Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(2):219-231. doi:10.7150/jca.22554 This issue Cite
Review
Break Breast Cancer Addiction by CRISPR/Cas9 Genome Editing
Haitao Yang1, MariaLynn Jaeger2, Averi Walker2, Daniel Wei3, Katie Leiker2, Tao Weitao2 ![]()
1. Laboratory for Cancer Genome Editing, Zhuhai Lifecode Medical Technologies. Inc. Department of Prenatal Diagnosis, Huizhou 2nd Hospital for Children and Women, #101 University Road, Tangjiawan, Zhuhai, 518900, Guangdong, China;
2. College of Science and Mathematics, Southwest Baptist University, 1600 University Avenue, Bolivar, Missouri 65613, USA;
3. University of Texas at Dallas, 800 W Campbell Rd, Richardson, TX 75080, USA.
Received 2017-8-26; Accepted 2017-9-25; Published 2018-1-1
Abstract
Breast cancer is the leading diagnosed cancer for women globally. Evolution of breast cancer in tumorigenesis, metastasis and treatment resistance appears to be driven by the aberrant gene expression and protein degradation encoded by the cancer genomes. The uncontrolled cancer growth relies on these cellular events, thus constituting the cancerous programs and rendering the addiction towards them. These programs are likely the potential anticancer biomarkers for Personalized Medicine of breast cancer. This review intends to delineate the impact of the CRSPR/Cas-mediated genome editing in identification and validation of these anticancer biomarkers. It reviews the progress in three aspects of CRISPR/Cas9-mediated editing of the breast cancer genomes: Somatic genome editing, transcription and protein degradation addictions.
Keywords: Breast cancer, tumorigenesis, metastasis, mutations, transcription, protein degradation, anticancer resistance, CRISPR/Cas9, genome.
Introduction
Cancer remains to be the second leading cause of death in the United States [1]. The mortality data collected by the National Center for Health Statistics have projected 1,688,780 new cancer cases and 600,920 cancer deaths for 2017 in the United States. For women, the top three cancers consist of breast, lung/bronchus and colorectal, accounting for 50% of all cases. Breast cancer alone comprises 30% of all new cancer diagnoses in women [1]. In fact, breast cancer has become the most common cancer among women globally. Metastatic breast cancer is still incurable with a prognosis dependent on its histopathological and molecular profiles [2-5]. The molecular profile of breast cancer unveils the heterogeneous nature of disease development and progression governed by the genes controlling cell growth, proliferation and differentiation [6, 7]. The evolution of breast cancer appears to be driven by the aberrant gene expression leading to gain of function or activation of downstream signal pathways [8] (Fig. 1). The insights into these gene expression profiles and genomic abnormities of cancer have started to revolutionize the classic paradigm of breast cancer treatment that relied on pathological and clinical manifestations [9]. New therapeutic principles of Personalized Medicine have emerged to match anti-cancer drugs with the molecular alterations unique to cancer cell proliferation [10]. Presently, while the principles have prompted development of the molecularly targeted drugs for clinical practice, the major challenge is identification of the anticancer biomarkers as targets for accurate treatment; thus the side effects associated with the off-target treatments and the overall cost can be minimized [11].
Genomic diversity of cancer dictates its addictions to major cellular events. These events include transcription, splicing, translation, protein folding/degradation, and cell proliferation. The busy cancer proliferation and tumorigenesis are not only fueled by the drivers and passengers but also encoded by the complex of the mutated genes and affected by other noncoding factors in cancer cells. This imposes “rush hours” on these molecular processes and renders the cancer cells addicted to them. These addictions can be disrupted by CRISPR-mediated genome editing (green arrows).
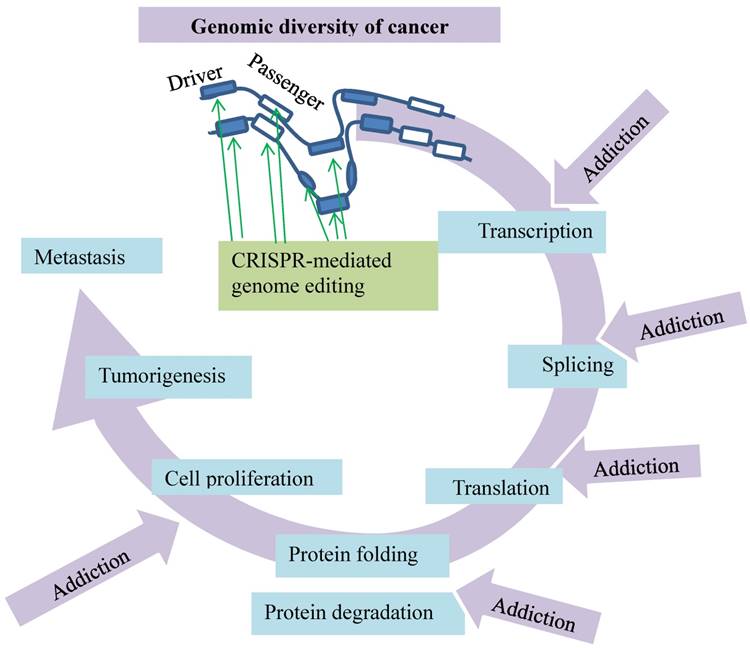
Fortunately, the advent of a genome editing technology with the clustered regularly interspaced short palindromic repeats (CRISPR) that are the associated protein nuclease (Cas9) or CRISPR/Cas9 system has facilitated the identification. CRISPR/Cas9 has two key components: a chimeric single guide RNA (gRNA) and the DNA endonuclease Cas9. Notably, gRNA carries Cas9 to a target genomic sequence [12, 13]. Specificity to the target is defined by complementarity of the 20 nucleotides at the 5′ end of the gRNA to the desired genomic sites. Cas9-mediated DNA cleavage at the target site is initiated by a protospacer adjacent motif (PAM) situated immediately downstream of the target sequence. Cas9-induced double-stranded breaks (DSBs) are subjected to the error-prone repair by nonhomologous end-joining [14], which introduces mutations to disrupt integrity and functions of the targeted genomic sites. This review intends to delineate the impact of the CRSPR/Cas9-mediated genome editing in searching and identifying molecular targets against breast cancer. It focuses on a theme of CRISPR/Cas9-mediated somatic genome editing, transcription and protein degradation addiction in breast cancer (Fig. 1).
CRISPR/Cas9-mediated somatic genome editing of breast cancer
The CRISPR/Cas9 approach is helping to revolutionize the diagnosis and treatment of breast cancer. For the diagnosis, we take an invasive lobular breast carcinoma as a model in which the CRISPR/Cas9-mediated somatic genome editing tool validates putative cancer drivers in vivo (Fig. 2) [15]. For the treatment, we discuss the inhibition of breast cancer cell proliferation through a dominant negative mutation generated by the CRSPR/Cas9-mediated targeting of oncogene HER2 (Fig. 3) [16]. Additionally, we delineate a study about revealing and verifying a culprit in rendering resistance of breast cancer to DNA-damage anticancer treatment (Fig. 4) [17].
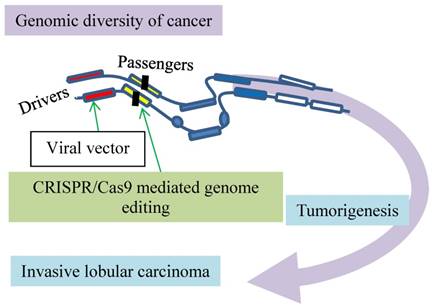
Modeling of invasive lobular carcinoma by CRISPR/Cas9 mediated genome editing. Putative cancer drivers (red boxes) can be validated in vivo. The green arrow at drivers indicates activation and at passengers inactivation of the genes as marked by vertical filled bars.
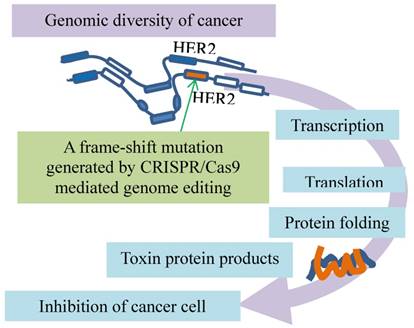
A dominant negative mutant phenotype resulting from incomplete CRISPR targeting of oncogene HER2 is a potential therapeutic target. The green arrow indicates inducing a mutation by CRISPR/Cas9 to one copy of HER2. Folding of the polypeptides from the mutated (orange) and intact (blue) genes leads to toxic products inhibits cancer growth.
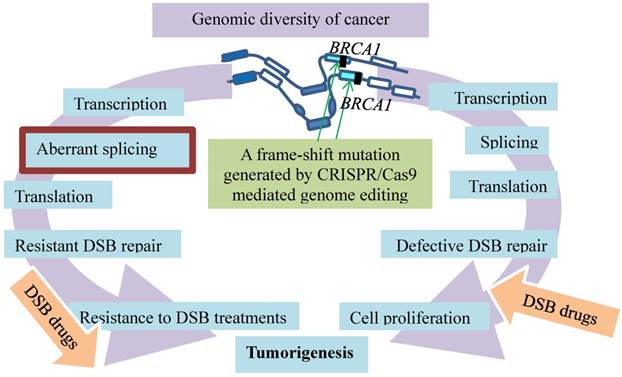
Resistance evolution of breast cancer by removal of disadvantageous germline BRCA1 mutations through somatic RNA splicing. Germline frame-shift mutations (vertical filled bars) in BRCA1 result in dysfunctional BRCA defective in DSB repair, which contributes to tumorigenesis and sensitivity to the DSB medication (right arc). The germline mutations are then bypassed by aberrant splicing under selection of DSB therapy. The resulting BRCA resists DSB treatment (left arc). An additional frame-shift mutation introduced by CRISPR/Cas9 genome editing (green arrows) restores the reading frame and avoids the outcomes of the aberrant splicing.
The advent of CRISPR/Cas9-mediated somatic genome editing facilitates modeling of invasive lobular carcinoma (ILC) to validate putative cancer drivers in vivo [15] (Fig. 2). Invasive lobular carcinoma, the second most common type of human breast cancer responsible for 8 - 14 % of all breast cancer cases [18-20], is characterized by discohesive epithelial cells invading the adjacent tissue in single-file patterns accompanied by copious fibroblasts and collagen deposition. Most human ILCs lose the cell-cell adhesion protein E-cadherin as a result of loss of heterozygosity, methylation of the CDH1 gene promoter [21-24] or compromised integrity of the E-cadherin-catenin membrane complex [25]. Enigmatically, the tissue-specific loss of E-cadherin in mammary epithelial cells leads to apoptosis [26] but not to mammary tumors in mice [26] [27, 28]. The combined loss of E-cadherin and p53 induces multifocal ILC [27, 28], suggestive of passenger and driver events in tumorigenesis.
With CRISPR/Cas9-mediated somatic gene editing, an in vivo validation of these two events is developed (Fig. 2). The procedure starts with intraductal injection of the lentiviral vector that expresses Cre recombinase. This recombinant vector is administered into female mice that have conditional alleles of the CDH1 gene encoding E-cadherin and the oncogenic AKT-E17K isoform. The local Lenti-Cre expression induces mammary gland-specific inactivation of E-cadherin but activation of AKT-E17K, leading to formation of a typical invasive lobular carcinoma. This approach with CRISPR/Cas9-mediated somatic gene editing is capable of detecting cancer driver candidates, such as the phosphatase and tensin homolog (Pten) gene, which is a negative regulator of the PI3K/AKT signaling pathway. Intraductal injection of a lentiviral vector encoding Cre and the CRISPR/Cas9 components with a sgRNA targeting Pten into the Cdh1F/F mice effectively induces ILC development. In contrast to the negative ATK regulator is the long non-coding RNAs (lncRNAs) recently identified as a positive regulator by a CRISPR/Cas9-based synergistic activation mediator screen with an AKT reporter [29]. This model can validate more putative cancer drivers in vivo, such as Pten and lncRNAs, implicated in breast cancer [15].
Another breast cancer driver is oncogene HER2 exons, which have multiple copies in breast cancer cells. Although the HER2 exons are targeted by CRISPR/Cas9, such induced mutations in one copy of the oncogene cannot disrupt the HER2 protein production from all other copies, indicative of incompleteness of the HER2 mutations. However, a mutant HER2 protein generated by a frame-shift mutation in HER2 exon12 is associated with the wildtype HER2 proteins, leading to a dominant negative mutant phenotype (Fig. 3). This alteration was also found to inhibit the MAPK/ERK axis of HER2 downstream signaling and cancer cell proliferation [16]. This inhibitory effect is enhanced by inhibitors of the poly ADP-ribose polymerase (PARP) that is involved in key cellular processes such as DNA repair and cell death [30], suggestive of synergistic anti-cancer effects of CRISPR/Cas9 in combination with DNA repair inhibition in the clinical setting. Thus, dominant negative mutants resulting from incomplete CRISPR targeting of oncogenes may be a potential therapeutic target [16].
Not only is CRSPR/Cas9 utilized to identify the therapeutic targets as above but also to investigate mechanisms of cancer resistance to the therapy. For example, germline mutations in the BRCA1 gene are bypassed by somatic alternative splicing that leads to resistance to the anticancer therapy, the finding that is verified by CRISPR/Cas9 [17] (Fig. 4). Germline mutations in the BRCA1 gene resulting in dysfunctional BRCA incur a high risk of breast and ovarian cancer development [31, 32]. BRCA1 mutations in exon 11 account for approximately 30% of the total mutation carriers who develop breast and ovarian cancer in the United States [33-35]. The BRCA1 protein plays an essential role in homologous recombination (HR)-mediated repair of DSB [36, 37]. As PARP inhibitors (PARPi) and platinum agents induce DSBs that are dependent on HR pathway for repair [38, 39], cells with dysfunctional BRCA1 or BRCA2 proteins in HR DNA repair are particularly sensitive to the treatments with these medications [40-43]. While such therapies can improve survival of breast cancer patients, the treatment with PARPi is not effective on many patients who carry germline BRCA1 or BRCA2 mutations [44-46]. Some of the patients, though initially showing improvements, eventually develop resistance and disease progression [47]. The resistance is developed through various somatic mechanisms, such as reversion BRCA2 mutations [48, 49], 53BP1 pathway activity loss [50, 51], hypomorphic BRCA1 expression [52, 53], and drug efflux [54].
Among these resistance mechanisms, the resistance promoted by the BRCA1-Δ11q alternative splice isoform is intriguing, as the splicing bypasses the germline mutations in BRCA1 [17] (Fig. 4). Alternative splicing generates BRCA1 mRNA isoforms. These isoforms that lack certain exons are expressed in cells and tissues [55-57]. One of them is the BRCA1 exon 11splice isoforms that exhibit the different levels during discrete phases of the cell cycle in both normal and cancer tissues [58-61]. These various isoforms range from full-length BRCA1 containing all coding exons to BRCA1 Δ11 skipping of exon 11 and BRCA1 Δ11q partial skipping of exon 11 [59, 60]. Since germline mutations of BRCA1 often lead to reading frameshifts and nonsense-mediated mRNA decay [62], such mutations plausibly affect splicing and expression of BRCA1-Δ11q. This premise is supported by the results showing that a BRCA1-Δ11q splice variant lacking most of exon 11 is produced from the cancer cell lines and tumors carrying mutations in exon 11 of BRCA1. The nature of the mutations is likely the frameshift mutations to exon 11 that cause nonsense-mediated mRNA decay of full-length but not the BRCA1-Δ11q isoform. This premise is verified with CRISPR/Cas9 by introducing the additional out-of-frame mutations or reversion mutations that restore the reading frame and avoid the nonsense-mediated mRNA decay. Interestingly, the BRCA1-Δ11q protein remains active in mediating partial PARPi and cisplatin resistance both in vitro and in vivo. The mechanism of the resistance appears to be the aberrant splicing because spliceosome inhibitors that decrease the BRCA1-Δ11q level sensitize the cancer cells harboring the exon 11 mutations to the PARPi treatment [17]. Therefore, selected by the anti-cancer treatments, the cell populations in breast cancer tend to evolve resistance by removal of disadvantageous germline BRCA1 mutations through somatic alternative mRNA splicing that generates isoforms coding for therapeutic resistance.
Altogether, the technology of CRISPR/Cas9-mediated somatic genome editing can help reveal new targets and previously speculated events in tumorigenesis for diagnosis and treatment of breast cancer. It can validate putative cancer drivers in vivo (Fig. 2) [15]. It can directly target oncogenes and impair breast cancer cell proliferation through a dominant negative mutation (Fig. 3) [16]. It can also help unveil mechanisms that mediate resistance of breast cancer to anticancer treatments (Fig. 4) [17]. Furthermore, it has paved a way to explore other cancerous events, such as addiction of breast cancer to transcription as discussed below.
Transcription addiction in breast cancer as validated by CRSPR/Cas9 genome editing
Cell- or tissue-specific transcriptional regulation is unveiled among nearly all species [63], in which organogenesis and neoplastic transformation are strictly controlled by the specific transcriptional programs. Tumor-specific gene expression programs validated with the CRSPR/Cas genome editing technology have been found in several tumors including breast basal-like, serous ovarian cancers and lung squamous cell carcinoma [64]. In this section, we first introduce a study showing that cancer cell specificity can be defined by certain transcription factors, the cell-specific transcription regulation. Second, we take triple-negative breast cancer (TNBC), an extremely aggressive form of breast cancer, as an example to manifest a transcriptional program entailing the exceedingly high levels of genetic heterogeneity [65, 66], fairly similar to the program of the solid tumors [67, 68] (Fig. 5). Third, we present a recent finding that the tumor-specific transcription programs appear affected epigenetically by a certain hormone signaling [69] (Fig. 6). Lastly, breast cancer appears to have conspicuous transcription programs, which are subjected to environment-responsive controls. The findings from these studies lead to the premise that breast cancer cells highly proliferative and metastatic are likely addicted to the genomic and transcriptional profiles; therefore, these programs acting as cancer drivers can be targeted by diagnostics and therapeutics, constituting the future personalized cancer medicine [70].
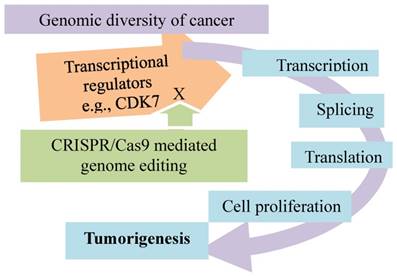
Addiction of triple-negative breast cancer to transcriptional programs promoted by transcriptional cyclin-dependent kinases. Knockout of CDK7 by CRISPR/Cas9 inhibits transcription in TNBC cells and impairs cancer growth.
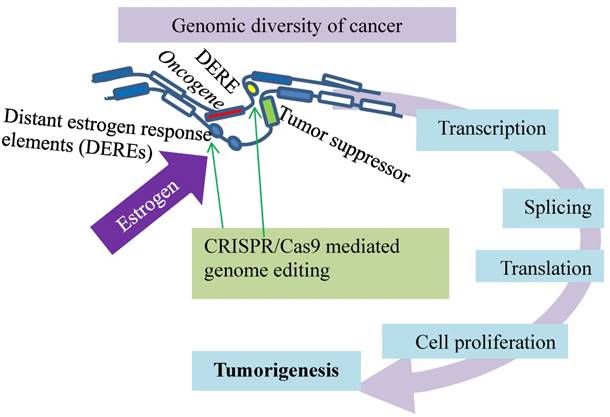
A spatiotemporal control of estrogen-responsive transcription in breast cancer. DEREs (oval) induce intra- or inter-chromatin interactions, through which the ERα-DERE (yellow oval) complex is brought to target promoters for transcriptional modulation: epigenetic stimulation of oncogenes (red box) and repression of tumor suppressors (green box). Deletion of certain DEREs by CRISPR/Cas9 (green arrows) attenuates proliferation-associated networks and decreases cancer cell growth.
Firstly, cancer cell specificity can be defined by certain transcription factors involved in the cell-specific transcription regulation. One of the transcription factors is forkhead box protein A1 (FOXA1) that regulates the organogenesis and cancer progression in the liver, breast, prostate, lung, and endometrium [71-78]. FOXA1 defines cancer cell specificity as evidenced from a study showing that FOXA1 marks cell-specific genomic signatures and regulates gene expression differentially in different human cancer cell lines, such as HepG2, LNCaP, MCF7, and T47D [79]. While the FOXA1-bound genes exist in these cancer cells, the functional FOXA1 target genes mostly are unique to each cancer cell type. This functioning of cell-specific FOXA1 targeting in human cancer cells likely relies on certain histone modifications. Such cell-specific FOXA1 regulation is validated with CRSPR/Cas9, specifying the unique FOXA1 binding, genetic variations, and potential epigenetic regulation [79].
Secondly, the triple-negative breast cancer is addicted to the incessant transcriptional programs promoted by some transcriptional cyclin-dependent kinases, and such a kinase-dependent addition is verified by the CRSPR/Cas genome editing technology (Fig. 5). Inhibition of CDK7, a transcriptional cyclin-dependent kinase, selectively targets the cancer cells, leading to apoptotic cell death. Loss of CDK7 via genetic editing of CDK7 gene in the TNBC cell lines by CRSPR/Cas9 selectively impairs TNBC cell growth and tumorigenesis. The results with CRISPR/Cas9 to knock out other known CDKs (8, 9, 12, 13, and 19) implicated in transcriptional regulation suggest that CDK7 is exclusively required for TNBC cell survival and proliferation. Of an "Achilles cluster" of genes specific to TNBC encoding super-enhancer-associated transcriptional regulators, the knockouts of EGFR, FOSL1, FOXC1, MYC and SOX9 by CRISPR/Cas9 impair the cancer cell proliferation. These observations suggest that these super-enhancers are cancer type-specific, and CDK7 is distinctively required for the cancer cell survival and proliferation. As these proteins play an important role in control of transcription, this type of breast cancer appears transcriptionally addicted [80]. This cancer-specific transcriptional addiction may be a new therapeutic target for the personalized cancer medicine.
Thirdly, the activated estrogen signaling induces nuclear translocation of the estrogen receptor-α (ERα) to bind estrogen-responsive elements near the target promoters for transcription [81-83]. Such a hormone-driven genomic event of ERα plays an essential role in stimulating abnormal proliferation of luminal breast cancers [81-85]. This estrogen stimulation also activates ERα association with distant estrogen response elements (DEREs) on chromosomes through genome looping (Fig. 6). Breast cancer cells positive for ERα display a spatiotemporal control of estrogen-responsive transcription through ERα association with DEREs, and the role of DEREs is confirmed by CRSPR/Cas genome editing technology (Fig. 6). Situated far away from target loci on the same or different chromosomes, DEREs can induce intra- or inter-chromatin interactions, through which the ERα-DERE complex is brought to the target promoters for transcriptional modulation [85-87]. DEREs originally clustered on 20q13 [the q terminal end (long arm) of chromosome 20] are frequently found in multiple chromosomal sites, leading to complex chromosomal rearrangements and amplification [87]. Transcription units are often gathered in a discrete focal site inside the nucleus to form a regulatory depot or a transcription factory [88-91] to which genes situated on different chromosomes are brought for transcriptional action [88-90]. Under estrogen stimulation, the scattered DEREs of 20q13 are mobilized by heterochromatin protein 1 (HP1) to form regulatory depots that modulate transcription of genes in target loci in two ways. The DERE regulatory depots synchronize gene expression of target loci and direct long-range epigenetic repression of estrogen-responsive tumor-suppressor genes. The CRISPR/Cas9-edited deletion of 20q13 DEREs attenuates the transcriptional modulation of proliferation-associated signaling networks, leading to a decrease in cancer cell growth but an increase in overall survival of the patients [69]. Disruption of the spatiotemporal control of the transcriptional program in cancer can be a new target for treating breast cancers and other malignancies.
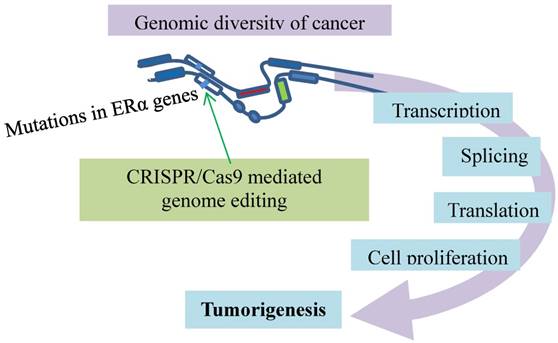
Lastly, the resistant mechanisms of metastatic breast cancer to the anti-ER therapies are studied with a CRISPR/Cas9-mediated knock-in mutational model (Fig. 7). As ERα is the major driver of breast cancer development and progression, inhibition of ER activity decreases relapse and increases patient survival [92, 93]. However, tumors become resistant against the anti-ER therapies in many cases, while they remain ER-positive and responsive to the treatments [94-97]. The resistance has raised a major clinical challenge. Mutations have been identified in the ER gene accounting for the resistance, though rare in primary breast cancer [98-100]. They are frequently present in the ER gene (ESR1) of the advanced breast cancer patients who have received endocrine therapies [101-104]. The rise after the treatments suggests that the resistance evolution occurs under the treatment-selective pressures for ESR1 mutations [105, 106]. This premise is tested with CRISPR/Cas9-mediated introduction of mutations in the ESR1 gene, which is a single allele knock-in of the most commonly mutated amino acid residue, tyrosine 537, in the estrogen-responsive MCF7 breast cancer cell line [107]. This mutation confers global constitutive ER activity, causing estrogen-independent cell growth, so that the tumor cells carrying the mutation become resistant to the anti-estrogen medications. Interestingly, the resistance involves transcriptional co-activator recruitment and histone modification to enhance the ligand-independent expression of the ER target genes. This ligand-independent gene expression appears associated with the ligand-independent phosphorylation of Serine 118 by cyclin-dependent kinase CDK7. The CDK7 inhibition that prevents the phosphorylation inhibits MCF7-Y537S cell growth. Additionally, CDK8 inhibition by CRISPR/CAS9 knockout suppresses estrogen-dependent transcription and the growth of estrogen receptor positive breast cancer [108]. These findings point to addiction of metastatic breast cancer to the cancer-specific transcriptional program that can be disrupted by CDK inhibition [107]. Further evidence comes from a recent model [109] in which novel target genes involved in metastasis-associated phenotypes are identified, and context-dependent activity and mutation-specific effect of the mutant receptor are noticed [109]. These studies highlight the utility of CRISPR/Cas9-mediated knock-in mutational models for exploring alternative anticancer approaches.
Metastatic breast cancer resistant to the anti-ER therapies has mutations conferring the resistance. The ERα genes (open boxes) responsible for cancer development and progression carry mutations (bars) for resistance to anti-ER therapies. This event is modeled with CRISPR/Cas9 (green arrow) that introduces a single allele knock-in mutation in the ESR1 gene. The mutation causes estrogen-independent growth and renders addiction to the cancer-specific transcriptional program.
Collectively, transcription addiction in breast cancer has been validated by CRSPR/Cas9 genome editing in several studies. Cancer cells display the cell-specific transcription regulation [79]; in fact, a certain hormone signaling modulates epigenetically the tumor-specific transcription programs [69] (Fig. 6). These lead to a discovery of addiction of the triple-negative breast cancer to the transcriptional programs mediated by some transcriptional cyclin-dependent kinases [80] (Fig. 5). In a CRISPR/Cas9-mediated knock-in mutational model, further investigation of the resistant mechanisms of metastatic breast cancer to the anti-ER therapies suggests addiction of the metastatic cancer to the cancer-specific transcriptional program. The program can be disrupted by inhibition of CDKs, such as CDK7, a potential therapy for the ER mutation-mediated endocrine-resistant breast cancer [107] (Fig. 7). Given the role of CDK7 in cancerous transcription regulation as discussed above in Fig. 5 and Fig. 7, inhibition of CDK7 not just selectively impairs the cell growth and tumorigenesis of TNBC (Fig. 5) but also the resistance evolution of the estrogen-responsive MCF7 breast cancer cells (Fig. 7). The role of these genomic and transcriptional profiles in tumorigenesis and therapeutic resistance of breast cancer can be further deciphered by the CRSPR/Cas-mediated genome editing (Fig. 7).
Protein degradation addiction in breast cancer as validated by CRSPR/Cas9 genome editing
Rapid proliferation of cancer cells often relies on an increased activity of protein degradation. The 26S proteasome, responsible for degradation of majority of cellular proteins in eukaryotes [110], confers cancer cells with addiction to proteasome activity (Fig. 1). Proteasomes and protein degradation have become targets for anticancer treatments and the therapy-resistance evolution of malignancies, such as multiple myeloma and certain solid cancers [111, 112]. In this section, we discuss how the CRISPR/Cas9 system validates the role of proteasome regulation in control of proliferation and tumorigenesis of breast cancer [113]. We also show how it aids in revealing the mechanisms by which breast cancer cells resist endocrine therapy via upregulation of the unfolded protein response that reduces degradation of ERα [114], and by which the E3-Ubiquitin Ligase UBR5, a key regulator of this response, acts as a driver for cancer growth and metastasis [115].
Proliferation and tumorigenesis of breast cancer is controlled by site-specific proteasome phosphorylation [113], a finding that is verified by the CRSPR/Cas9 genome editing. Among 33 subunits of the 26S proteasome, 14 (α1-7 and β1-7) constitute the 20S core particle (CP), and 1 barrel-shaped structure encloses 3 types of peptidase activities. The remaining 19 (Rpt1-6, Rpn1-3, 5-13 and 15) form the 19S regulatory particle (RP). RP that caps CP on one or both ends captures and processes protein substrates. These substrates are then threaded into CP for proteolysis. Both Rpt1-6 (the ATPase subunits) and CP (involving in gate opening) play important roles in substrate engagement, unfolding, and translocation [112, 116, 117]. Regulation of the 26S proteasome is exerted at various levels by many mechanisms, such as cell cycle regulation [118], transcriptional control and post-translational modifications or phosphorylation of proteasome subunits [119-124]. About the phosphorylation regulation, the 26S proteasome has more than 300 phosphorylation sites. Many proteasome phosphorylation events occur at various cell cycle stages [125-128]. Thr 25 of the 19S subunit Rpt3 is dynamically phosphorylated during the cell cycle, and the phosphorylation is carried out by the dual-specificity tyrosine-regulated kinase 2 (DYRK2). The CRISPR/Cas9 system was used to generate the homozygous T25A knock-in and DYRK2 knock-out mutations. Both mutations disrupt tumorigenesis of the proteasome-addicted human breast cancer cells in mice [113]. Therefore, proteasome regulation plays a role in control of cell proliferation and tumorigenesis (Fig. 1).
The mechanism of antiestrogen resistance of breast cancer cells also involves upregulation of the unfolded protein response that delays ERα degradation [114], the conclusion validated by the CRSPR/Cas9 genome editing (Fig. 1). Breast cancers positive for ERα can be treated with endocrine therapy that includes both inhibition of estrogen production by aromatase inhibitors and competition against estrogen binding to ERα by tamoxifen and fulvestrant/Faslodex/ICI 182,780 (ICI). Under such a therapeutic selection, advanced metastatic breast cancer become resistant and outgrown as ERα mutations ERαY537S and ERαD538G occur frequently [102-104, 129]. These mutations were investigated in cell lines derived from circulating tumor cells, with some results to suggest the mutations responsible for resistance to aromatase inhibitors [102-104, 129-132] and the others to tamoxifen and fulvestrant/ICI [129, 133, 134]. The uncertainty about the mutations was further investigated in an ERα positive breast cancer cell model in which the cells expressing the mutated ERα and wild-type ERα were compared. This model was created with the CRISPR/Cas9 genome editing and homology-directed repair system with which one or both wild-type ERα genes were replaced with ERαY537S or ERαD538G. Both ERαY537S and ERαD538G cells have constitutive expression of the estrogen-ERα target genes and partial resistance to antiestrogen, manifesting estrogen-independent growth. The antiestrogen resistance of ERαY537S and ERαD538G cells appears associated with upregulation of the unfolded protein response that reduces the ERα degradation [114].
Further studies into the unfolded protein response reveal the E3-Ubiquitin Ligase UBR5, a key regulator of this pathway and also a driver for growth and metastasis of Triple-Negative Breast Cancer [115]. UBR5, originally identified in a screen for progestin-regulated genes in human breast cancer cells as a tumor suppressor gene [135], is a nuclear phosphoprotein, overexpressed in many TNBC samples [136]. It modulates both ERα-induced gene expression and cell proliferation via its ubiquitin ligase activity in human breast cancer cell lines [137]. The results from a recent study reveal strong gene amplifications and overexpression of UBR5 in TNBC [115]. CRISPR/Cas9-mediated knockout of UBR5 impairs tumor growth and metastasis in vivo as shown in an experimental murine mammary carcinoma model of TNBC. These phenotypes are restored by reconstitution with wild-type UBR5. The growth phenotype stems from a disrupted angiogenesis inside the tumor, aligned with the increased apoptosis, necrosis, and growth arrest. The metastasis phenotype results from aberrant epithelial-to-mesenchymal transition mainly through rescinded expression of E-cadherin. Moreover, the UBR5-facilitated tumor growth relies on immune cells in the microenvironment, but the UBR5-promoted metastasis depends on tumor cells. Therefore, UBR5 plays a role in the growth and metastasis of TNBC. This makes UBR5 a potential therapeutic target for the treatment of highly aggressive breast and ovarian cancers [115].
Taken together, addiction of breast cancer to protein degradation has been validated by CRSPR/Cas9 genome editing in recent studies. The site-specific proteasome phosphorylation affects breast cancer proliferation and tumorigenesis, rending the cancer addiction to proteasome [113]. This conclusion suggests a role of turnover of unfolded and cancerous proteins in cancer proliferation and tumorigenesis. Aligned with this premise is upregulation of the unfolded protein response that delays ERα degradation, the mechanism that mediates antiestrogen resistance of breast cancer [114]. A key regulator of this unfolded protein response, the E3-Ubiquitin Ligase UBR5, acts as a driver for growth and metastasis of TNBC [115]. All these conclusions, verified by CRSPR/Cas9 genome editing, provide insights about potential therapeutic targets for the treatment of breast cancer.
CRISPR/Cas9 geno/immuno-toxicity and the potential remedy
Genome editing with CRISPR/Cas9 to target the coding and non-coding sites related to carcinogenesis on the chromosomes has provided a useful tool for both basic research and clinical application on breast cancer. However, CRISPR/Cas9 poses both genotoxicity and immunotoxicity. The genotoxicity entails off-target effects. This technical problem has hampered development of the CRISPR/Cas9-based therapeutics because the off-target activities cause mutations that may be carcinogenic to the patients. The off-target effects are controllable in animal studies as the undesired traits from the non-target chromosomes can be excluded. The off-target effects are due to inaccurate Cas9 cleavage of an imperfect heteroduplex between sgRNA and PAM-labeled target DNA where mismatches are allowed. The type, number, and location of these mismatches affect the frequency of the on-target and off-target cleavages [138-140]. Base pairing between the sgRNA and the off-target DNA occurs with multiple mismatches, leading to cleavage at the off-target site [140, 141]. The immunotoxicity includes host immune response to the bacterial Cas9 protein, the delivery viral vector and the targeted cells. As shown in Fig. 2, the invasive lobular carcinoma has been modeled by CRISPR/Cas9 mediated genome editing to detect a cancer driver, Pten, which acts as a negative regulator of the PI3K/AKT signaling pathway. While it was not reported in the breast cancer modeling, the immunotoxicity was observed in a liver disease modeling in mice in which the same Pten gene was targeted by adenovirus-mediated somatic genome editing [142].
On the positive side, both types of CRISPR-related toxicity have become a focus of research effort to seek solutions. As Cas9 cleaves DNA only at recognition sites of its cognate sgRNA, it does not possess inherent nuclease activity in the absence of sgRNA [143]. Furthermore, there are the several approaches to reduce the potential off-target effects. The first is the stringent selection of sgRNAs through computational efforts to improve sgRNA design and to predict the potential off-target sites in the chromosomes [144, 145]. The second is use of a shorter sgRNA, which may reduce the off-target effects [146]. The third is optimal selection PAMs. The availability of various PAMs provides more sgRNA options for the choice of minimal off-target activity. The discovery of species-specific Cas9s offers alternative PAMs [147]. Cas9 can be engineered to have altered PAM specificities [148]. The last is modification of Cas9. The engineered Cas9 can nick the opposite DNA strand of the heteroduplex between sgRNAs and target DNA leading to DSB that decreases the off-target activity [149]. Such engineering can further increase specificity of Cas9 variants, which decrease the off-target effects [150]. A fusion chimera of enzymatically dead Cas9 (dCas9) from bacteria with the dimerization-dependent FokI nuclease domain has been constructed into the RNA-guided FokI-nucleases (RFNs) [151, 152]. The dimerization-dependent cleavage increases specificity and reduces the off-target activity [151-153]. The RFN chimeras have been reengineered for even higher cleavage efficiency and fidelity with broader genome targetability for genome editing in human cells [154]. By comparison to the progress in the off-target research, the battle against the immunotoxicity is not just beginning but complicated. The delivery viral vector can induce the CRISPR/Cas9-specific immune responses [142], pointing to the need to attenuate such immunogenicity of the CRISPR/Cas9 components. Now that delivery viral vectors are immunogenic, new delivery methods have been developed, such as microinjection, lipofection, and electroporation [155]. Altogether, CRISPR/Cas9 poses threats of geno- and immuno-toxicity, the technical drawbacks that have hindered progress of the clinical application against breast cancer but have spurred research efforts for solutions.
Conclusive remarks
The nature of cancer genomic diversity generates leverages for carcinogenesis, metastasis and resistance evolution, which can be broken by CRISPR/Cas9. The incessant proliferation of cancer cells is not only encoded by the diverse sets of the mutated genes but also affected by epigenetic factors in cancer cells. This burdens overwhelmingly such major cellular events as transcription, splicing, translation, protein folding and degradation (Fig. 1), rendering the cancer growth addicted to these events. Behind the uncontrolled cancer proliferation are the drivers and passengers on the train of the cellular events. When encountering anticancer treatments, the cancer cells carrying such a genomic diversity are capable of evolving resistance by multiple mechanisms, such as mutations, aberrant transcription, splicing, protein folding and degradation. Such targets as ERα or CDK7 appear to link a chain of actions. The activated estrogen signaling triggers nuclear translocation of ERα to bind estrogen-responsive elements near target promoters for cancerous transcription (Fig. 6) [81-83]. This leads to development of the antiestrogen therapies against the ER activity that reduce relapse and increase survival of the breast cancer patient [92, 93], but the antiestrogen resistance evolves with the mechanism involving upregulation of the unfolded protein response that delays ERα degradation (Fig. 1) [114]. The CRISPR/Cas9-mediated knockout of UBR5 encoding a key regulator of the unfolded protein response in an experimental murine mammary carcinoma model of TNBC impairs tumor growth and metastasis in vivo [115]. Additionally, CDK7 inhibition selectively impairs the cell growth and tumorigenesis of TNBC (Fig. 5) and the resistance evolution of the estrogen-responsive MCF7 breast cancer cells (Fig. 7). While such genetic and epigenetic diversity of cancer presents a pressing challenge for humans to defeat cancers, utilizing genome editing technology such as the CRISPR/Cas9 can help identify the targets at these cancerous events. Only with the potential targets unveiled in breast cancer patients can more accurate, efficient, and economic cancer therapies be prescribed.
Acknowledgements
We thank Todd Sukany for comments and proofreading.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: A Cancer Journal for Clinicians. 2016;67:7-30
2. Zeichner SB, Herna S, Mani A. et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Research and Treatment. 2015;153:617-624
3. Mauri D, Polyzos NP, Salanti G. et al. Multiple-Treatments Meta-analysis of Chemotherapy and Targeted Therapies in Advanced Breast Cancer. JNCI: Journal of the National Cancer Institute. 2008;100:1780-1791
4. Cnossen JA, Heinemann V, Laessig D. et al. Long-term survival with metastatic breast cancer (MBC): Results of a retrospective, single-centre analysis from 2000-2005. Journal of Clinical Oncology. 2008;26:1128-1128
5. Minicozzi P, Bella F, Toss A. et al. Relative and disease-free survival for breast cancer in relation to subtype: a population-based study. Journal of Cancer Research and Clinical Oncology. 2013;139:1569-1577
6. Sledge Jr GW, Miller KD. Exploiting the hallmarks of cancer: the future conquest of breast cancer. European Journal of Cancer. 2003;39:1668-1675
7. Ingvarsson S. Genetics of breast cancer. Drugs Today (Barc). 2004;40:991-1002
8. Toss A, Venturelli M, Peterle C. et al. Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? International Journal of Molecular Sciences. 2017;18:85-109
9. Cardoso F, Harbeck N, Fallowfield L. et al. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2012;23:vii11-vii19
10. Sonnenblick A, Pondé N, Piccart M. Metastatic breast cancer: The Odyssey of personalization. Molecular Oncology. 2016;10:1147-1159
11. Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Research: BCR. 2015;17:60
12. Gasiunas G, Barrangou R, Horvath P. et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences. 2012;109:E2579-E2586
13. Jinek M, Chylinski K, Fonfara I. et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816-821
14. Wyman C, Kanaar R. DNA Double-Strand Break Repair: All's Well that Ends Well. Annual Review of Genetics. 2006;40:363-383
15. Annunziato S, Kas SM, Nethe M. et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes & Development. 2016;30:1470-1480
16. Wang H, Sun W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Letters. 2017;385:137-143
17. Wang Y, Bernhardy AJ, Cruz C. et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Research. 2016;76:2778-2790
18. Martinez V, Azzopardi JG. Invasive lobular carcinoma of the breast: incidence and variants. Histopathology. 1979;3:467-488
19. Borst. M.J. I, J.A.. Metastatic patterns of invasive lobular versus invasive ductal carcinoma of the breast. Surgery. 1993;114:637-641
20. Wong H, Lau S, Cheung P. et al. Lobular breast cancers lack the inverse relationship between ER/PR status and cell growth rate characteristic of ductal cancers in two independent patient cohorts: implications for tumor biology and adjuvant therapy. BMC Cancer. 2014;14:826
21. Moll R, Mitze M, Frixen UH. et al. Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. The American Journal of Pathology. 1993;143:1731-1742
22. Vos CB, Cleton-Jansen AM, Berx G. et al. E-cadherin inactivation in lobular carcinoma in situ of the breast: an early event in tumorigenesis. British Journal of Cancer. 1997;76:1131-1133
23. Droufakou S, Deshmane V, Roylance R. et al. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. International Journal of Cancer. 2001;92:404-408
24. Ciriello G, Gatza Michael L, Beck Andrew H. et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506-519
25. Rakha EA, Patel A, Powe DG. et al. Clinical and Biological Significance of E-cadherin Protein Expression in Invasive Lobular Carcinoma of the Breast. The American Journal of Surgical Pathology. 2010;34:1472-1479
26. Boussadia O, Kutsch S, Hierholzer A. et al. E-cadherin is a survival factor for the lactating mouse mammary gland. Mechanisms of Development. 2002;115:53-62
27. Derksen PWB, Liu X, Saridin F. et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437-449
28. Derksen PWB, Braumuller TM, van der Burg E. et al. Mammary-specific inactivation of E-cadherin and p53 impairs functional gland development and leads to pleomorphic invasive lobular carcinoma in mice. Disease Models & Mechanisms. 2011;4:347-358
29. Koirala P, Huang J, Ho T-T. et al. LncRNA AK023948 is a positive regulator of AKT. Nature Communications. 2017;8:14422
30. Jubin T, Kadam A, Jariwala M. et al. The PARP family: insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Proliferation. 2016;49:421-437
31. Szabo CI, King MC. Inherited breast and ovarian cancer. Human Molecular Genetics. 1995;4:1811-1817
32. Friedman LS, Ostermeyer EA, Szabo CI. et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994;8:399-404
33. Thompson D, Easton D. Variation in BRCA1 Cancer Risks by Mutation Position. Cancer Epidemiology Biomarkers & Prevention. 2002;11:329-336
34. Risch HA, McLaughlin JR, Cole DEC. et al. Population BRCA1 and BRCA2 Mutation Frequencies and Cancer Penetrances: A Kin-Cohort Study in Ontario, Canada. JNCI: Journal of the National Cancer Institute. 2006;98:1694-1706
35. Risch HA, McLaughlin JR, Cole DEC. et al. Prevalence and Penetrance of Germline BRCA1 and BRCA2 Mutations in a Population Series of 649 Women with Ovarian Cancer. The American Journal of Human Genetics. 2001;68:700-710
36. Moynahan ME, Cui TY, Jasin M. Homology-directed DNA Repair, Mitomycin-C Resistance, and Chromosome Stability Is Restored with Correction of a Brca1 Mutation. Cancer Research. 2001;61:4842-4850
37. Scully R, Chen J, Ochs RL. et al. Dynamic Changes of BRCA1 Subnuclear Location and Phosphorylation State Are Initiated by DNA Damage. Cell. 1997;90:425-435
38. Synthetic Lethality and Cancer Therapy. Lessons Learned from the Development of PARP Inhibitors. Annual Review of Medicine. 2015;66:455-470
39. Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381-1388
40. Bryant HE, Schultz N, Thomas HD. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913-917
41. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-921
42. Kennedy RD, Quinn JE, Mullan PB. et al. The Role of BRCA1 in the Cellular Response to Chemotherapy. JNCI: Journal of the National Cancer Institute. 2004;96:1659-1668
43. Quinn JE, Kennedy RD, Mullan PB. et al. BRCA1 Functions as a Differential Modulator of Chemotherapy-induced Apoptosis. Cancer Research. 2003;63:6221-6228
44. Gelmon KA, Tischkowitz M, Mackay H. et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology. 2011;12:852-861
45. Sandhu SK, Schelman WR, Wilding G. et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. The Lancet Oncology. 2013;14:882-892
46. Ang JE, Gourley C, Powell CB. et al. Efficacy of Chemotherapy in BRCA1/2 Mutation Carrier Ovarian Cancer in the Setting of PARP Inhibitor Resistance: A Multi-Institutional Study. Clinical Cancer Research. 2013;19:5485-5493
47. Ledermann J, Harter P, Gourley C. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. The Lancet Oncology. 2014;15:852-861
48. Edwards SL, Brough R, Lord CJ. et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111-1115
49. Sakai W, Swisher EM, Karlan BY. et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116-1120
50. Bouwman P, Aly A, Escandell JM. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688-695
51. Bunting SF, Callén E, Wong N. et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell. 2010;141:243-254
52. Johnson N, Johnson SF, Yao W. et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proceedings of the National Academy of Sciences. 2013;110:17041-17046
53. Drost R, Bouwman P, Rottenberg S. et al. BRCA1 RING Function Is Essential for Tumor Suppression but Dispensable for Therapy Resistance. Cancer Cell. 2011;20:797-809
54. Rottenberg S, Jaspers JE, Kersbergen A. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences. 2008;105:17079-17084
55. Colombo M, Blok MJ, Whiley P. et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. Human Molecular Genetics. 2014;23:3666-3680
56. Thomassen M, Blanco A, Montagna M. et al. Characterization of BRCA1 and BRCA2 splicing variants: a collaborative report by ENIGMA consortium members. Breast Cancer Research and Treatment. 2012;132:1009-1023
57. Romero A, García-García F, López-Perolio I. et al. BRCA1 Alternative splicing landscape in breast tissue samples. BMC Cancer. 2015;15:219
58. Orban TI, Olah E. Expression Profiles of BRCA1 Splice Variants in Asynchronous and in G1/S Synchronized Tumor Cell Lines. Biochemical and Biophysical Research Communications. 2001;280:32-38
59. Tammaro C, Raponi M, Wilson David I. et al. BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochemical Society Transactions. 2012;40:768-772
60. Raponi M, Smith LD, Silipo M. et al. BRCA1 exon 11 a model of long exon splicing regulation. RNA Biology. 2014;11:351-359
61. Orban TI, Olah E. Emerging roles of BRCA1 alternative splicing. Molecular Pathology. 2003;56:191-197
62. Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D. et al. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Human Molecular Genetics. 2002;11:2805-2814
63. Odom DT, Dowell RD, Jacobsen ES. et al. Tissue-Specific Transcriptional Regulation has Diverged Significantly between Human and Mouse. Nature genetics. 2007;39:730-732
64. Hoadley KA, Yau C, Wolf DM. et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell. 2014;158:929-944
65. Abramson VG, Lehmann BD, Ballinger TJ. et al. Subtyping of triple-negative breast cancer: implications for therapy. Cancer. 2015;121:8-16
66. The Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490:61-70
67. Parker JS, Mullins M, Cheang MCU. et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. Journal of Clinical Oncology. 2009;27:1160-1167
68. Perou CM, Sorlie T, Eisen MB. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-752
69. Hsu PY, Hsu HK, Hsiao TH. et al. Spatiotemporal control of estrogen-responsive transcription in ER[alpha]-positive breast cancer cells. Oncogene. 2016;35:2379-2389
70. Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17:297-303
71. Kaestner KH. The FoxA Factors in Organogenesis and Differentiation. Current opinion in genetics & development. 2010;20:527-532
72. Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12:381-385
73. Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Bioscience Reports. 2012;32:113-130
74. Hurtado A, Holmes KA, Ross-Innes CS. et al. FOXA1 is a critical determinant of Estrogen Receptor function and endocrine response. Nature genetics. 2011;43:27-33
75. Cowper-Sal·lari R, Zhang X, Wright JB. et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nature genetics. 2012;44:1191-1198
76. Li Z, Tuteja G, Schug J. et al. Foxa1 and Foxa2 Are Essential for Sexual Dimorphism in Liver Cancer. Cell. 2012;148:72-83
77. Carroll JS, Liu XS, Brodsky AS. et al. Chromosome-Wide Mapping of Estrogen Receptor Binding Reveals Long-Range Regulation Requiring the Forkhead Protein FoxA1. Cell. 2005;122:33-43
78. Lupien M, Eeckhoute J, Meyer CA. et al. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell. 2008;132:958-970
79. Zhang G, Zhao Y, Liu Y. et al. FOXA1 defines cancer cell specificity. Science Advances. 2016;2:e1501473
80. Wang Y, Zhang T, Kwiatkowski N. et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163:174-186
81. Brisken C, O'Malley B. Hormone Action in the Mammary Gland. Cold Spring Harbor Perspectives in Biology. 2010;2:a003178. doi: 10.1101/cshperspect.a003178
82. Thomas C, Gustafsson J-Å. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597-608
83. Cicatiello L, Mutarelli M, Grober OMV. et al. Estrogen Receptor alpha Controls a Gene Network in Luminal-Like Breast Cancer Cells Comprising Multiple Transcription Factors and MicroRNAs. The American Journal of Pathology. 2010;176:2113-2130
84. Carroll JS, Meyer CA, Song J. et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289-1297
85. Hsu P-Y, Hsu H-K, Singer GAC. et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Research. 2010;20:733-744
86. Fullwood MJ, Liu MH, Pan YF. et al. An oestrogen-receptor-[agr]-bound human chromatin interactome. Nature. 2009;462:58-64
87. Hsu P-Y, Hsu H-K, Lan X. et al. Amplification of Distant Estrogen Response Elements Deregulates Target Genes Associated with Tamoxifen Resistance in Breast Cancer. Cancer Cell. 2013;24:197-212
88. Sutherland H, Bickmore WA. Transcription factories: gene expression in unions? Nat Rev Genet. 2009;10:457-466
89. Mitchell JA, Fraser P. Transcription factories are nuclear subcompartments that remain in the absence of transcription. Genes & Development. 2008;22:20-25
90. Deng B, Melnik S, Cook PR. Transcription factories, chromatin loops, and the dysregulation of gene expression in malignancy. Seminars in Cancer Biology. 2013;23:65-71
91. Papantonis A, Cook PR. Transcription Factories: Genome Organization and Gene Regulation. Chemical Reviews. 2013;113:8683-8705
92. Cuzick J, Sestak I, Baum M. et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. The Lancet Oncology. 2010;11:1135-1141
93. Osborne CK. Tamoxifen in the Treatment of Breast Cancer. New England Journal of Medicine. 1998;339:1609-1618
94. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2:101-112
95. Ali S, Buluwela L, Coombes RC. Antiestrogens and Their Therapeutic Applications in Breast Cancer and Other Diseases. Annual Review of Medicine. 2011;62:217-232
96. Johnston SRD, Dowsett M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer. 2003;3:821-831
97. Ali S, Buluwela L, Coombes RC. Mechanisms of Endocrine Resistance in Breast Cancer. Annual Review of Medicine. 2011;62:233-247
98. Herynk MH, Fuqua SAW. Estrogen Receptor Mutations in Human Disease. Endocrine Reviews. 2004;25:869-898
99. Oesterreich S, Davidson NE. The search for ESR1 mutations in breast cancer. Nat Genet. 2013;45:1415-1416
100. Segal CV, Dowsett M. Estrogen Receptor Mutations in Breast Cancer—New Focus on an Old Target. Clinical Cancer Research. 2014;20:1724-1726
101. Jeselsohn R, Yelensky R, Buchwalter G. et al. Emergence of Constitutively Active Estrogen Receptor-α Mutations in Pretreated Advanced Estrogen Receptor-Positive Breast Cancer. Clinical Cancer Research. 2014;20:1757-1767
102. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A. et al. D538G Mutation in Estrogen Receptor-α: A Novel Mechanism for Acquired Endocrine Resistance in Breast Cancer. Cancer Research. 2013;73:6856-6864
103. Robinson DR, Wu Y-M, Vats P. et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446-1451
104. Toy W, Shen Y, Won H. et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439-1445
105. Guttery DS, Page K, Hills A. et al. Noninvasive Detection of Activating Estrogen Receptor 1 (ESR1) Mutations in Estrogen Receptor-Positive Metastatic Breast Cancer. Clinical Chemistry. 2015;61:974-982
106. Schiavon G, Hrebien S, Garcia-Murillas I. et al. Analysis of <em>ESR1</em> mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Science Translational Medicine. 2015;7:313ra182-313ra182
107. Harrod A, Fulton J, Nguyen VTM. et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene. 2016
108. McDermott MSJ, Chumanevich AA, Lim C-u. et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget. 2017;8:12558-12575
109. Bahreini A, Li Z, Wang P. et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Research: BCR. 2017;19:60
110. Coux O, Tanaka K, Goldberg AL. Structure and Functions of the 20S and 26S Proteasomes. Annual Review of Biochemistry. 1996;65:801-847
111. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438-444
112. Orlowski RZ, Kuhn DJ. Proteasome Inhibitors in Cancer Therapy: Lessons from the First Decade. Clinical Cancer Research. 2008;14:1649-1657
113. Guo X, Wang X, Wang Z. et al. Site-specific Proteasome Phosphorylation Controls Cell Proliferation and Tumorigenesis. Nature cell biology. 2016;18:202-212
114. Mao C, Livezey M, Kim JE. et al. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific Reports. 2016;6:34753
115. Liao L, Song M, Li X. et al. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Research. 2017;77:2090-2101
116. Bhattacharyya S, Yu H, Mim C. et al. Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol. 2014;15:122-133
117. Ehlinger A, Walters KJ. Structural Insights into Proteasome Activation by the 19S Regulatory Particle. Biochemistry. 2013;52:10.1021 /bi400417a
118. Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Current Opinion in Cell Biology. 1997;9:788-799
119. Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009;10:104-115
120. Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochimica et biophysica acta. 2014;1843:10.1016 /j.bbamcr.2013.1008.1012
121. Radhakrishnan SK, Lee CS, Young P. et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Molecular cell. 2010;38:17-28
122. Tomko RJ, Hochstrasser M. Molecular Architecture and Assembly of the Eukaryotic Proteasome. Annual review of biochemistry. 2013;82:10.1146 /annurev-biochem-060410-150257
123. Wang X, Chen C-F, Baker PR. et al. Mass Spectrometric Characterization of the Affinity-Purified Human 26S Proteasome Complex. Biochemistry. 2007;46:3553-3565
124. Wang X, Huang L. Identifying Dynamic Interactors of Protein Complexes by Quantitative Mass Spectrometry. Molecular & Cellular Proteomics. 2008;7:46-57
125. Dephoure N, Zhou C, Villén J. et al. A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10762-10767
126. Olsen JV, Vermeulen M, Santamaria A. et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Science Signaling. 2010;3:ra3-ra3
127. Nagano K, Shinkawa T, Mutoh H. et al. Phosphoproteomic analysis of distinct tumor cell lines in response to nocodazole treatment. PROTEOMICS. 2009;9:2861-2874
128. Kettenbach AN, Schweppe DK, Faherty BK. et al. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-Like Kinase Activities in Mitotic Cells. Science signaling. 2011;4:10.1126 /scisignal.2001497
129. Zhang Q-X, Borg Å, Wolf DM. et al. An Estrogen Receptor Mutant with Strong Hormone-independent Activity from a Metastatic Breast Cancer. Cancer Research. 1997;57:1244-1249
130. Spoerke JM, Gendreau S, Walter K. et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nature Communications. 2016;7:11579
131. Fribbens C, O'Leary B, Kilburn L. et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. Journal of Clinical Oncology. 2016;34:2961-2968
132. Schiavon G, Hrebien S, Garcia-Murillas I. et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Science Translational Medicine. 2015;7:313ra182-313ra182
133. Yu M, Bardia A, Aceto N. et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216-220
134. Paoletti C, Larios JM, Muñiz MC. et al. Heterogeneous estrogen receptor expression in circulating tumor cells suggests diverse mechanisms of fulvestrant resistance. Molecular Oncology. 2016;10:1078-1085
135. Callaghan MJ, Russell AJ, Woollatt E. et al. Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene. 1998;17:3479-3491
136. Clancy JL, Henderson MJ, Russell AJ. et al. EDD, the human orthologue of the hyperplastic discs tumour suppressor gene, is amplified and overexpressed in cancer. Oncogene. 2003;22:5070-5081
137. Bolt M, Stossi F, Callison A. et al. Systems level-based RNAi screening by high content analysis identifies UBR5 as a regulator of estrogen receptor-α protein levels and activity. Oncogene. 2015;34:154
138. Hsu PD, Scott DA, Weinstein JA. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotech. 2013;31:827-832
139. Mali P, Yang L, Esvelt KM. et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823-826
140. Fu Y, Foden JA, Khayter C. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotech. 2013;31:822-826
141. Lin Y, Cradick TJ, Brown MT. et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Research. 2014;42:7473-7485
142. Wang D, Mou H, Li S. et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Human Gene Therapy. 2015;26:432-442
143. Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet. 2016;17:300-312
144. Cradick TJ, Qiu P, Lee CM. et al. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Molecular Therapy - Nucleic Acids. 2014;3:e214
145. Montague TG, Cruz JM, Gagnon JA. et al. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 2014;42:W401-W407
146. Tsai SQ, Zheng Z, Nguyen NT. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotech. 2015;33:187-197
147. Ran FA, Cong L, Yan WX. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186-191
148. Kleinstiver BP, Prew MS, Tsai SQ. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481-485
149. Ran FA, Hsu Patrick D, Lin C-Y. et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380-1389
150. Slaymaker IM, Gao L, Zetsche B. et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84-88
151. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature biotechnology. 2014;32:577-582
152. Tsai SQ, Wyvekens N, Khayter C. et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature biotechnology. 2014;32:569-576
153. Wyvekens N, Topkar VV, Khayter C. et al. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Human Gene Therapy. 2015;26:425-431
154. Havlicek S, Shen Y, Alpagu Y. et al. Re-engineered RNA-Guided FokI-Nucleases for Improved Genome Editing in Human Cells. Molecular Therapy. 2017;25:342-355
155. Jacobi AM, Rettig GR, Turk R. et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods. 2017;121:16-28
Author contact
![]() Corresponding authors: Haitao Yang, hsyang8com. MariaLynn Jaeger, wholocked.2014com. Averi Walker, s696341edu. Daniel Zhuangyao Wei, danielzw96com. Katie Leikier, s646651edu. Tao Weitao, tweiedu.
Corresponding authors: Haitao Yang, hsyang8com. MariaLynn Jaeger, wholocked.2014com. Averi Walker, s696341edu. Daniel Zhuangyao Wei, danielzw96com. Katie Leikier, s646651edu. Tao Weitao, tweiedu.