Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(2):331-345. doi:10.7150/jca.18188 This issue Cite
Research Paper
MiR-378 and MiR-1827 Regulate Tumor Invasion, Migration and Angiogenesis in Human Lung Adenocarcinoma by Targeting RBX1 and CRKL, Respectively
Chai San Ho1, Suzita Mohd Noor2, Noor Hasima Nagoor3 ![]()
1. Institute of Biological Sciences, Division of Genetics and Molecular Biology, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia;
2. Department of Biomedical Science, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia;
3. Institute of Biological Sciences, Division of Genetics and Molecular Biology, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia; Centre for Research in Biotechnology for Agriculture (CEBAR), University of Malaya, 50603 Kuala Lumpur, Malaysia.
Received 2016-11-3; Accepted 2017-5-3; Published 2018-1-1
Abstract
MicroRNAs (miRNAs) have been extensively studied over the decades and have been proposed as potential molecular targets for cancer treatment. Studies have shown that miR-378 participates in numerous biological processes in various cancers; whereas miR-1827 has only been reported in pediatric glioma. The mechanism of how miRNAs modulate lung cancer metastasis remains unclear. Our previous study demonstrated that miR-378 is up-regulated while miR-1827 is down-regulated in high invasive lung cancer sub-cell lines, and their biological functions have been described. Here, we report that miR-378 and miR-1827 modulate lung cancer cell invasion and migration via epithelial-mesenchymal transition (EMT). We also demonstrated that cells treated with miR-378 inhibitors or miR-1827 mimics had reduced number of metastases and ectopic vessels in the zebrafish embryo model. We then showed that miR-378 promoted invasion and miR-1827 suppressed migration by targeting RBX1 and CRKL, respectively. Restored protein expression in miRNA-overexpressed/ miRNA-suppressed cells attenuated the inhibitory/ inducing effect of the miRNA on lung cancer cells. Collectively, our findings highlight that miR-378 and miR-1827 could serve as novel therapeutic targets in lung cancer.
Keywords: MicroRNAs, Lung Cancer, Invasion, Migration, Angiogenesis, EMT.
Introduction
Lung cancer continues to be the leading cause of cancer mortality worldwide [1]. The overall five-year survival rate for lung cancer patients has been stagnant in spite of the introduction of modern therapeutic interventions such as chemotherapy, radiotherapy and surgical resection [2]. This is likely due to the late presentation of the disease, where tumor cells have migrated to the other parts of the body, known as metastasis [3]. Therefore, there is an urgent need to develop more effective molecular targeted therapy that halts lung cancer tumorigenesis.
MicroRNAs are short endogenous non-coding RNAs that inhibit protein translation by binding most commonly to the 3'-untranslated regions (3'-UTRs) of their target mRNAs [4]. Accumulating evidence has indicated that miRNAs are dysregulated in various human cancers including lung cancer and that they regulate processes such as cell proliferation, apoptosis, angiogenesis, and metastasis, acting as either oncogenes or tumor suppressors [5-8]. Therefore, identification of specific miRNAs that play crucial roles in tumorigenesis offers value for miRNAs as diagnosis, prognosis and therapeutic tools.
Previously, we found that miR-378 is overexpressed while miR-1827 is under-expressed in high invasive A549 and SK-LU-1 non-small cell lung cancer (NSCLC) sub-cell lines. Moreover, we revealed their specific functions in controlling cell invasion, migration and angiogenesis [9]. However, the effects of these miRNAs in vivo and the detailed mechanism of the biological regulation by these miRNAs were yet to be evaluated. In this study, we report that miR-378 inhibitors and miR-1827 mimics significantly inhibited tumor dissemination and ectopic vessel formation of high invasive A549 cells in the zebrafish embryo model. In addition, we also show that RBX1 and CRKL are directly targeted by miR-378 and miR-1827 respectively, to mediate invasion or migration in lung cancer cells, and that these two processes involve EMT. Taken together, our data reveal a novel regulatory function for lung cancer metastasis and propose that miR-378 and miR-1827 might serve as potential targets for lung cancer therapy.
Materials and Methods
Cell Lines
Human lung adenocarcinoma epithelial cell lines, A549 (American Type Culture Collection, ATCC, VA, USA) was cultured in Roswell Park Memorial Institute 1640 (HyClone, UT, USA), while SK-LU-1 (ATCC, VA, USA) was cultured in minimum essential medium α (Nacalai Tesque, Kyoto, Japan). All media were supplemented with fetal bovine serum (FBS) (HyClone, Northumberland, UK) and penicillin/ streptomycin (Lonza, MD, USA). Human umbilical vein endothelial cells (HUVEC) (ATCC, VA, USA) were cultured in Medium 200 (Gibco, CA, USA) supplemented with large vessel endothelial supplement (Gibco, CA, USA) and used below passage-7. All cells were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Cell Line Authentication
Human lung adenocarcinoma epithelial cell lines, A549 and SK-LU-1, were authenticated by short tandem repeat (STR) profiling by Genetica DNA Laboratories, NC, USA. Fifteen STR loci and the gender identity locus amelogenin were profiled using PowerPlex 16 HS (Promega, WI, USA). Comparison to the ATCC database of A549 and SK-LU-1 cell lines reference profiles was carried out and 100% match was obtained.
Serial Selection of Low and High Invasive Sub-cell Lines
Serial selection of low and high invasive A549 and SK-LU-1 sub-cell lines from their parental populations was described previously [9]. Briefly, selection was performed using transwell invasion assay protocol, with the difference being both the invasive cells at the bottom membrane and non-invasive cells on the top membrane were harvested and subjected to sequential selection up to the seventh generation. High invasive sub-cell lines were denoted as A549-I7 and SK-LU-1-I7 while low invasive sub-cell lines were denoted as A549-NI7 and SK-LU-1-NI7.
Transfection
MiRIDIAN hsa-miR-378 and hsa-miR-1827 mimics and hairpin inhibitors, as well as ON-TARGET plus SMARTpool small interfering RNAs (siRNAs) targeting human ring-box 1 gene (RBX1) and v-crk avian sarcoma virus CT10 oncogene homolog-like proto-oncogene (CRKL) were purchased from Dharmacon, CO, USA. MiRIDIAN non-targeting mimics and hairpin inhibitors, and ON-TARGET plus SMARTpool non-targeting siRNAs were used as negative controls. Mammalian expression plasmids (pCMV-AC-RBX1-GFP and pCMV-AC-CRKL-GFP) containing the open reading frames (ORFs) of RBX1 and CRKL without the 3'-UTRs were purchased from OriGene, MD, USA. Empty plasmids (pCMV-AC-GFP) served as negative control. MicroRNA mimics, microRNA hairpin inhibitors, siRNAs, plasmids and negative controls were transfected into the cells using DharmaFECT 1 transfection reagent (Dharmacon, CO, USA), according to the manufacturer's instructions.
Protein Isolation and Western Blot
Total protein was extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, MA, USA) and the protein concentration was determined using BCA Protein Assay Kit (Thermo Scientific, MA, USA), according to the manufacturer's protocols. Equal amount of protein was fractionated using SDS-PAGE before transfer to nitrocellulose membranes (Bio-Rad Laboratories, CA, USA). Membranes were blocked with bovine serum albumin (AMRESCO, OH, USA) or non-fat skim milk (Merck, Hesse, Germany) and were then incubated with primary antibodies: β-catenin rabbit monoclonal antibody (1:1000) (Cell Signaling Technology, MA, USA), vimentin rabbit monoclonal antibody (1:1000) (Cell Signaling Technology, MA, USA), RBX1 rabbit monoclonal antibody (1:000) (Cell Signaling Technology, MA, USA) or CRKL mouse monoclonal antibody (1:1000) (Cell Signaling Technology, MA, USA). GAPDH rabbit monoclonal antibody (1:10000) (Cell Signaling Technology, MA, USA) was used as endogenous control. Protein expression was detected with WesternBright Quantum HRP substrate (Advansta, CA, USA), visualized on FUSION FX7 Image and Analytics System (Vilber Lourmat, Eberhardzell, Germany), and quantified using ImageJ v1.49.
Experimental Animals
Wild type zebrafish embryos (Danio rerio) at 2 days post fertilization (dpf) were used in the experiments. All animal procedures were performed according to protocols approved by the Faculty of Medicine Institutional Animal Care and Use Committee (IACUC), University of Malaya. Wild type zebrafish adults (Danio rerio) were housed in the Zebrafish Laboratory (Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited), University of Malaya, under standard conditions (14 h: 10 h light: dark cycle, regulated conductivity, pH and temperature), in a ZebTEC zebrafish housing system (Tecniplast, VA, Italy). Embryos were incubated and allowed to develop at 28.5 °C in system water containing methylene blue (Sigma Aldrich, MO, USA) and transferred into system water added with N-Phenylthiourea (PTU) (Sigma Aldrich, MO, USA) at 1 dpf to inhibit melanization.
In Vivo Metastasis Model
Transfected A549-I7 cells (hsa-miR-378 inhibitors, hsa-miR-1827 mimics, scrambled inhibitors and scrambled mimics) were labeled with 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate (DiI) stain (Invitrogen, CA, USA) and resuspended at a density of 2 × 105 cells/mL. Two-dpf zebrafish embryos were dechorionated and anesthetized with benzocaine (Sigma Aldrich, MO, USA) before injection. Approximately 100 DiI-labeled A549-I7 cells were injected into the middle of the embryonic yolk sac region using Eppendorf FemtoJet and InjectMan NI 2 (Eppendorf, Hamburg, Germany) with 20µm TransferTip (Eppendorf, Hamburg, Germany), aided by the fully automated Leica M205 A stereo microscope (Leica Microsystems, Wetzlar, Germany). The embryos were maintained in system water containing PTU at 28.5 °C. After confirmation of visible fluorescent cell mass at the injection site, embryos were transferred to a 37 °C incubator. At 24 hours post injection (hpi), living zebrafish embryos were anesthetized using benzocaine and mounted with VECTASHIELD (Vector Laboratories, CA, USA). Serial sections were captured using Leica TCS SP5 II confocal microscope (Leica Microsystems, Wetzlar, Germany) at low magnification (5×) to analyze the tumor cell dissemination pattern throughout the fish. The Fiji (Fiji is Just ImageJ) software package (National Institutes of Health, NIH, MD, USA) [10] was used for cell counting. A 190-255 fluorescence intensity threshold was set to select cells.
Whole-Mount Alkaline Phosphatase (ALP) Staining
Briefly, the 24-hpi zebrafish embryos were euthanized with an overdose of benzocaine and fixed in 4% paraformaldehyde (Sigma Aldrich, MO, USA) overnight at 4 °C. Fixed embryos were subsequently permeabilized with serial dehydration and rehydration in methanol (Merck, Hesse, Germany), and then equilibrated with ALP buffer (100mM NaCl, 100mM Tris-HCl, pH 9.5, 50mM MgCl2, 0.1% Tween 20), followed by incubation in staining solution (110µg/mL NBT, 55µg/mL BCIP) at 37 °C until the required staining was attained. The reaction was terminated with stop buffer (0.25mM EDTA in PBST, pH 5.5). The embryos were mounted with glycerol (Fisher Scientific, MA, USA) and photographed under a stereo microscope. Fiji was used to analyze the number and length of ectopic vessels [11].
Prediction of Candidate Target Genes
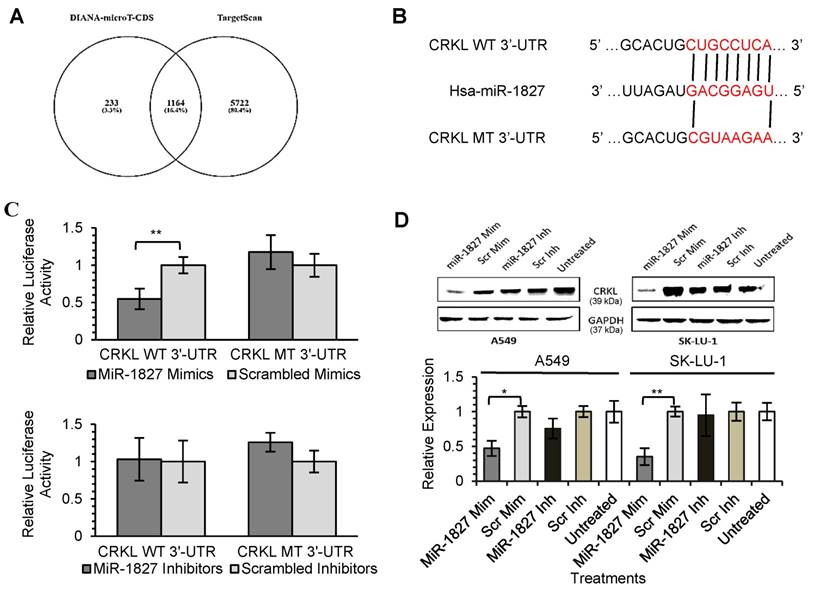
Putative hsa-miR-378 and hsa-miR-1827 targets were generated using publicly available programs: DIANA-microT-CDS v5.0 (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index) [12] and TargetScan v7.1 (http://www.targetscan.org/vert_71/) [13]. Hypothetical miRNA targets were subjected to gene-annotation enrichment analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 (https://david-d.ncifcrf.gov/) [14]. We summarized the results from these programs as candidate targets of hsa-miR-378 and hsa-miR-1827 using Venny v2.1 (http://bioinfogp.cnb.csic.es/tools/venny/) [15].
Luciferase Plasmid Construction and Dual Luciferase Reporter Assay
The 3'-UTRs of RBX1 and CRKL that contain the predicted hsa-miR-378 or hsa-miR-1827 binding site (wild type, WT) were amplified by PCR. Wild type 3'-UTR was cloned downstream of the firefly luciferase gene of the pmirGLO dual luciferase miRNA target expression vector (Promega, WI, USA) to generate pmirGLO-RBX1 WT 3'-UTR and pmirGLO-CRKL WT 3'-UTR vectors. These plasmids were mutated (MT) at the nucleotides that interact with the seed region of hsa-miR-378 or hsa-miR-1827 using QuikChange Multi Site-Directed Mutagenesis Kit (Agilent Technologies, CA, USA), to produce pmirGLO-RBX1 MT 3'-UTR and pmirGLO-CRKL MT 3'-UTR. All constructs were verified by sequencing. A549-I7 cells were seeded prior to co-transfection of hsa-miR-378 mimics/ inhibitors or hsa-miR-1827 mimics/ inhibitors or scrambled mimics/ inhibitors and WT/ MT 3'-UTR plasmids. Cell lysates were harvested 48 hours later and luciferase activities were measured on Dual Luciferase Reporter Assay System (Promega, WI, USA), according to the manufacturer's instructions. Firefly luciferase activity was normalized to Renilla luciferase activity, which was used as an internal control.
Transwell Invasion Assay
Cell invasion assay was performed using transparent PET membrane inserts (Corning, NY, USA). In brief, cells in serum free medium were seeded on the upper compartment of the chamber pre-coated with Matrigel (Corning, NY, USA). The lower chamber was filled with medium with 20% FBS as a chemo-attractant. After being incubated for 22 hours, the non-invasive cells on the upper surface were removed by scraping with a cotton swab. Invaded cells on the lower membrane surface were fixed in ethanol (Fisher Scientific, MA, USA) and stained with methylene blue. Cell invasion was quantified by counting invaded cells in each well under Nikon ECLIPSE TS100 inverted microscope (Nikon, Tokyo, Japan) using a 20× objective.
Wound Healing Assay
A scratch was created across the center of the well containing 70-80% confluent cells and cells were allowed to migrate for 28 hours. Images were captured under the inverted microscope at 100× magnification. The gap distance was evaluated using TScratch v7.8 [16].
In vitro Tube Formation Assay
HUVEC were added to plate pre-coated with Geltrex LDEV-free reduced growth factor basement membrane matrix (Gibco, CA, USA) and incubated with tumor-conditioned medium (TCM) for 15 hours. Tubes formed were stained using calcein-AM dye (Invitrogen, CA, USA) and photographed with Nikon ECLIPSE TS100 inverted fluorescence microscope (Nikon, Tokyo, Japan) using a 5× objective. The images were analyzed using WimTube (Wimasis, Munich, Germany) to quantify angiogenesis.
Statistical Analyses
All experiments were performed in at least triplicates and data are expressed as mean ± standard error of mean (SEM). The statistical significance of the difference between two groups of data was evaluated using Student's t-test. P < 0.05 (*) and P < 0.01 (**) were considered statistically significant.
Results
MiR-378 and MiR-1827 Regulate Lung Cancer Metastasis via EMT
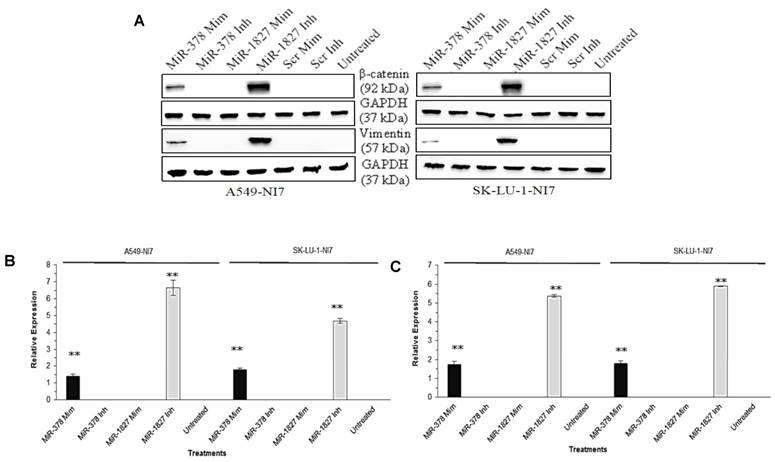
We have thus far demonstrated that miR-378 could induce invasion and angiogenesis while miR-1827 could suppress migration and angiogenesis of lung cancer cells in vitro [9]. While EMT is considered as an early stage in tumor metastasis, we aimed to examine the effects of miR-378 and miR-1827 on EMT. EMT is characterized by marker changes, such as dissociation of β-catenin from E-cadherin and accumulation in the cytosol before translocation into the nucleus, as well as induction of vimentin. As shown in Fig. 1, overexpression of miR-378 significantly increased the expression of β-catenin and vimentin in A549-NI7 and SK-LU-1-NI7. Likewise, inhibition of endogenous miR-1827 amplified the expression of β-catenin and vimentin in low invasive A549 and SK-LU-1 cells.
MiR-378 and miR-1827 regulate the EMT. (A) Western blot for the expression of β-catenin and vimentin in A549-NI7 and SK-LU-1-NI7 transfected with miR-378 mimics/ inhibitors or miR-1827 mimics/ inhibitors or scrambled mimics/ inhibitors. GAPDH served as an internal control. (B) Expression of β-catenin (epithelial marker) post transfection. MiR-378 induces while miR-1827 represses EMT. (C) The protein level of vimentin (mesenchymal marker) was determined by Western blot after transfection.
MiR-378 Promotes while MiR-1827 Inhibits Metastasis and Angiogenesis In Vivo
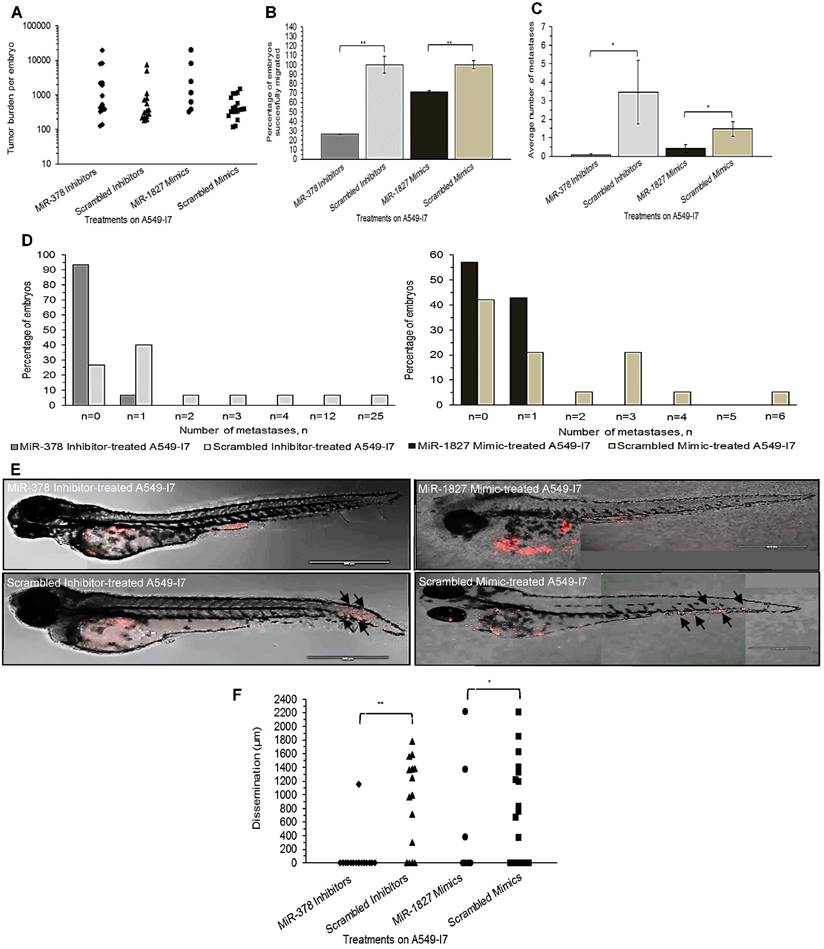
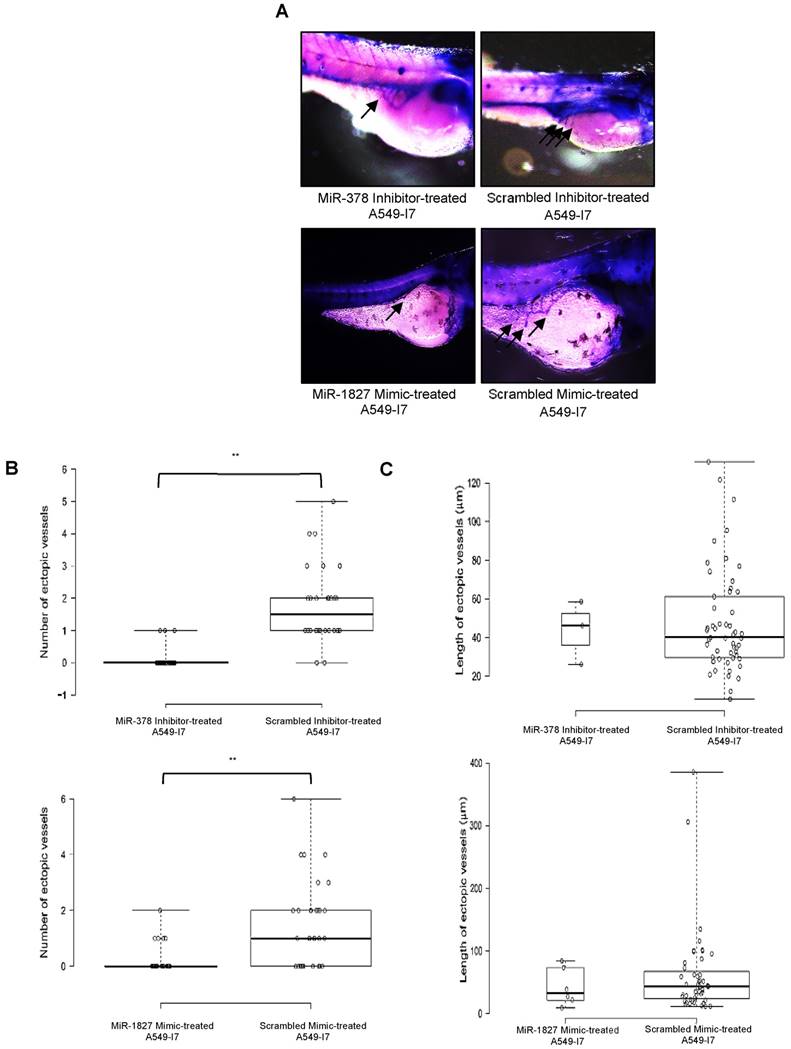
We next investigated whether these miRNAs would have a similar function in vivo using the zebrafish embryo xenograft model. We injected A549-I7 cells transfected with either miR-378 inhibitors/ miR-1827 mimics or negative control miRNAs (miR-NC) into the yolk sacs of 2-dpf zebrafish embryos and monitored tumor dissemination at 24 hpi. The number of embryos with successfully migrated tumor cells and the mean number of metastases in the miR-378 inhibitors and miR-1827 mimics groups were significantly lower than that in the miR-NC groups (Fig. 2). When the zebrafish embryos were stained for endogenous alkaline phosphatase present in endothelial cells, the mean number of tumor-induced ectopic vessels in the miR-378 inhibitors and miR-1827 mimics groups was significantly lower than those from the miR-NC groups (Fig. 3). These observations indicated that anti-miR-378 and miR-1827 could suppress lung cancer metastasis and angiogenesis in vivo.
MiR-378 inhibitor- and miR-1827 mimic-treated A549-I7 cells had reduced number of metastases throughout the body of the zebrafish embryos. (A) Tumor burden per zebrafish embryo of four treatment groups, calculated by multiplying the number of cell clusters and the mean area of the cell clusters present in the embryo. (B) Number of embryos bearing successfully migrated tumor cells, expressed in percentage, was significantly lower in the miR-378 inhibitor- and miR-1827 mimic-treated groups than the scrambled inhibitor- and scrambled mimic-treated groups. (C) Metastasis was defined when there were ≥ 5 cells outside the yolk sac region. Average number of metastases was significantly higher in the scrambled inhibitor- and scrambled mimic-treated groups. (D) Bar charts detailing the percentage of embryos bearing a particular number of metastases. (E) Confocal imaging showing dispersion of treated A549-I7 cells (red) throughout the fish body. MiR-378 inhibitor- and miR-1827 mimic-treated cells were mostly confined in the yolk sac while scrambled inhibitor- and scrambled mimic-treated cells can be seen to have spread to the tail. (F) Migration was quantified by calculating the average distance between each cluster of tumor cells and site of injection (SOI). NMiR-378 inhibitor-treated = 15; NScr inhibitor-treated = 15; NMiR-1827 mimic-treated = 7; NScr mimic-treated = 19
MiR-378 inhibitor- and miR-1827 mimic-treated A549-I7 cells mitigated tumor angiogenesis in embryonic zebrafish. (A) Representative images of the zebrafish embryos subjected to whole-mount ALP staining. Arrows in black depict the ectopic vessel(s) induced by tumor cells implanted. (B) Quantification of number of ectopic vessels originating from the developing SIV in each embryo of four treatment groups. Scrambled inhibitor- and scrambled mimic-treated cells induced robust neovascular response, whereas miR-378 inhibitor- and miR-1827 mimic-treated cells were less angiogenic. (C) Quantification of length of ectopic vessels originating from the developing SIV in each embryo of four treatment groups. NMiR-378 inhibitor-treated = 44; NScr inhibitor-treated = 30; NMiR-1827 mimic-treated = 30; NScr mimic-treated = 34
Identification of RBX1 and CRKL as a Direct Target of MiR-378 and MiR-1827, Respectively
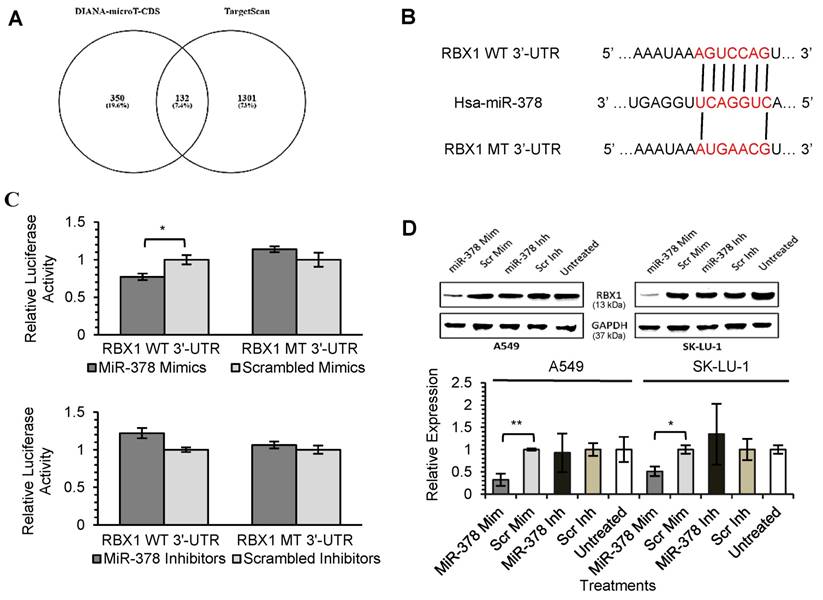
As miRNAs are known to exert their functions by hindering the translation of mRNAs into proteins, we attempted to identify the targets directly regulated by miR-378 and miR-1827. Using 2 different online algorithms (DIANA-microT-CDS and TargetScan) (Fig. 4A & 5A) together with enrichment analysis by DAVID, we identified common targets predicted by all 3 methods. In total, 8 (miR-378) and 59 (miR-1827) putative target genes were identified (Table S1 & S2). RBX1 and CRKL were chosen based on the scores and novelty in interaction with miR-378 and miR-1827. The putative miR-378 or miR-1827 binding sites in RBX1 and CRKL mRNAs were illustrated in Fig. 4B & 5B. We then performed dual luciferase reporter assay to determine whether their expression could indeed be directly regulated by corresponding miRNA. Luciferase reporter plasmids encoding the WT or MT 3'-UTR regions of RBX1 and CRKL mRNAs were constructed. MT 3'-UTR was achieved by substituting the binding site. They were co-transfected along with miR-378 mimics/ inhibitors or miR-1827 mimics/ inhibitors or scrambled mimics/ inhibitors into A549-I7 cells. As shown in Fig. 4C, luciferase activities in cells co-transfected with miR-378 mimics and pmirGLO-RBX1 WT 3'-UTR were significantly lower than the combination containing either pmirGLO-RBX1 MT 3'-UTR or scrambled mimics. We found similar changes in luciferase activities where CRKL WT 3'-UTR was significantly repressed by miR-1827 (Fig. 5C). No significant differences in luciferase activities was observed between cells co-transfected with miRNA inhibitors and pmirGLO-WT 3'-UTR versus scrambled inhibitors and pmirGLO-WT 3'-UTR, or between cells co-transfected with miRNA inhibitors and pmirGLO-MT 3'-UTR versus scrambled inhibitors and pmirGLO-MT 3'-UTR (Fig. 4C & 5C).
MiR-378 directly targets RBX1. (A) Diagram illustrating overlapping gene targets predicted by two online algorithms (DIANA-microT-CDS and TargetScan) for miR-378. (B) Schematic representation of the putative miR-378 target site in 3'-UTR of RBX1 mRNA. (C) Luciferase assay results from A549-I7 cells co-transfected with the RBX1 WT 3'-UTR or RBX1 MT 3'-UTR reporter plasmids and either miR-378 mimics/ inhibitors or scrambled mimics/ inhibitors. Firefly luciferase activity was normalized to Renilla luciferase activity (Firefly/ Renilla). (D) Overexpression of miR-378 down-regulated the expression of RBX1, in both A549 and SK-LU-1 cells. The expression was normalized to GAPDH.
MiR-1827 directly targets CRKL. (A) Diagram illustrating overlapping gene targets predicted by two online algorithms (DIANA-microT-CDS and TargetScan) for miR-1827. (B) Schematic representation of the putative miR-1827 target site in 3'-UTR of CRKL mRNA. (C) Luciferase assay results from A549-I7 cells co-transfected with the CRKL WT 3'-UTR or CRKL MT 3'-UTR reporter plasmids and either miR-1827 mimics/ inhibitors or scrambled mimics/ inhibitors. Firefly luciferase activity was normalized to Renilla luciferase activity (Firefly/ Renilla). (D) Overexpression of miR-1827 down-regulated the expression of CRKL, respectively, in both A549 and SK-LU-1 cells. The expression was normalized to GAPDH.
To further demonstrate the regulation of RBX1 and CRKL by miR-378 and miR-1827, Western blot analyses were conducted to detect alterations in their protein expression level after miRNA transfection. RBX1 protein expression was significantly reduced following miR-378 overexpression in A549 and SK-LU-1 (Fig. 4D). In addition, we found that similar CRKL protein down-regulation occurred in both A549 and SK-LU-1 when miR-1827 was overexpressed (Fig. 5D). Taken together, these results suggested that RBX1 and CRKL are directly targeted and negatively regulated by miR-378 and miR-1827.
MiR-378 Regulates Lung Cancer Cell Invasion by Down-regulating RBX1
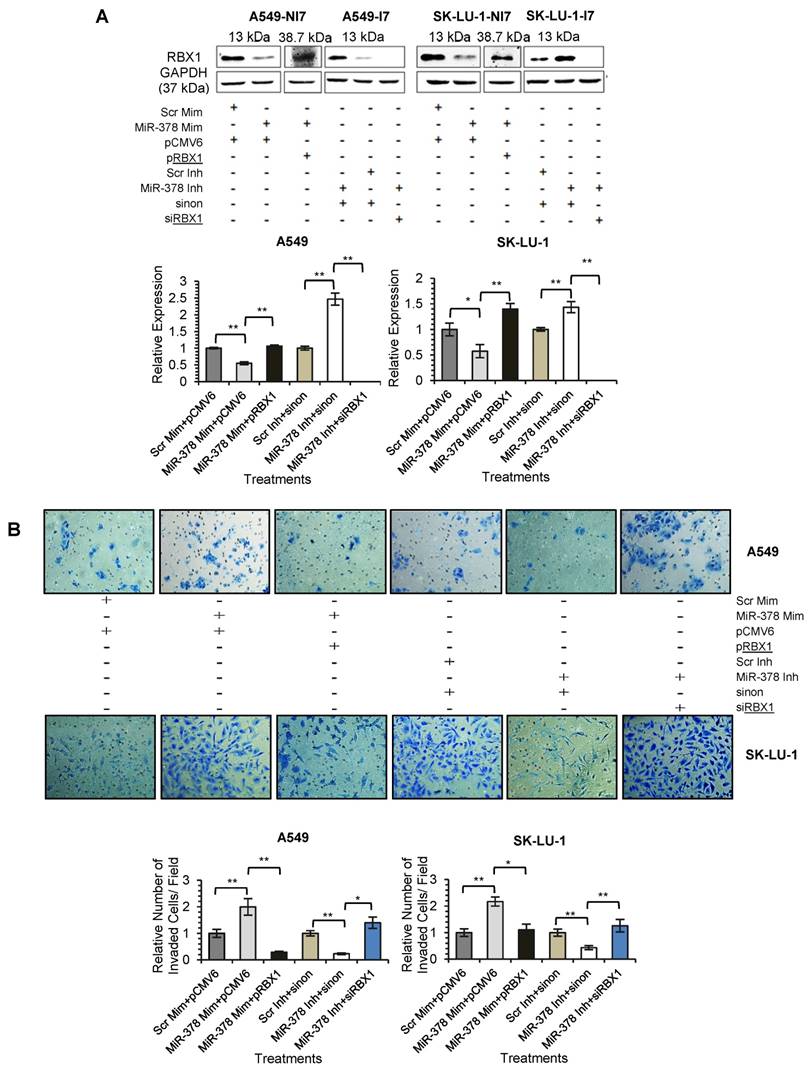
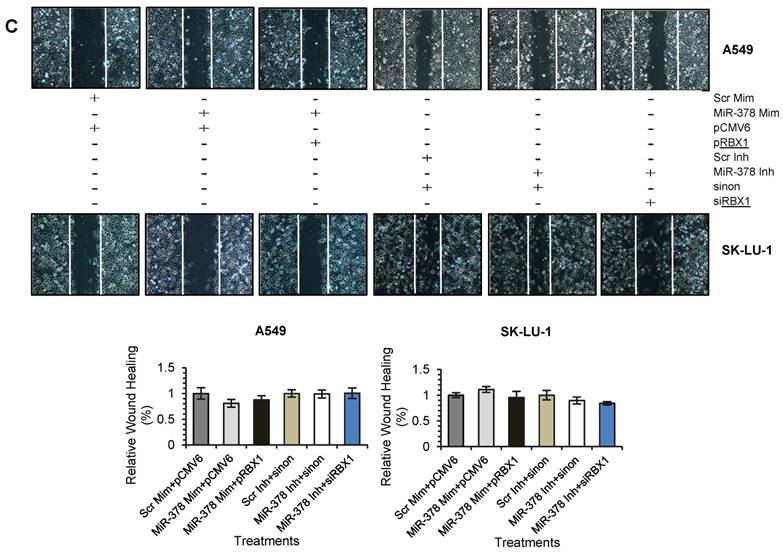
We next assessed the functional role of RBX1 in cell invasion under miR-378 regulation. We carried out co-transfection of miR-378 mimics and pCMV-AC-RBX1-GFP or empty vector (pCMV-AC-GFP) into A549-NI7 and SK-LU-1-NI7. Co-transfection of scrambled mimics and pCMV-AC-GFP was used for normalization. Knockdown experiment was also performed by co-transfecting miR-378 inhibitors and siRBX1 or non-targeting siRNA (sinon) while co-transfection of scrambled inhibitors and sinon was used as negative control. The transfection efficiencies were verified by 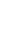 Western blot and subsequently transwell invasion assay and wound healing assay were performed with the treated lung cancer cells. The overexpression of RBX1, which lacks 3'-UTR, significantly reversed the inhibition of endogenous RBX1 by miR-378 in A549-NI7 and SK-LU-1-NI7 (Fig. 6A). Furthermore, ectopic RBX1 expression successfully restored the non-aggressiveness but not the non-migratory ability of A549-NI7. Similar results were observed in SK-LU-1-NI7 (Fig. 6B & 6C). In contrast, RBX1 silencing abolished the effect of miR-378 knockdown in A549-I7 and SK-LU-1-I7 (Fig. 6A). Consistent with this, the non-invasive effect induced by miR-378 inhibitors could be rescued (Fig. 6B). However, no significant effect was seen in scratch assay (Fig. 6C). Collectively, our data revealed that RBX1 participates in the miR-378-dependent regulation of lung cancer cell invasion.
Western blot and subsequently transwell invasion assay and wound healing assay were performed with the treated lung cancer cells. The overexpression of RBX1, which lacks 3'-UTR, significantly reversed the inhibition of endogenous RBX1 by miR-378 in A549-NI7 and SK-LU-1-NI7 (Fig. 6A). Furthermore, ectopic RBX1 expression successfully restored the non-aggressiveness but not the non-migratory ability of A549-NI7. Similar results were observed in SK-LU-1-NI7 (Fig. 6B & 6C). In contrast, RBX1 silencing abolished the effect of miR-378 knockdown in A549-I7 and SK-LU-1-I7 (Fig. 6A). Consistent with this, the non-invasive effect induced by miR-378 inhibitors could be rescued (Fig. 6B). However, no significant effect was seen in scratch assay (Fig. 6C). Collectively, our data revealed that RBX1 participates in the miR-378-dependent regulation of lung cancer cell invasion.
RBX1 participates in miR-378-mediated lung cancer cell invasion. (A) Western blot analyses of RBX1 expression in A549 and SK-LU-1 cells after rescue with RBX1 overexpression plasmids or siRBX1. (B) Transwell invasion assay performed to evaluate the reversal of cell invasion by RBX1 overexpression plasmids and siRBX1 in A549 and SK-LU-1 cells. (C) Wound healing assay to evaluate the restorative effects of RBX1 overexpression plasmids and siRBX1 on cell migration in A549 and SK-LU-1 cells.
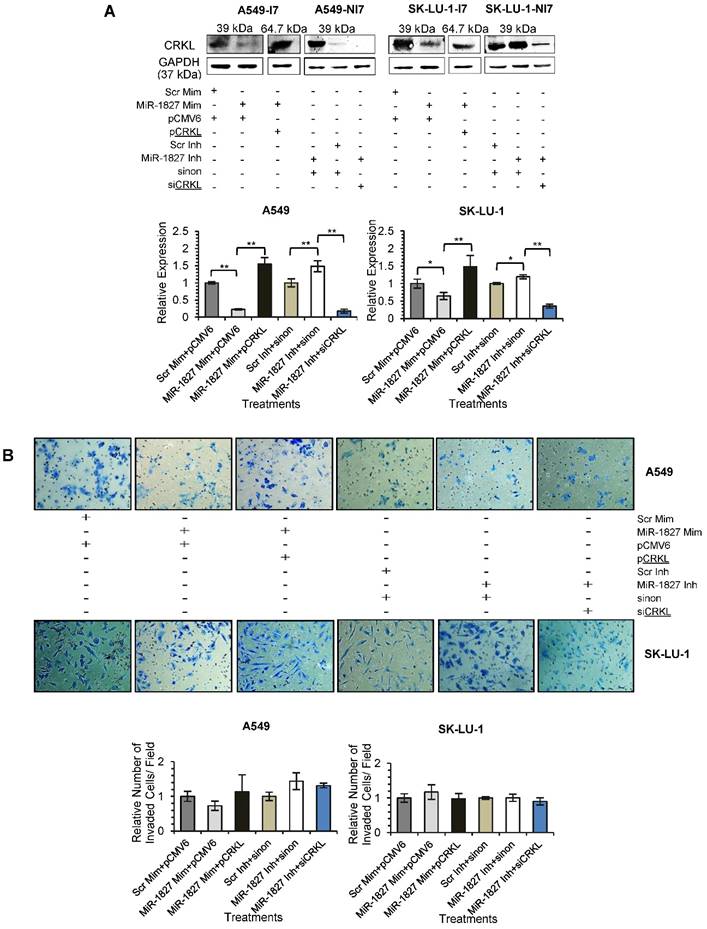
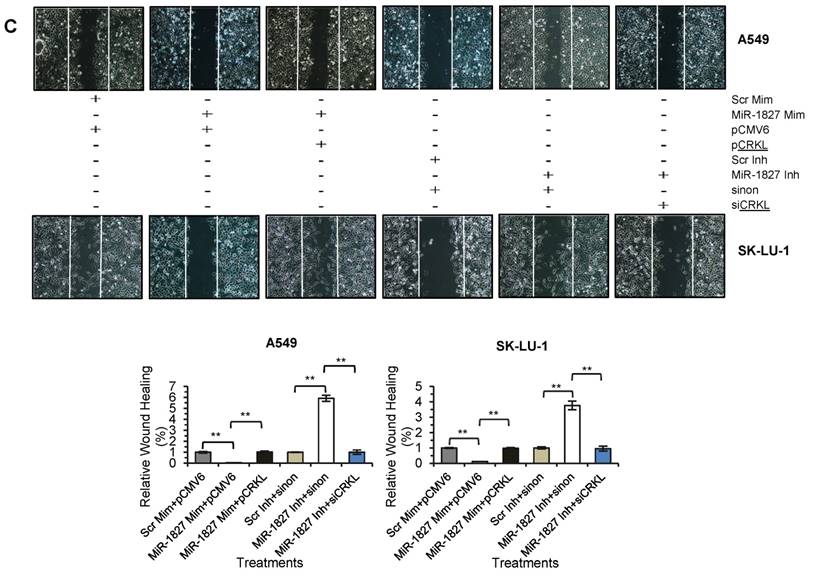
Repression of CRKL is required for the MiR-1827-mediated Inhibition of Cell Migration in Lung Cancer
To confirm that down-regulation of CRKL reduces lung cancer cell motility, overexpression and silencing experiments were conducted in A549 and SK-LU-1. Our results indicated that CRKL overexpression overcame miR-1827 inhibition on CRKL protein expression (Fig. 7A) and cell movement (Fig. 7C). However, exogenous CRKL expression showed no significant effect on cell invasion in miR-1827 mimic-transfected A549-I7 and SK-LU-1-I7 (Fig. 7B). Conversely, knockdown of CRKL compromised the effect of miR-1827 inhibitors in A549-NI7 and SK-LU-1-NI7. The restoration of CRKL protein expression (Fig. 7A) in miR-1827-suppressed cells was accompanied by decreased migrated distance (Fig. 7C) in scratch assay. Unfortunately, lung cancer cells remained low invasive (Fig. 7B). Together, our data supported the idea that miR-1827 represses cell migration by targeting CRKL in lung cancer.
CRKL participates in miR-1827-mediated lung cancer cell migration. (A) Western blot analyses of CRKL expression in A549 and SK-LU-1 cells after rescue with CRKL overexpression plasmids or siCRKL. (B) Transwell invasion assay performed to evaluate the reversal of cell invasion by CRKL overexpression plasmids and siCRKL in A549 and SK-LU-1 cells. (C) Wound healing assay to evaluate the restorative effects of CRKL overexpression plasmids and siCRKL on cell migration in A549 and SK-LU-1 cells.
RBX1 and CRKL are not associated with Angiogenesis in Lung Cancer
As miR-378 and miR-1827 were also proven to modulate tumor angiogenesis in lung cancer [9], we looked at the involvement of their targets, RBX1 and CRKL in repressing or activating HUVEC tube formation using TCM collected from treated lung cancer cells in all of the overexpression and knockdown rescue experiments. Our data showed that both the transfection of RBX1 or CRKL overexpression plasmids and siRNAs against RBX1 or CRKL partially reversed the effects of miRNAs, compared to their empty vector control (Fig. S1), suggesting that they do not play a major role in angiogenesis signaling pathway in lung cancer.
Discussion
MicroRNA-378 was discovered as an independent transcription product of intron 1 of the gene encoding master metabolic regulator, peroxisome proliferator-activated receptor gamma, co-activator 1 beta (PPARGC1B) on chromosome 5 [17, 18]. Earlier findings have indicated that miR-378 could be up-regulated [19-25] or down-regulated [26-29] in cancers. This suggests that miR-378 could act as both oncogene and tumor suppressor in various types of human cancers depending on the origin of the tumor. MicroRNA-1827, located on chromosome 12, was first identified in HeLa cervical carcinoma cells [30], and was reported to be up-regulated only in glioma [31] while miR-1827 binding to MYCL1 3'-UTR with single nucleotide polymorphisms (SNPs) affects predisposition to small cell lung cancer [32]. Our previous report demonstrated that the expression of miR-378 (up-regulated) and miR-1827 (down-regulated) are dysregulated in high invasive lung cancer cells compared to low invasive control [9]. The highest fold change of miR-378 and poorly understood underlying molecular mechanism of miR-1827 in lung cancer warrant further investigation.
In the present study, miR-378 and miR-1827 were further demonstrated to affect EMT in NSCLC. In the presence of miR-378 mimics or miR-1827 inhibitors, cadherin-bound β-catenin was down-regulated to cytosolic β-catenin. The disruption of linkage of cadherin-β-catenin-actin cytoskeleton signifies loss of cadherin and eliminates cell adhesion, a key step in EMT leading to metastasis. The down-regulation of epithelial marker was accompanied by induction of the intermediate filament protein vimentin (mesenchymal marker). High level of vimentin correlated with invasive and motile behavior of A549-NI7 and SK-LU-1-NI7 cells when transfected with miR-378 mimics and miR-1827 inhibitors, respectively. The function of vimentin during cell invasion and migration is thought to be mediated through invadopodia and lamellipodia formation as well as maintenance of cell polarity [33].
The roles of miR-378 and miR-1827 in lung cancer metastasis and angiogenesis were also confirmed in vivo using the zebrafish embryo model. The zebrafish embryo xenograft model provides higher statistical power than spontaneous metastasis assay and the metastatic potential of tumor cells evaluated using this model correlated with the invasion ability in vitro [34]. We showed that the abilities of high invasive lung cancer cells to metastasize and induce angiogenesis were greatly suppressed when treated with miR-378 inhibitors or miR-1827 mimics. Consistent with our findings, others have provided evidence that miR-378 functions as an oncogene by promoting invasion, migration and angiogenesis in the nude mice xenograft model [25, 35, 36].
To elucidate the mechanism by which miR-378 and miR-1827 regulate lung cancer metastasis, we predicted their target genes and validated these predictions by performing dual luciferase assay and Western blot. Our results demonstrated that miR-378 negatively regulates RBX1 expression while miR-1827 directly targets CRKL. RBX1, also known as regulator of cullins-1 (ROC1), is the RING component of the cullin-based RING ligase (CRL), the largest family of E3 ubiquitin ligases [37]. The association of RBX1 with lung cancer remains elusive and little is known about how RBX1 is regulated at the post-transcriptional level. RBX1 has only been reported to be regulated by miR-194 to modulate cell proliferation, invasion and migration in gastric cancer [38]. Here, we found that RBX1 was down-regulated in high invasive lung cancer cells, which is in contrast with past findings that implicated RBX1 as being frequently up-regulated in human tissues [39] and cancers [40]. In addition, we showed that RBX1 is directly targeted by miR-378 and ectopic expression of RBX1 could overcome increased invasion driven by miR-378 in lung cancer cells. We have thus shown that RBX1 is functionally involved in miR-378-mediated tumor cell invasion, revealing a novel role for RBX1 in suppressing invasion in lung cancer besides promoting cell growth [41], migration and epithelial-mesenchymal transition (EMT) [42] in other types of human cancers. However, the detailed mechanism of miR-378/ RBX1 signaling merits further research.
On the other hand, CRKL is a member of CRK family and acts as an adaptor protein participating in intra-cellular signal transduction. It has been reported previously that CRKL is up-regulated in human cancers including NSCLC [43-45] and CRKL has been proven to be negatively regulated by miRNAs such as miR-126 [43] and miR-200b/200c/429 [45, 46]. Interestingly, we identified CRKL as the first target for miR-1827 and the pro-migratory effect achieved by anti-miR-1827 was reversed via CRKL silencing in lung cancer cells, which is in accordance with previous findings that CRKL is involved in tumor cell migration [46, 47]. To the best of our knowledge, this is the first report detailing the function of miR-1827 as a tumor suppressor in lung cancer.
As much as miR-378 and miR-1827 were shown to regulate lung cancer cell invasion, migration and angiogenesis, their targets RBX1 and CRKL were not revealed to be major players in angiogenesis. This could be due to the need for CRL to be activated by the dissociation of CAND1 (cullin-associated and neddylation-dissociated-1) that sequesters cullin/RBX1 heterodimers upon binding to substrate-recognition module (SRM) followed by the neddylation of the cullin, thereby mediating the angiogenic effects in lung cancer cells along with adaptor and substrate receptor proteins [48, 49]. The result is the insignificant elevation of the amount of angiogenic factors secreted into TCM, rather than silencing the gene in HUVEC itself [50]. RBX1 also plays a role in HIF-1 pathway to produce VEGF to induce angiogenesis during hypoxia and our experimental conditions might have been the cause for partial restoration of angiogenic effects. There are as yet no reports that postulate the role of CRKL as an angiogenic modulator.
In summary, this study demonstrates that increased expression of miR-378 and decreased expression of miR-1827 in human lung cancer might be potentially significant in the acquisition of an aggressive phenotype, through the regulation of RBX1 and CRKL. This might serve as a novel prognostic and therapeutic biomarker for lung cancer.
Supplementary Material
Supplementary figure and tables.
Acknowledgements
This work was supported by the University of Malaya High Impact Research Grant (UM.C/625/1/HIR/MOE/CHAN/016) and the CEBAR Grant (RU015-2016).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C. et al. GLOBOCAN 2012 v1.1, Cancer incidence and mortality worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer. 2014
2. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y. et al. International association for the study of lung cancer/ American Thoracic Society/ European Respiratory Society international multi-disciplinary-classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244-85
3. Isobe T, Herbst RS, Onn A. Current management of advanced non-small cell lung cancer: Targeted therapy. Semin Oncol. 2005;32:315-28
4. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228-34
5. Deng B, Wang B, Fang J, Zhu X, Cao Z, Lin Q. et al. MiRNA-203 suppresses cell proliferation, migration and invasion in colorectal cancer via targeting of EIF5A2. Sci Rep. 2016;6:28301
6. Cheng Y, Xiang G, Meng Y, Dong R. MiRNA-183-5p promotes cell proliferation and inhibits apoptosis in human breast cancer by targeting the PDCD4. Reprod Biol. 2016;16:225-33
7. Wu SY, Rupaimoole R, Shen F, Pradeep S, Pecot CV, Ivan C. et al. A miR-192-EGR1-HOXB9 regulatory network controls the angiogenic switch in cancer. Nat Commun. 2016;7:11169
8. Rice MA, Ishteiwy RA, Magani F, Udayakumar T, Reiner T, Yates TJ. et al. The microRNA-23b/-27b cluster suppresses prostate cancer metastasis via Huntingtin-interacting protein 1-related. Oncogene. 2016;35:4752-61
9. Ho CS, Yap SH, Phuah NH, In LLA, Hasima N. MicroRNAs associated with tumour migration, invasion and angiogenic properties in A549 and SK-Lu1 human lung adenocarcinoma cells. Lung Cancer. 2014;83:154-62
10. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: An open source platform for biological-image analysis. Nat Methods. 2012;9:676-82
11. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G. et al. Cell migration: Integrating signals from front to back. Science. 2003;302:1704-9
12. Paraskevopoulou MD, Georgakilas G, Kostulas N, Vlachos IS, Vergoulis T, Reczko M. et al. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;419:169-73
13. Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005
14. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4:44-57
15. Oliveros JC. Venny. An interactive tool for comparing lists with Venn's diagrams. 2015
16. Geback T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265-74
17. Eichner LJ, Perry MC, Dufour R, Bertos N, Park M, St-Pierre J. et al. MiR-378 (*) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010;12:352-61
18. Hupkes M, Sotoca AM, Hendriks JM, van Zoelen EJ, Dechering KJ. MicroRNA miR-378 promotes BMP2-induced osteogenic differentiation of mesenchymal progenitor cells. BMC Mol Biol. 2014;15:1
19. Liu H, Zhu L, Liu B, Yang L, Meng X, Zhang W. et al. Genome-wide microRNA profiles identify miR-378 as a serum biomarker for early detection of gastric cancer. Cancer Lett. 2012;316:196-203
20. Redova M, Poprach A, Nekvindova J, Iliev R, Radova L, Lakomy R. et al. Circulating miR-378 and miR-451 in serum are potential biomarkers for renal cell carcinoma. J Transl Med. 2012;10:55
21. Ferrarelli LK. Deciphering the signatures of bone metastases. Sci Signal. 2013;6:ec257
22. Qian J, Lin J, Qian W, Ma JC, Qian SX, Li Y. et al. Overexpression of miR-378 is frequent and may affect treatment outcomes in patients with acute myeloid leukemia. Leuk Res. 2013;37:765-8
23. Chan JK, Kiet TK, Blansit K, Ramasubbaiah R, Hilton JF, Kapp DS. et al. MiR-378 as a biomarker for response to anti-angiogenic treatment in ovarian cancer. Gynecol Oncol. 2014;133:568-74
24. Yu BL, Peng XH, Zhao FP, Liu X, Lu J, Wang L. et al. MicroRNA-378 functions as an onco-miR in nasopharyngeal carcinoma by repressing TOB2 expression. Int J Oncol. 2014;44:1215-22
25. Chen LT, Xu SD, Xu H, Zhang JF, Ning JF, Wang SF. MicroRNA-378 is associated with non-small cell lung cancer brain metastasis by promoting cell migration, invasion and tumor angiogenesis. Med Oncol. 2012;29:1673-80
26. Fei B, Wu H. MiR-378 inhibits progression of human gastric cancer MGC-803 cells by targeting MAPK1 in vitro. Oncol Res. 2012;20:557-64
27. Zhang GJ, Zhou H, Xiao HX, Li Y, Zhou T. MiR-378 is an independent prognostic factor and inhibits cell growth and invasion in colorectal cancer. BMC Cancer. 2014;14:109
28. Li H, Dai S, Zhen T, Shi H, Zhang F, Yang Y. et al. Clinical and biological significance of miR-378a-3p and miR-378a-5p in colorectal cancer. Eur J Cancer. 2014;50:1207-21
29. Avgeris M, Stravodimos K, Scorilas A. Loss of miR-378 in prostate cancer, a common regulator of KLK2 and KLK4, correlates with aggressive disease phenotype and predicts the short-term relapse of the patients. Biol Chem. 2014;395:1095-104
30. Friedlander MR, Chen W, Adamidi C, Maaskola J, Eispanier R, Knespel S. et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407-15
31. Liu F, Xiong Y, Zhao Y, Tao L, Zhang Z, Zhang H. et al. Identification of aberrant microRNA expression pattern in pediatric gliomas by microarray. Diagn Pathol. 2013;8:158
32. Xiong F, Wu C, Chang J, Yu D, Xu B, Yuan P. et al. Genetic variation in an miRNA-1827 binding site in MYCL1 alters susceptibility to small-cell lung cancer. Cancer Res. 2011;71:5175-81
33. Kidd ME, Shumaker DK, Ridge KM. The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol. 2014;50:1-6
34. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER. et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022-9
35. Ma J, Lin J, Qian J, Qian W, Yin J, Yang B. et al. MiR-378 promotes the migration of liver cancer cells by down-regulating Fus expression. Cell Physiol Biochem. 2014;34:2266-74
36. Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. PNAS. 2007;104:20350-5
37. Wei D, Sun Y. Small RING finger proteins RBX1 and RBX2 of SCF E3 ubiquitin ligases: The role in cancer and as cancer targets. Genes Cancer. 2010;1:700-7
38. Chen X, Wang Y, Zang W, Du Y, Li M, Zhao G. MiR-194 targets RBX1 gene to modulate proliferation and migration of gastric cancer cells. Tumour Biol. 2015;36:2393-401
39. Swaroop M, Gosink M, Sun Y. SAG/ROC2/Rbx2/Hrt2, a component of SCF E3 ubiquitin ligase: Genomic structure, a splicing variant, and two family pseudogenes. DNA Cell Biol. 2001;20:425-34
40. Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis. and senescence. Cancer Res. 2009;69:4974-82
41. Wang W, Liu Z, Qu P, Zhou Z, Zeng Y, Fan J. et al. Knockdown of regulator of cullins-1 (ROC1) expression induces bladder cancer cell cycle arrest at the G2 phase and senescence. PLoS ONE. 2013;8:e62734
42. Wang W, Chen H, Liu Z, Qu P, Lan J, Chen H. et al. Regulator of cullins-1 expression knockdown suppresses the malignant progression of muscle-invasive transitional cell carcinoma by regulating mTOR/DEPTOR pathway. Br J Cancer. 2016;114:305-13
43. Wang J, Chen X, Li P, Su L, Yu B, Cai Q. et al. CRKL promotes cell proliferation in gastric cancer and is negatively regulated by miR-126. Chem Biol Interact. 2013;206:230-8
44. Cheung HW, Du J, Boehm JS, He F, Weir BA, Wang X. et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1:608-25
45. Tamura M, Sasaki Y, Kobashi K, Takeda K, Nakagaki T, Idogawa M. et al. CRKL oncogene is downregulated by p53 through miR-200s. Cancer Sci. 2015;106:1033-40
46. Ye ZB, Ma G, Zhao YH, Xiao Y, Zhan Y, Jing C. et al. MiR-429 inhibits migration and invasion of breast cancer cells in vitro. Int J Oncol. 2015;46:531-8
47. Lin Q, Sun MZ, Guo C, Shi J, Chen X, Liu S. CRKL overexpression suppresses in vitro proliferation, invasion and migration of murine hepatocarcinoma Hca-P cells. Biomed Pharmacother. 2015;69:11-7
48. Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-liagases by neddylation and dimerization. Cell Mol Life Sci. 2009;66:1924-38
49. Bulatov E, Ciulli A. Targeting cullin-RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem J. 2015;467:365-86
50. Tao WT, Wu JF, Yu GY, Wang R, Wang K, Li LH. et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014;5:e1059
Author contact
![]() Corresponding author: Institute of Biological Sciences, Division of Genetics and Molecular Biology, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia. Tel.: +603-79675921 Fax: +603-79675908 E-mail address: hasimaedu.my
Corresponding author: Institute of Biological Sciences, Division of Genetics and Molecular Biology, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia. Tel.: +603-79675921 Fax: +603-79675908 E-mail address: hasimaedu.my