penetrating the artificial basement membrane obviously increased in those treated with dapivirine (P‹ 0.0001) (Fig. 1E). Western blotting analysis in U87 cells showed that the level of N-cadherin, a mesenchymal marker, was increased significantly after treatment with dapivirine in a time-dependent manner (Fig. 1F). We also detected the expression of E-cadherin and T-cadherin, two classical epithelial markers. As shown in Fig. 1F, T-cadherin was decreased in a time- and drug dosage-dependent manner in U87 cells treated with dapivirine. However, neither dapivirine treated-U87 cells or controls showed significant expression of E-cadherin (not shown in figures).
Dapivirine promotes autophagy in U87 cells
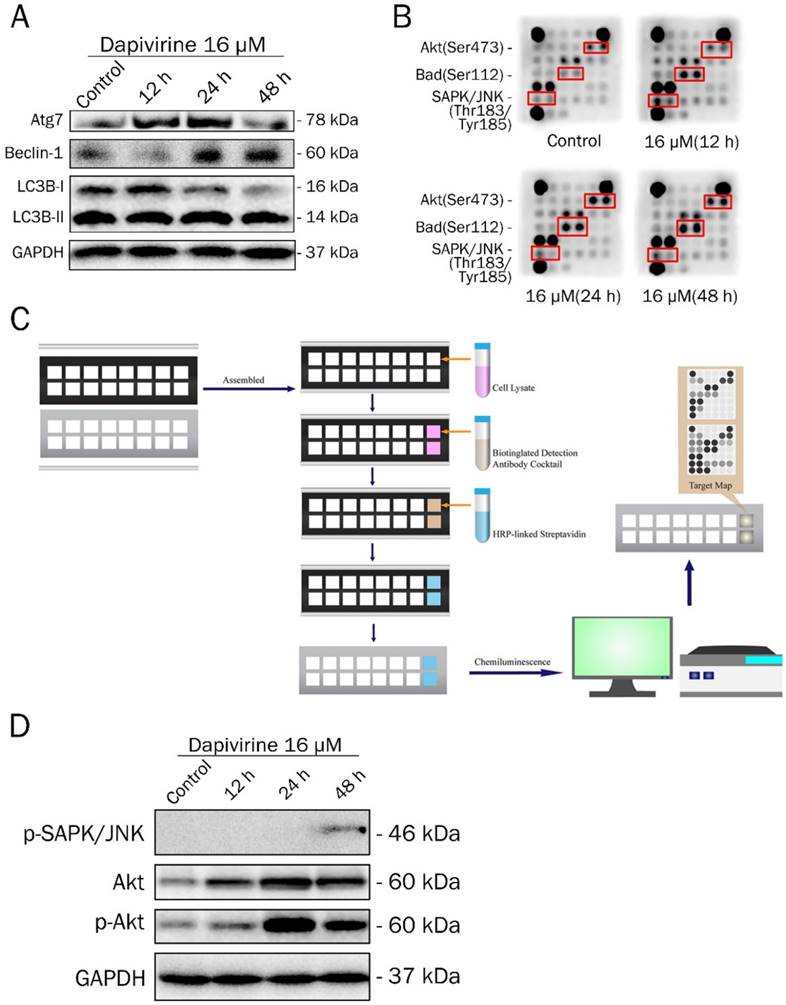
Autophagy activity was analyzed by western blotting to examine the expression of key proteins, after 12 h, 24 h and 48 h exposure to dapivirine (16 μM) (Fig. 3A). With the exposure time of dapivirine increasing, there was a reduction in LC3A/B-I, and the ratio of LC3A/B-II to LC3A/B-I was increased. The expression of Atg7 and Beclin-1 were increased and there was a highest level after 24 h exposure.
Dapivirine regulates autophagy and induced Akt, Bad and SAPK/JNK activations. (A) Western blotting was used to detect the expression of autophagy-related proteins in U87 glioblastoma cells treated with dapivirine (16 μM) for 12 h, 24 h and 48 h. (B, C) PathScan® Intracellular Signaling Array Kit (Chemiluminescent Readout) was used to detect important and well-characterized signaling molecules that were relevant to dapivirine (16 μM for 12 h, 24 h and 48 h) in U87 glioblastoma cells. (B) The dark dots indicate that there was a significant increase in some proteins. Images were acquired by briefly exposing the slide to standard chemiluminescent film. (C) Schematic drawing of operating procedure for PathScan® Intracellular Signaling Array Kit. Cell extracts were prepared and analyzed using the PathScan® kit and images were acquired by exposing the slide to chemiluminescent film. (D) Western blotting was used to verify the results of PathScan® intracellular signaling.
Dapivirine induces Akt, Bad and SAPK/JNK activations
To further illuminate the molecular mechanisms of dapivirine affecting U87 cell growth, a Path Scan intracellular signaling array kit was used to detect the changes of signaling molecules in U87 cells with or without dapivirine treatment (Fig. 3B and 3C). Cell extracts of U87 cells treated with dapivirine and controls were prepared and analyzed using the PathScan Intracellular Signaling Array Kit and images were acquired by briefly exposing the slide to standard chemiluminescent film. The PathScan Intracellular Signaling Array Kit could be used to monitor the expression of 18 signaling molecules that are phosphorylated in response to signal-transduction pathway activation. The dark dots indicate that there was a significant activation of some proteins. As shown in Fig. 3B, the phosphorylation of Akt (Ser473), Bad (Ser112) and SAPK/JNK (Thr183/Tyr185) were accumulated in dapivirine-treated cells. Similar to PathScan, western blotting revealed that phospho-SAPK/JNK, Akt and phospho-Akt were elevated by dapivirine (16 μM for 12h, 24h, 48h) treatment (Fig. 3D). The data indicates that dapivirine could significantly affect the function of U87 cells via stimulation of Bad while Akt and JNK are responsible.
Effiency of dapivirine of human glioblastoma cells in vivo
Dapivirine inhibits the proliferation of glioblastoma in vivo
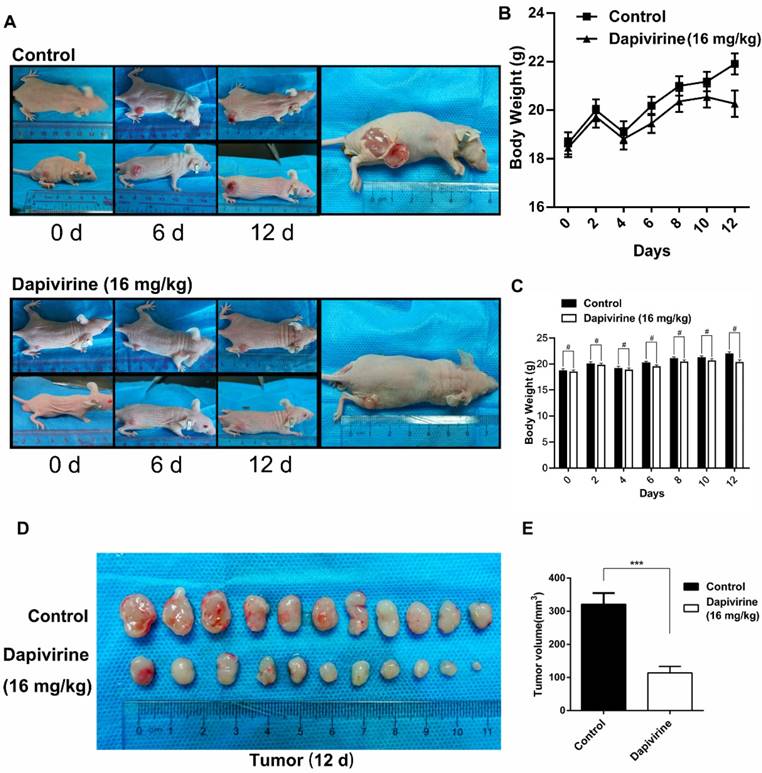
The U87 cells were subcutaneously injected into the nude mice (5×107 cells per mouse) to evaluate the effect of dapivirine on GBM growth in vivo. Following tumor formation, about 14 days after cell injection, the mice were treated with either vehicle (DMSO) or dapivirine (16 mg/kg) once every 3 days for 12 days. Next, tumors were dissected from mice to measure volume at the end of study, 28 days post-U87 cell implantation (Fig. 4A). As shown in Fig. 4B and 4C, increased growth trends of mice weight were in both dapivirine-treated mice and controls, and no significant difference of weight was observed between dapivirine-treated mice and controls. The tumor volumes of dapivirine-treated cells were significantly decreased in mice than controls (Fig. 4D and 4E). These results indicate that cancer proliferation is inhibited by dapivirine in vivo. In addition, proliferation status of tumor tissues was evaluated by Ki67, a marker for proliferating cells that is over-expressed in many cancers. Immunohistochemistry experiments performed on the sections of U87 tumor tissues showed a significant decrease in Ki67 staining in sections of dapivirine-treated tumors compared to tumors from vehicle-treated mice (Fig. 5E-F). Specifically, quantification of Ki67 positive cells revealed that dapivirine treatment reduced tumor growth by 2.8fold and reduced cell proliferation (Ki67) by 2.6fold with in U87.
Dapivirine inhibits tumor growth of U87 glioblastoma cells in vivo. (A) Subcutaneous tumors of mice injected with U87 cells. Either vehicle or dapivirine (16 mg/kg) was treated once every 3 days for 12 days, and mice were then sacrificed. (B, C) Body weights of mice in 2 groups were measured every 2 days. (D, E) Tumors were dissected, volumes were measured.
Inhibition of proliferation and induction of apoptosis by dapivirine in a mouse model. (A, B) Representative tumor regions in routinely H&E staining. (C, D) Analysis of apoptosis (TUNEL) by immunohistochemistry. Each sample was taken pictures by the microscope and the ratio of apoptotic cells (TUNEL-positive cells) were counted. (E, F) Analysis of cell proliferation (Ki67) by immunohistochemistry. The ratio of Ki-67 positive cells were counted. (G) Expression of apoptosis-related proteins, cell proliferation-related proteins and Epithelial-Mesenchymal Transition-related proteins were detected by Western blotting.
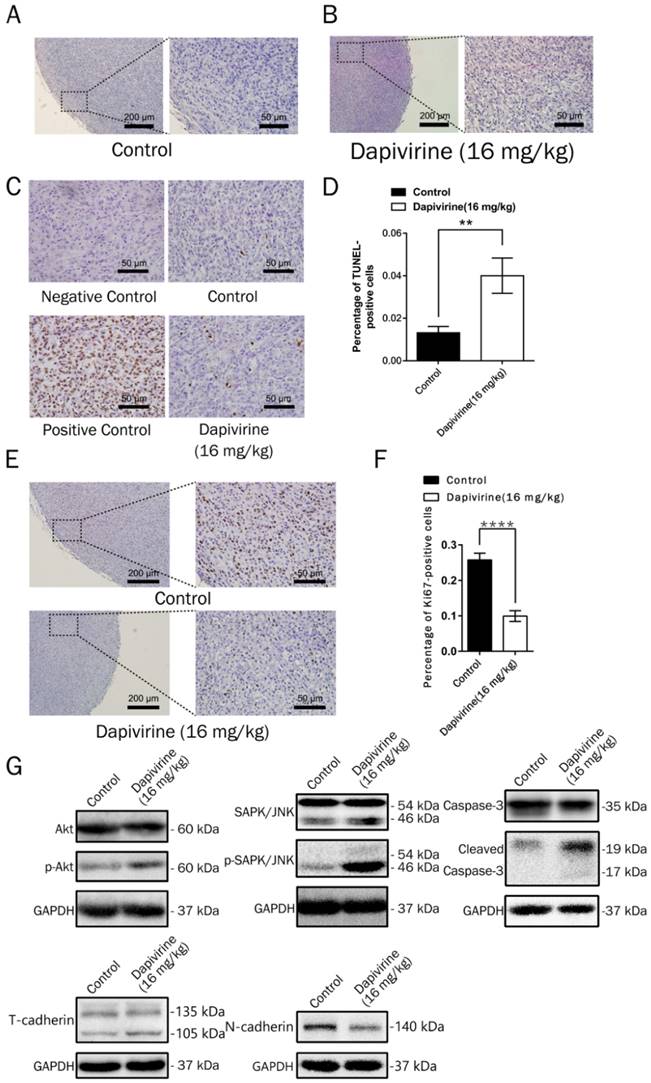
Dapivirine induces apoptosis of glioblastoma in vivo
On the H&E sections, there was no obvious necrosis in the tumor tissues (Fig. 5A and 5B), either controls or dapivirine treatment. The extent of apoptosis in tumor tissues was further examined using the TUNEL assay (Fig. 5C). The quality of the staining was nuclear, and increased TUNEL labeling was observed in the tumor tissues treated with dapivirine (16 mg/kg). There was also an increase in the expression of cleaved caspase-3, which is consistent with the results in vitro with dapivirine treatment (Fig. 5G).
Dapivirine up-regulates expression of SAPK/JNK, Akt in glioblastoma
Western blotting of tumors disstected from mice was used to detect the expression of SAPK/JNK and phospho-SAPK/JNK, Akt and phospho-Akt. As shown in Fig. 5G, the phosphorylation of Akt, SAPK/JNK and phospho-SAPK/JNK were accumulated in dapivirine-treated tumors.
Toxicity analysis
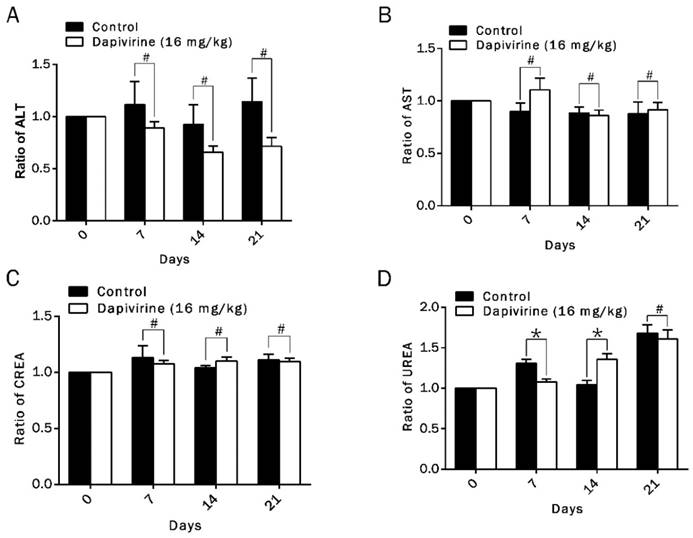
Toxicity analysis (including hepatic and renal function indexes) was carried out in all groups of tumor-bearing nude mice with dapivirine (16 mg/kg) or vehicle treatment. It was carried out on day 0, day 7, day 14 and day 21 after drug injection. Function indexes of liver (ALT, AST) and kidney (CREA, UREA) were detected to evaluate the hepatic and renal damage after drug treatment (Fig. 6A-D). Hepatic function index of group of dapivirine revealed no obvious difference with group vehicle (p > 0.05). Although renal function index of dapivirine group showed that values of CREA were higher than group vehicle on day 7, there was no obvious effect on kidney when toxicity analysis was carried out on day14 or day 21. Our data suggests no signs of liver damage after dapivirine treatment, and dapivirine might have a temporary effect on kidney with the concentration, 16 mg/kg.
Toxicity analysis of dapivirine. (A-D) Hepatic and renal function indexes in all of tumor-bearing nude mice with vehicle- or dapivirine-treatment at day 0, day 7, day 14 and day 21. Function indexes of liver (ALT, AST) and kidney (CREA, UREA) were measured to evaluate the hepatic and renal damage after drug injection.
Discussion
Our study took task to explore the effects of dapivirine on GBM, and whether the antiretroviral drug has an antineoplastic activity. We first observed that dapivirne markedly inhibited human GBM cell proliferation. The results of flow cytometry showed that dapivirine induces apoptosis in U87 cells. Western blotting revealed that full length caspase-3 was reduced and cleaved form was increased with dapivirine treatment. Caspases are intracellular proteases that work during apoptosis to disassemble the cell into apoptotic bodies. Initiator caspases, including caspase-8 and caspase-9, activate executioner caspases, including caspase-3, which are responsible for carrying out final steps in the disassembling process. Elevated levels of active forms of the caspases are considered markers of cellular apoptosis [17]. The observation confirms that inhibition of cell proliferation occurs and apoptosis may be one of the reasons. However, we found that neither the 12 h incubation with dapivirine at 16 μM- nor incubation for a longer time (24 h/48 h)-have much higher apoptosis rate than 3-fold of the control group, which did not exactly match up to the results of CCK-8 assay. Thus, there must be other mechanisms responsible for the proliferation inhibition.
The autophagy-apoptosis puzzle in the tumor context seems to have an extremely complex role. Autophagy (referring to macroautophagy) is an evolutionarily conserved cellular mechanism for the clearance of damaged or superfluous macro-complexes and organelles in eukaryotic cells [18-20]. During autophagy, a process of dynamic flux, autophagosomes engulf cytoplasmic components, including cytosolic proteins and organelles. Concomitantly, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine to form LC3-phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. Autophagosomes fuse with lysosomes to form autolysosomes, and intra-autophagosomal components are degraded by lysosomal hydrolases. At the same time, LC3-II in autolysosomal lumen is degraded [19]. In our study, we further indicate that autophagy was stimulated by dapivirine treatment. We observed that dapivirine treatment firstly increased expression of autophagy-related proteins, Beclin-1 [21, 22] and ATG7. And as time goes on, their levels were down-regulated. Furthermore, LC3-I and LC3-II were decreased in a time-dependent manner, with increased ratio of LC3-II to LC3-I. These changes suggest that autophagic flux was stimulated by dapivirine in a short term. With dapivirine exposure on U87 GBM cells in 48 h, cell proliferation was significantly inhibited with a moderate cell death, induced by apoptosis. Therefore, we hypothesize that when U87 cells are treated with dapivirine, cell death might serve as a late response in the event that autophagy induction fails to provide effective protection to the injured U87 cells. It is consistent with a previous report showing the role of autophagy both as a tumor suppressor as well as a factor in tumor cell survival [23]. It was confirmed that dapivirine significantly induces apoptosis in vivo by TUNEL test, however, results of western blotting showed that there was insignificant difference of autophagy related proteins between tumors treated with dapivirine and controls (data not shown in manuscript) in vivo. Therefore, increased autophagy induced by dapivirine seems to be a temporary phenomenon in U87 cells. In murine models, the mice were treated with dapivirine for 12 days, which is longer than in vitro assays.
Interestingly, we observed that dapivirine treatment contributed to cell motility in U87 cells. Western blotting showed up-regulation of N-cadherin and down-regulation of T-cadherin, which are importantly associated with GBM invasion and metastasis [24]. Previous study has shown that induction of autophagy increased cell migration and infiltration in GBM cells [25]. Our results indicated that stimulation of the invasion by dapivirine may occur via the stimulation of autophagy. Moreover, invasiveness was insignificantly enhanced in tumors of nude mice, which suggests that the increased invasiveness induced by autophagy is a temporary phenomenon.
To illuminate the molecular mechanism that how dapivirine alters the maintenance of GBM, we detected modifications of some molecules associated with cell growth, survival and cell cycle control in U87 cells after dapivirine treatment. The present study demonstrated that activation of Bad (Ser112), Akt (Ser473) and SAPK/JNK (Thr183/Tyr185) may be associated with dapivirine-induced apoptosis, invasion and autophagy.
Recently, it has been demonstrated that stresses activate JNK, inducing autophagy to counteract apoptosis in mesenchymal stem cells [26]. PI3K/Akt pathway is the major signaling pathway related to progression and invasion of cancer [27], and activation of Akt and JNK pathway contribute to the protective effect against stress [28].On the one hand, the promotion of invasion in U87 cells treated with dapivirine, which may be correlated with the increased expression of p-Akt and Akt. Choy, Y.Y., et al showed that Akt inhibited the intrinsic mitochondrial pathway by phosphorylating Bad at Ser136, which prevents Bad translocation to the mitochondria [29].
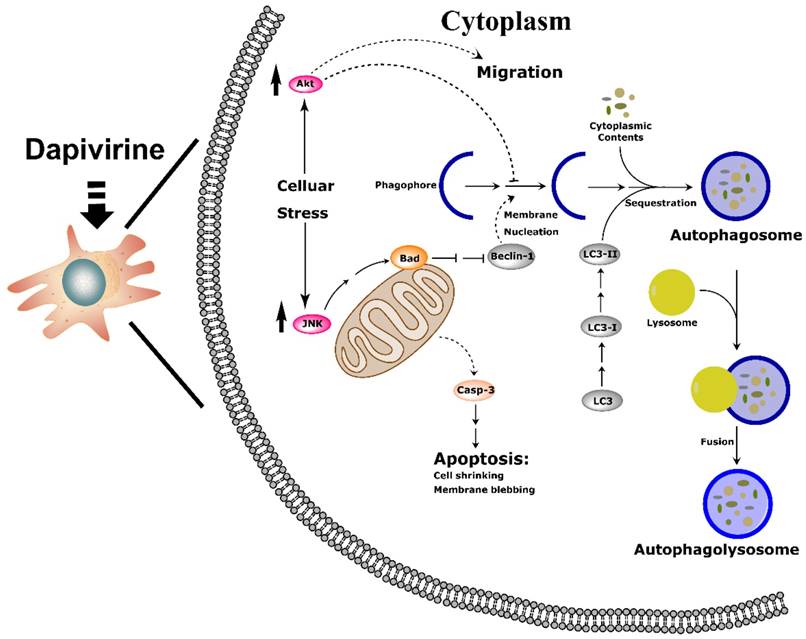
Taken together, our research suggests that dapivirine exposure induces stress, leading to JNK and PI3K/Akt pathway activation, which diminishes the inhibition of apoptosis and autophagy cascade in U87 GBM cells. As a result, dapivirine inhibits cell growth and stimulates cell invasion (Fig. 7).
Schematic drawing of the molecular mechanism of dapivirine affecting U87 cells. (A) Dapivirine exposure induces stress, resulting in JNK and PI3K/Akt pathway activation through diminished inhibition coupled with activation of the apoptosis and autophagy cascade in U87 GBM cells, which inhibits cell growth and stimulates cell invasion.
Abbreviations
NNRTIs, Non-nucleoside reverse transcriptase inhibitors; Dapivirine, 4-[[4-(2, 4, 6-trimethylphenyl) amino]-2-pyrimidinyl]amino]-benzonitrile; GBM, glioblastoma; HAART, Highly active antiretroviral therapy; AIDS, Immune Deficiency Syndrome; CCK-8, Cell Counting Kit 8; IR, Inhibition Rate; Akt, protein kinase B, PKB; SAPK, stress-activated protein kinase; JNK, c-Jun N-terminal kinase; Bad, BCL-2/BCL-XL-associated death promoter; PI3K, phosphatidylinositol 3-kinase; TMZ, Temozolomide; Caspase, cysteinyl aspartate specific proteinase; ATG, Autophagy Related Gene; Ki-67, nuclear- associated antigen; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81472315, 81302229), the Natural Science Foundation of Guangdong Province (2014A030313167) and National Key Technology Research and Development Program of the Ministry of Science and Technology of China (2014BAI04B01). We thank for their technical assistance, advice and for help with statistical analysis. We are also grateful to all members of Laboratory for Precision Neurosurgery, Nanfang hospital, Southern Medical University, for their support for this study.
Ethics Approval and Consent to Participate
All appropriate guidelines and legislation in conducting the study were followed. Nanfang hospital, Southern Medical University approved this study. The use of animals in experiments will have observed the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, Ad Hoc Animal Research Committee.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8
2. Dehdashti AR, Hegi Me Fau - Regli L, Regli L Fau - Pica A, Pica A Fau - Stupp R, Stupp R. New trends in the medical management of glioblastoma multiforme: the role of temozolomide chemotherapy. Neurosurgical focus. 2006;20:E6
3. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. Jama. 2013;310:1842-50
4. Bondarev IE, Khavinson VK. Suppression of alternative telomere lengthening in cancer cells with reverse transcriptase inhibitors. Advances in gerontology = Uspekhi gerontologii. 2016;29:218-21
5. Wang H, Zhou J, He Q, Dong Y, Liu Y. Azidothymidine inhibits cell growth and telomerase activity and induces DNA damage in human esophageal cancer. Mol Med Rep. 2017;15:4055-60
6. Zhou FX, Liao ZK, Dai J, Xiong J, Xie CH, Luo ZG. et al. Radiosensitization effect of zidovudine on human malignant glioma cells. Biochemical and biophysical research communications. 2007;354:351-6
7. Li H, Song T, Xu W, Yu Y, Xin X, Hui D. Effect of 3`-Azido-3`-Deoxythymidine (AZT) on Telomerase Activity and Proliferation of HO-8910 Cell Line of Ovarian Cancer. International journal of biomedical science: IJBS. 2006;2:34-40
8. Ji HJ, Park KH, Kim TS, Rha SY, Yoo NC, Kim JM. et al. Changes of Telomerase Activity and Proliferation by Inhibition of Reverse Transcriptase Activity in Human Cancer Cell. Cancer research and treatment: official journal of Korean Cancer Association. 2002;34:223-33
9. Brown T, Sigurdson E, Rogatko A, Broccoli D. Telomerase inhibition using azidothymidine in the HT-29 colon cancer cell line. Annals of surgical oncology. 2003;10:910-5
10. Murakami J, Nagai N, Shigemasa K, Ohama K. Inhibition of telomerase activity and cell proliferation by a reverse transcriptase inhibitor in gynaecological cancer cell lines. European Journal of Cancer. 1999;35:1027-34
11. Yuen CK, Chan CP, Fung SY, Wang PH, Wong WM, Tang HM. et al. Suppression of Type I Interferon Production by Human T-Cell Leukemia Virus Type 1 Oncoprotein Tax through Inhibition of IRF3 Phosphorylation. Journal of virology. 2016;90:3902-12
12. Bregnard C, Guerra J, Dejardin S, Passalacqua F, Benkirane M, Laguette N. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-inflammatory Cytokine Production. EBioMedicine. 2016;8:184-94
13. Srirangam A, Milani M Fau - Mitra R, Mitra R Fau - Guo Z, Guo Z Fau - Rodriguez M, Rodriguez M Fau - Kathuria H, Kathuria H Fau - Fukuda S. et al. The human immunodeficiency virus protease inhibitor ritonavir inhibits lung cancer cells, in part, by inhibition of survivin. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2011;6:661-70
14. Choy W, Lagman C, Lee SJ, Bui TT, Safaee M, Yang I. Impact of Human Immunodeficiency Virus in the Pathogenesis and Outcome of Patients with Glioblastoma Multiforme. Brain tumor research and treatment. 2016;4:77-86
15. Winceslaus SJ. Dapivirine Vaginal Ring for HIV-1 Prevention. The New England journal of medicine. 2017;376:994-5
16. das Neves J, Martins JP, Sarmento B. Will dapivirine redeem the promises of anti-HIV microbicides? Overview of product design and clinical testing. Advanced drug delivery reviews. 2016;103:20-32
17. Unsain N, Barker PA. New Views on the Misconstrued: Executioner Caspases and Their Diverse Non-apoptotic Roles. Neuron. 2015;88:461-74
18. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Developmental cell. 2004;6:463-77
19. Tanida I, Ueno T Fau - Kominami E, Kominami E. LC3 and Autophagy. Autophagosome and Phagosome. 2008;445:77-88
20. Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861-73
21. Liang XH, Jackson S Fau - Seaman M, Seaman M Fau - Brown K, Brown K Fau - Kempkes B, Kempkes B Fau - Hibshoosh H, Hibshoosh H Fau - Levine B. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672-6
22. Zhu H, Wu H Fau - Liu X, Liu X Fau - Li B, Li B Fau - Chen Y, Chen Y Fau - Ren X, Ren X Fau - Liu C-G. et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816-23
23. Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12:1528-34
24. Yan X, Yan L, Liu S, Shan Z, Tian Y, Jin Z. N-cadherin, a novel prognostic biomarker, drives malignant progression of colorectal cancer. Mol Med Rep. 2015;12:2999-3006
25. Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q. et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11:2213-32
26. Liu GY, Jiang XX, Zhu X, He WY, Kuang YL, Ren K. et al. ROS activates JNK-mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta pharmacologica Sinica. 2015;36:1473-9
27. Wang SC, Chai DS, Chen CB, Wang ZY, Wang L. HPIP promotes thyroid cancer cell growth, migration and EMT through activating PI3K/AKT signaling pathway. Biomed Pharmacother. 2015;75:33-9
28. Hu YR, Ma H, Zou ZY, He K, Xiao YB, Wang Y. et al. Activation of Akt and JNK/Nrf2/NQO1 pathway contributes to the protective effect of coptisine against AAPH-induced oxidative stress. Biomed Pharmacother. 2017;85:313-22
29. Choy YY, Fraga M, Mackenzie GG, Waterhouse AL, Cremonini E, Oteiza PI. The PI3K/Akt pathway is involved in procyanidin-mediated suppression of human colorectal cancer cell growth. Mol Carcinogen. 2016;55:2196-209
Author contact
![]() Corresponding authors: liuyaweiedu.cn (Y. Liu), qisongtaonfyycom (S. Qi). Telephone number: 86-020-61641806 Address: Department of Neurosurgery, Nanfang hospital, Southern Medical University, Guangzhou Dadao Bei Street 1838#, Guangzhou 510515, P.R. China
Corresponding authors: liuyaweiedu.cn (Y. Liu), qisongtaonfyycom (S. Qi). Telephone number: 86-020-61641806 Address: Department of Neurosurgery, Nanfang hospital, Southern Medical University, Guangzhou Dadao Bei Street 1838#, Guangzhou 510515, P.R. China