Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(2):263-268. doi:10.7150/jca.20059 This issue Cite
Review
Cancer vaccine: learning lessons from immune checkpoint inhibitors
ZhenLong Ye, Qiming Qian, HuaJun Jin ![]() , QiJun Qian
, QiJun Qian ![]()
Shanghai Engineering Research Center for Cell Therapy, 75 Qianyang Road, Shanghai 201805, China.
Received 2017-3-13; Accepted 2017-8-28; Published 2018-1-1
Abstract
Cancer vaccines have been exclusively studied all through the past decades, and have made exceptional achievements in cancer treatment. Few cancer vaccines have been approved by the US Food and Drug Administration (FDA), for instance, Provenge, which was approved for the treatment of prostate carcinoma in 2012. Moreover, more recently, T-VEC got approval for the treatment of melanoma. While, the overall therapeutic effects of cancer vaccines have been taken into consideration as below expectations, low antigenicity of targeting antigen and tumor heterogeneity are the two key limiting barriers encountered by the cancer vaccines. Nonetheless, recent developments in cancer immune-therapies together with associated technologies, for instance the unparalleled achievements bagged by immune checkpoint inhibitor based therapies and neo-antigen identification tools, envisage potential improvements in cancer vaccines in respect to the treatments of malignancies. This review brings forth measures for the purpose of refining therapeutic cancer vaccines by learning lessons from the success of PD-1 inhibitor based immune-therapies.
Keywords: Cancer vaccine, immune checkpoint inhibitor, PD-1, combination therapy, immunotherapy
Introduction
Different from preventive vaccines, put to use on healthy individuals for the prevention of diseases, therapeutic cancer vaccines are directly used on cancer patients for the purpose of eliminating cancer cells through strengthening patients' own immune responses, particularly CD8+ T cell mediated responses, with the assistance of suitable adjuvants [1-3]. Since pioneered by Dr. William Coley for the stimulation of patient's immune system with the use of intratumoral injection of Coley's Toxin (inactivated Streptococcus pyogenes and Serratia marcescens) in 1890s, the field of cancer vaccine has been quite active, introducing several kinds of cancer vaccines, for instance DC cell based vaccines [4, 5], peptide/protein vaccines [6, 7], genetic vaccines [8] and tumor cell vaccines [9], targeting various cancer cell antigens, including cancer testis antigens, differentiation antigens, oncofetal antigens, EMT (Epithelial-Mesenchymal Transition) factors and TME (Tumor Microenvironment) factors [10]. Determination of the therapeutic efficacy of cancer vaccines is made by taking into account many factors, include differential expression of targets between tumor cells and normal cells, the immunogenicity of vaccines and the antigenicity of targets within tumor microenvironment [7, 11, 12]. Thus, the paucity of TSAs (tumor specific antigens), immune suppressive effect of tumor microenvironment and tumor heterogeneity pose to be the key limiting barriers encountered by cancer vaccines [13-16].
By targeting immune suppressive microenvironment for the release of cytotoxic T cells, immune checkpoint inhibitors have attained unparalleled success as regards the treatment of cancers [17-19]. Ipilimumab, an anti-CTLA-4 antibody, received approval from the FDA for the treatment of melanoma in 2011 [20, 21]. Nivolumab and pembrolizumab were approved in 2014 for the treatment of melanoma and squamous non-small cell lung cancer (NSCLC) [22-24]. The mechanistic research works brought to light the fact that the therapeutic efficacy of anti-PD-1/PD-L1 antibodies was associated with somatic mutation load of tumor tissue and subsequent neo-antigen number through the comparison of mis-match repair (MMR) proficient and MMR deficient patients [25-29]. The neo-antigens are now recognized as determinants for immune response of numerous immune-therapies [30-32]. The development in associated technologies, for instance neo-antigen predicting tools and antigenicity assessing tools, together with the decreasing cost of the next-generation sequencing, make scientist capable of identifying tumor specific neo-antigens, responsible for immune responses during immune-therapies [33-35].
Thus, in this review, we primarily throw discussion on the barriers that are limiting the applications of cancer vaccines. Moreover, thereafter, it would proceed with exploring the neo-antigens and lessons from the success of immune checkpoint inhibitor based immune-therapies for refining the cancer vaccines.
Obstacles limiting cancer vaccines
As stated earlier, the therapeutic efficacy of cancer vaccine is dependent on the differential expression of target antigens by tumor cells as well as normal cells [11]. That is why TSAs are theoretically given preference to TAAs in cancer vaccine design [14]. Since the first immunogenic antigen MAGE-1 was brought to light, several immunogenic antigens have been reported [36, 37]. But majority of them is shared TAAs that are expressed by both tumor cells and normal cells, despite the fact that, at a relative lower level, the on-target/off tumor effects pose to be potential threats [38, 39]. Furthermore, shared TAAs can be classified into three groups: 1) cancer-testis antigens; 2) tissue differentiation antigens; and 3) over-expressed antigens (table. 1) [40]. Conversely, TAAs are not taken into consideration as the optimal choices for cancer vaccine because of the two key reasons other than being shared by normal tissues: 1) low antigenicity, implying that TAAs are typically tolerated even as “self”, in this way, majority of vaccines targeting TAAs are put to application in means of combination with immunogenicity enhancers, like co-stimulatory cytokine granulocyte-macrophage colony stimulating factor (GM-CSF) together with TLR agonist, for instance, provenge is developed by loading DCs in vitro through co-culturing with prostatic acid phosphatase (PAP) and GM-CSF fusion protein [41, 42]; 2) heterogeneity, heterogeneity among tumor cells within the same tissue is likely to lead to the selection of TAA negative tumor cells, heterogeneity among patients are likely to lead to personalized cancer vaccine requirement that is with constrained application in clinic because of elevated cost and intense labor in personalized epitope identification [43, 44].
Classification of tumor antigens in cancer vaccine and related limitations
| Target type | Example | Limitations | Reference | |
|---|---|---|---|---|
| Shared TAAs | Cancer-testis antigens | NY-ESO-1, MAGE-A1, SSX-2 | 1. Low antigenicity; 2. Activate limited type of T cell responses; 3. Non-driver mutation resources; 4. On-target side effect on normal tissues. | [11] |
| Differentiation antigens | Gp100, Mart-1, PSA | [1] | ||
| Over-expressed antigens | hTERT, surviving, MUC1 | [27] | ||
| TSAs | Neo-antigens | EGFRVIII, ERBB2IPE805G, KRASG12D BRAFV600E | 1. Low antigenicity; 2. Activate limited type of T cell responses; 3. Non-driver mutation resources; 4. Difficult to identify. | [45-48] |
TAAs: tumor associated antigens, TSAs: tumor specific antigens, NY-ESO-1: New York esophageal carcinoma antigen 1, MAGE-A1: Melanoma-associated antigen 1, SSX2: synovial sarcoma X breakpoint 2; gp100: glycoprotein 100, Mart-1: melanoma antigen recognized by T-cells 1, PSA: prostate-specific antigen, hTERT: Human telomerase reverse transcriptase, MUC1: Mucin 1; EGFRVIII: epidermal growth factor receptor-variant VIII.
In theory, the ideal antigens for cancer vaccines are clonal immunogenic TSAs that are specifically expressed and shared by all the tumor cells together with being efficient in eliciting immune responses from hosts [11]. Nonetheless, with the paucity of TSAs, numerous alternative approaches have been suggested whereby one is targeting multiple TAAs or even full-length protein in combination with immune response enhancers for the purpose of better eliciting both CD4+ and CD8+ T cell responses, for instance PANVAC targets CEA and MUC-1 antigens and encodes enhancing sequences for both targets [49]. Despite that fact, the cancer vaccine is still encountering the challenges of low-antigenicity and heterogeneity. Thus, novel strategies are in desperately required to enhance the efficiency of cancer vaccines.
Lessons learned from immune checkpoint inhibitors
The immune checkpoint inhibitors based immunotherapies have attained exceptional achievements in addition to refreshing the field of cancer treatment [50]. The underlying mechanisms of checkpoint inhibitors and cancer vaccines are to some level similar, harnessing patients' own immune system to fight against cancers [51, 52]. In this way, numerous lessons can be learned from immune checkpoint inhibitor based immunotherapy to cancer vaccines.
Tumors are considered to have evolved from thousands of somatic mutations that develop tumor cells growth as well as survival benefits over normal cells [53, 54]. All through the development of tumors, tumor cells adopt numerous mechanisms for the purpose of escaping from the surveillance of host immune system, a major one is developing immune suppressive microenvironment in order to suppress the function of immune effectors, for instance CD8+ T cells, through expressing immune checkpoints, like CTLA-4, LAG-3, Tim-3 and PD-1 [55]. Through the discovery of this mechanism, there have been developed numerous for the purpose of targeting those immune checkpoints, such as PD-1 inhibitors, Nivolumab and Pembrolizumab that have been both appraised and approved by the FDA in respect of the treatment of melanoma, non-small cell lung cancer (NSCLC) and melanoma respectively [50]. Furthermore, the therapeutic efficiency of PD-1 inhibitors was reported having association with the somatic mutation load, which is coupled with the dysfunction of MMR system [38]. It implies that the higher somatic mutation load of the patients together with the higher number of neo-antigens will be encoded, and the higher response rate of the patients will be attained from PD-1 inhibitors [56]. In this way, the diversity of neo-antigens within the tumor tissues of patients poses to be a key determinant for immunotherapy response.
Targeting Immunogenic Neo-antigens
It has been revealed by several research works that, among neo-antigens, many are immunogenic, effective in eliciting immune response from host in vitro and in vivo [57, 58]. That is why identification of immunogenic neo-antigens possesses critical importance for the application of neo-antigens to cancer vaccines. With the development of associated technologies, for instance Next-generation Sequencing (NGS), peptide manufacturing and peptide immunogenicity in silico prediction, targeting tumor specific neo-antigen is now turning out to be a sound reality [33]. For the purpose of overcoming the low antigenicity issue, cancer vaccines can be designed in order to target immunogenic neo-antigens. For the purpose of tackling the heterogeneity issue, numerous immunogenic neo-antigens are advised to target in the meantime, in case of the selection of targeting neo-antigen negative tumor cells [56]. Among these somatic mutations that encode immunogenic neo-antigens, there are some considered to be driver mutations that are defined essential for the development of tumors [59]. This is how cancer vaccines can be further designed to target immunogenic neo-antigens that are derived from driver mutations. This point was affirmed by a recent research work, throwing light on the fact that the heterogeneity of neo-antigens within a single tumor tissue determines the immuno-reactivity as well as sensitivity to immune checkpoint inhibitors [60]. All through the development of tumors, the number of driver mutations boosts up. In this way, the early staged NSCLC patients possess higher number of clonal neo-antigens, clonally encoded and shared by tumor cells, together with the lower neo-antigen heterogeneity, associated with improved clinical performance of immune checkpoint inhibitor based immunotherapies [61]. Some scientists even proposed cancer immuno-prevention by cancer vaccines to individuals without cancers, but at high risk of having cancers [62].
Together with targeting multiple immunogenic neo-antigens or immunogenic clonal neo-antigens, it determines the efficiency of immune checkpoint inhibitors based immune-therapies. Another lesson can be learned, among cancers, melanoma is taken into account to be the most somatic mutation loaded cancer [59]. Furthermore, speculation can be drawn from this that melanoma patients possess the biggest number of neo-antigens, and perhaps the clonal neo-antigens, derived from driver mutations, some of which are shared by different types of cancers [63]. Consequently, the whole tumor cell derived from patients, containing shared clonal neo-antigens can be put to application as autologous cancer vaccines or even allogeneic cancer vaccines catering to HLA matching scenario. In the same fashion, autologous cancer vaccines or even allogeneic cancer vaccines can be made out of tumor tissues from the majority of responsive patients for the purpose of treating the same cancers or even different types of cancers under HLA matching scenario.
To conclude, there are at least three lessons that can be learned from immune checkpoint inhibitor based immunotherapies to cancer vaccines for the purpose of overcoming the low antigenicity and heterogeneity issues by targeting neo-antigens: 1) targeting multiple immunogenic neo-antigens; 2) targeting immunogenic clonal neo-antigens; 3) deriving tumor cell based cancer vaccines from the most immunogenic clonal neo-antigens loaded patients.
Combine with immune checkpoint inhibitors
Cancer vaccines are designed to bring forth the immunogenic antigens to excite patients' own immune system, particular tumor specific CD8+ T cell responses [1]. Immune checkpoint inhibitors are designed for the release of the patients' own effects or cells from suppressed state. Majority of patients is not responsive to immune checkpoint inhibitors because of the lack of tumor specific effector cells [64-66]. While, cancer vaccine has been revealed with the ability to elicit diverse neo-antigen specific effector cells [67, 68]. This is how it is quite adequate to apply cancer vaccines providing tumor specific T cells before immune checkpoint inhibitor based immunotherapies [67, 69]. It has been brought to light by the studies that this combination showcases more effectiveness in comparison with either mono-therapy by promotion of cytotoxic T cell activity, facilitation of effector T cell infiltration and accumulation of memory precursor CD8+ T Cells [70-72]. Therefore, cancer vaccines are considered to be perfectly matching immune checkpoint inhibitors [73]. Moreover, there are several ongoing clinical trials (table 2).
Part of clinical trials investigating combination of cancer vaccine with checkpoint inhibitors
| Agent | Malignance | Phase | Status/ results | NCT Identifier |
|---|---|---|---|---|
| GVAX+Nivolumab | Pancreatic cancer | I/II | Recruiting | NCT02451982 |
| DC AML Vaccine+CT-011 | Acute myelogenous leukemia | II | Recruiting | NCT01096602 |
| pTVG-HP Plasmid DNA Vaccine+ Pembrolizumab | Hormone-Resistant, Metastatic Prostate Cancer | I/II | Recruiting | NCT02499835 |
| DC Vaccines+ Nivolumab | Recurrent Grade III and Grade IV Brain Tumors | I | Recruiting | NCT02529072 |
| Provenge+ CT-011 | Advanced Prostate Cancer | II | Recruiting | NCT01420965 |
| GVAX+CRS207 With or Without Nivolumab | Metastatic Adenocarcinoma of the Pancreas | II | Recruiting | NCT02243371 |
| TLPLDC Vaccine+ checkpoint inhibitors | Metastatic Melanoma | I/II | Recruiting | NCT02678741 |
| Vigil™ vaccine+ Pembrolizumab | Advanced Melanoma | I | Recruiting | NCT02574533 |
| 6MHP+ Ipilimumab | Melanoma | I/II | Recruiting | NCT02385669 |
GVAX: Granulocyte-macrophage Colony-stimulating Factor (GM-CSF) Gene-transfected Tumor Cell Vaccine; DC: Dendritic Cell; AML: Acute Myelocytic Leukemia; CT-011: Pidilizumab, Pembrolizumab and Nivolumab, programmed cell death 1 blockade inhibitors; pTVG-HP: DNA vaccine encoding Prostatic acid phosphatase (PAP); CRS-207: live-attenuated Listeria vaccine expressing mesothelin; TLPLDC: tumor lysate particle-loaded dendritic cellvaccine; Vigil™: GMCSF/bi-shRNA furin DNA engineered autologous tumor cell product; 6MHP:six melanoma-associated helper peptides vaccine; Ipilimumab: cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) antibody.
For the purpose of further delivering diversely enough CD8+ T cells with cancer vaccines, numerous approaches can be followed in order to improve the efficiency of the combination of cancer vaccines and immune checkpoint inhibitors. It has been well documented that IFN-γ is capable of inducing the expression of MHC molecules from tumor cells [74, 75]. On these bases of this notion, in whole tumor cells design, for the purpose of better providing stimulations from diversely enough antigens, tumor cells can be cultured with suitable amount of IFN-γ possessing medium or genetically manipulated to express IFN-γ prior to the application in combination with immune checkpoint inhibitors. Moreover, the expression of PD-L1 on tumor cells that can be further induced by IFN-γ, is likely to stand for another factor that adversely influences the efficiency of tumor cell vaccines [76]. In this way it is interesting to carry out the investigation of the influence of knock-down or knock-out the expression of PD-L1 of tumor cell vaccines on the combination of cancer vaccines with immune checkpoint inhibitors.
Hence, in addition to targeting neo-antigens, there are still numerous lessons that can be learned from immune checkpoint inhibitor based immuno-therapies to cancer vaccines by combining with immune checkpoint inhibitors; 1) culturing tumor cells with IFN-γ containing medium prior to application as cancer vaccines in combination with immune checkpoint inhibitors; 2) genetically manipulating tumor cells to secrete IFN-γ prior to application as cancer vaccines in combination with immune checkpoint inhibitors; 3) ablating the expression of PD-L1 on tumor cells prior to application as cancer vaccines in combination with immune checkpoint inhibitors (table 3).
Lessons can be learned from immune checkpoint inhibitors to cancer vaccines
| Cancer Vaccine | Major Challenges | Strategies |
| Low antigenicity | Targeting neo-antigens: 1. targeting multiple immunogenic neo-antigens; 2. targeting clonal neo-antigens; 3. develop cancer vaccines from immune checkpoint inhibitor responsive tumor tissues. Combine with immune checkpoint inhibitors: 4. culturing tumor cells with IFN-γ containing medium ahead; 5. genetically manipulate tumor cells to secret IFN-γ ahead; 6. delete the expressing of PD-L1 on tumor cell vaccine. | |
| Heterogeneity |
Conclusion
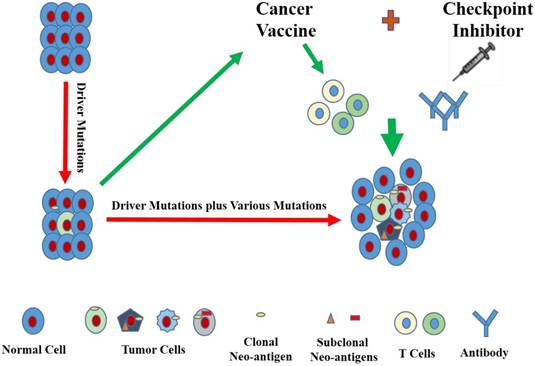
With the development of associated technologies, lessons can be learned to target neo-antigens of cancer vaccines from immune checkpoint inhibitors, and those neo-antigens should better be clonal and immunogenic. For the purpose of better provoking the patients' own immune responses, cancer vaccines together with immune checkpoint inhibitor can be perfectly combined with each other. Furthermore, additional modifications, such as knocking down the expression of PD-L1 or addition of IFN-γ secretion to tumor cell-based vaccines, can also be incorporated to better equip this combination (Fig. 1). Moreover, it goes without saying that the type of cancer vaccines, the reliability of neo-antigen identification tools, vector type, ratio dosage of cancer vaccines and immune checkpoint inhibitors and other factors are also required to be reckoned with.
Cancer vaccines can be designed in a way to target immunogenic neo-antigens, even clonal neo-antigens that are encoded by driver mutations. Furthermore, cancer vaccines can be put to application in combination with immune checkpoint inhibitors.
Acknowledgements
This work was received the support from the Capacity Building Project of the Science and Technology Commission of Shanghai Municipality (16DZ2281000).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Butterfield LH. Cancer vaccines. BMJ. 2015;350:h988
2. Zahm CD, Colluru V, McNeel DG. Vaccination with High-Affinity Epitopes Impairs Antitumor Efficacy by Increasing PD-1Expression on CD8+ T Cells. Cancer immunology research. 2017
3. Mahdavi M, Moreau V, Kheirollahi M. Identification of B and T cell epitope based peptide vaccine from IGF-1 receptor in breast cancer. Journal of molecular graphics & modelling. 2017;75:316-21
4. Filley AC, Dey M. Dendritic cell based vaccination strategy: an evolving paradigm. Journal of neuro-oncology. 2017
5. Small EJ, Lance RS, Gardner TA, Karsh LI, Fong L, McCoy C. et al. A Randomized Phase II Trial of Sipuleucel-T with Concurrent versus Sequential Abiraterone Acetate plus Prednisone in Metastatic Castration-Resistant Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:3862-9
6. Lilleby W, Gaudernack G, Brunsvig PF, Vlatkovic L, Schulz M, Mills K. et al. Phase I/IIa clinical trial of a novel hTERT peptide vaccine in men with metastatic hormone-naive prostate cancer. Cancer immunology, immunotherapy: CII. 2017
7. Obara W, Eto M, Mimata H, Kohri K, Mitsuhata N, Miura I. et al. A phase I/II study of cancer peptide vaccine S-288310 in patients with advanced urothelial carcinoma of the bladder. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28:798-803
8. Yamasaki S, Miura Y, Davydova J, Vickers SM, Yamamoto M. Intravenous genetic mesothelin vaccine based on human adenovirus 40 inhibits growth and metastasis of pancreatic cancer. International journal of cancer. 2013;133:88-97
9. Inoges S, Tejada S, de Cerio AL, Gallego Perez-Larraya J, Espinos J, Idoate MA. et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. Journal of translational medicine. 2017;15:104
10. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Advances in cancer research. 2013;119:421-75
11. Schlom J, Hodge JW, Palena C, Tsang KY, Jochems C, Greiner JW. et al. Therapeutic cancer vaccines. Advances in cancer research. 2014;121:67-124
12. Ye Z, Li Z, Jin H, Qian Q. Therapeutic Cancer Vaccines. Advances in experimental medicine and biology. 2016;909:139-67
13. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S. et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nature reviews Clinical oncology. 2014;11:509-24
14. Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Human vaccines & immunotherapeutics. 2014;10:3332-46
15. Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J. et al. Immune resistance orchestrated by the tumor microenvironment. Immunological reviews. 2006;213:131-45
16. The 150 most important questions in cancer research and clinical oncology series. questions 31-39: Edited by Chinese Journal of Cancer. Chinese journal of cancer. 2017;36:48
17. Morris VK, Salem ME, Nimeiri H, Iqbal S, Singh P, Ciombor K. et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. The Lancet Oncology. 2017;18:446-53
18. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H. et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. The New England journal of medicine. 2016;375:1845-55
19. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J. et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. The Lancet Oncology. 2017;18:312-22
20. Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6958-62
21. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A. et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. The New England journal of medicine. 2016;375:143-53
22. Postow MA. Managing immune checkpoint-blocking antibody side effects. American Society of Clinical Oncology educational book / ASCO American Society of Clinical Oncology Meeting. 2015:76-83
23. Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ. et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer immunology research. 2016;4:345-53
24. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF. et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. The Lancet Oncology. 2016;17:1497-508
25. Lee V, Le DT. Efficacy of PD-1 blockade in tumors with MMR deficiency. Immunotherapy. 2016;8:1-3
26. Kelderman S, Schumacher TN, Kvistborg P. Mismatch Repair-Deficient Cancers Are Targets for Anti-PD-1 Therapy. Cancer cell. 2015;28:11-3
27. Asaoka Y, Ijichi H, Koike K. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;373:1979
28. Diaz LA Jr, Le DT. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;373:1979
29. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;372:2509-20
30. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. The Journal of clinical investigation. 2015;125:3413-21
31. Franzese O, Torino F, Fuggetta MP, Aquino A, Roselli M, Bonmassar E. et al. Tumor immunotherapy: drug-induced neoantigens (xenogenization) and immune checkpoint inhibitors. Oncotarget. 2017;8:41641-69
32. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S. et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-oncology. 2017
33. Desrichard A, Snyder A, Chan TA. Cancer Neoantigens and Applications for Immunotherapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:807-12
34. Lutz ER, Jaffee EM. Can we predict mutant neoepitopes in human cancers for patient-specific vaccine therapy? Cancer immunology research. 2014;2:518-21
35. Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. Journal of immunological methods. 2011;374:26-34
36. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643-7
37. Zhou F, Shang W, Yu X, Tian J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Medicinal research reviews. 2017
38. Bright RK, Bright JD, Byrne JA. Overexpressed oncogenic tumor-self antigens. Human vaccines & immunotherapeutics. 2014;10:3297-305
39. Kaelin WG Jr. Common pitfalls in preclinical cancer target validation. Nature reviews Cancer. 2017;17:425-40
40. Ilyas S, Yang JC. Landscape of Tumor Antigens in T Cell Immunotherapy. J Immunol. 2015;195:5117-22
41. Arlen PM, Wood LV. Prostate cancer vaccines: moving therapeutic vaccination forward in the post-Provenge era. Expert review of vaccines. 2012;11:287-302
42. Kumai T, Kobayashi H, Harabuchi Y, Celis E. Peptide vaccines in cancer-old concept revisited. Current opinion in immunology. 2017;45:1-7
43. Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C. et al. Toward understanding and exploiting tumor heterogeneity. Nature medicine. 2015;21:846-53
44. Sayour EJ, Mitchell DA. Manipulation of Innate and Adaptive Immunity through Cancer Vaccines. Journal of immunology research. 2017;2017:3145742
45. Neagu MR, Reardon DA. An Update on the Role of Immunotherapy and Vaccine Strategies for Primary Brain Tumors. Current treatment options in oncology. 2015;16:54
46. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641-5
47. Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P. et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clinical and translational gastroenterology. 2016;7:e157
48. Ritterhouse LL, Barletta JA. BRAF V600E mutation-specific antibody: A review. Seminars in diagnostic pathology. 2015;32:400-8
49. Baxevanis CN. Outlining novel scenarios for improved therapeutic cancer vaccines: the PANVAC paradigm. Expert review of vaccines. 2012;11:275-7
50. Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell. 2016;165:272-5
51. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. The Journal of clinical investigation. 2015;125:3401-12
52. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450-61
53. Yates LR, Campbell PJ. Evolution of the cancer genome. Nature reviews Genetics. 2012;13:795-806
54. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J. et al. Exploiting the mutanome for tumor vaccination. Cancer research. 2012;72:1081-91
55. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl). 2015
56. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483-9
57. Kreiter S, Castle JC, Tureci O, Sahin U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology. 2012;1:768-9
58. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387-90
59. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
60. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-9
61. Hutchinson L. Immunotherapy: Clonal neoantigens and immune response: a balancing act. Nature reviews Clinical oncology. 2016
62. Finn OJ, Beatty PL. Cancer immunoprevention. Current opinion in immunology. 2016;39:52-8
63. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H. et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic acids research. 2015;43:D805-11
64. Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME. et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Annals of oncology: official journal of the European Society for Medical Oncology. 2015;26:2375-91
65. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nature reviews Clinical oncology. 2016;13:143-58
66. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017
67. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803-8
68. Zhang X, Sharma PK, Peter Goedegebuure S, Gillanders WE. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine. 2017;35:1094-100
69. Kleponis J, Skelton R, Zheng L. Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer biology & medicine. 2015;12:201-8
70. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y. et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1-11
71. Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer immunology research. 2016;4:95-100
72. Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW. et al. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer research. 2014;74:2974-85
73. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205-14
74. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. International reviews of immunology. 2009;28:239-60
75. Giroux M, Schmidt M, Descoteaux A. IFN-gamma-induced MHC class II expression: transactivation of class II transactivator promoter IV by IFN regulatory factor-1 is regulated by protein kinase C-alpha. J Immunol. 2003;171:4187-94
76. Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K. et al. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. British journal of cancer. 2015;112:1501-9
Author contact
![]() Corresponding authors: Qi-Jun Qian, Tel: + 86-21-81875371; Fax: + 86-21-65580677; E-mail: qianqjcn; Hua-Jun Jin, Tel: + 86-21-81875372; E-mail: hj-jincom
Corresponding authors: Qi-Jun Qian, Tel: + 86-21-81875371; Fax: + 86-21-65580677; E-mail: qianqjcn; Hua-Jun Jin, Tel: + 86-21-81875372; E-mail: hj-jincom