Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(2):358-366. doi:10.7150/jca.20266 This issue Cite
Research Paper
Clinical Significance of Glycoprotein Non-metastatic B and Its Association with EGFR/HER2 in Gastrointestinal Cancer
Jesse Yu Tajima1, Manabu Futamura1 ![]() , Siqin Gaowa1, Ryutaro Mori1, Toshiyuki Tanahashi1, Yoshihiro Tanaka1, Nobuhisa Matsuhashi1, Takao Takahashi1, Kazuya Yamaguchi1, Tatsuhiko Miyazaki2, Kazuhiro Yoshida1
, Siqin Gaowa1, Ryutaro Mori1, Toshiyuki Tanahashi1, Yoshihiro Tanaka1, Nobuhisa Matsuhashi1, Takao Takahashi1, Kazuya Yamaguchi1, Tatsuhiko Miyazaki2, Kazuhiro Yoshida1
1. Department of Surgical Oncology, Graduate School of Medicine, Gifu University, Gifu, Japan;
2. Division of Pathology, Gifu University Hospital, Gifu, Japan.
Received 2017-3-26; Accepted 2017-11-29; Published 2018-1-1
Abstract
Glycoprotein non-metastatic B (GPNMB), a type I transmembrane glycoprotein, is overexpressed in melanoma and breast cancer and promotes cancer-cell invasion and motility. We previously reported cross-talk between GPNMB and human epidermal growth factor receptor 2 (HER2) in breast cancer, suggesting that GPNMB might play an important role in resistance to anti-HER2 therapy in breast cancer. Here, we clarified the association between GPNMB and HER-family proteins in gastrointestinal cancer by examining their relationships using gastric and colorectal cancer cell lines. We found that GPNMB depletion of by small-interfering RNA increased epidermal growth factor receptor (EGFR) expression and phosphorylation through AKT8 virus oncogene cellular homolog (AKT) and mitogen-activated protein kinase (MAPK) pathways. Additionally, treatment with cetuximab (CTX) also increased GPNMB expression, and combination therapy consisting of GPNMB depletion and CTX treatment significantly suppressed cell growth in colorectal cancer cell lines, but not in gastric cancer cell lines. Furthermore, we also evaluated changes in GPNMB expression in vivo, with immunohistochemistry detecting GPNMB overexpression in a colorectal cancer patient following anti-EGFR therapy. These results suggested possible cross-talk between GPNMB and EGFR, and that GPNMB might play an important role in resistance to anti-EGFR therapy in gastrointestinal cancer.
Keywords: GPNMB, gastrointestinal cancer, cross-talk, molecular-targeted therapy, drug resistance
Introduction
Gastrointestinal (GI) cancer is among the most common causes of cancer death worldwide. Despite recent advances in the development of diagnostic and therapeutic strategies, many patients are often diagnosed at an advanced stage or relapse after curative surgery, and >1.4 million patients still die of gastric cancer (GC) or colorectal cancer (CRC) annually [1]. Recently, molecular-targeted therapies, such as anti-epidermal growth factor receptor (EGFR) therapy in CRC and anti-human epidermal growth factor receptor type 2 (HER2) therapy in GC, were shown to inhibit tumor progression and prolong survival in combination with chemotherapy [2-5]. However, resistance or non-reactivity to these drugs might be a problem, and the mechanisms associated with drug resistance currently remain unclear [6].
Glycoprotein non-metastatic B (GPNMB) is a type I transmembrane glycoprotein expressed in various normal cells, including melanocytes, osteoblasts, and antigen-presenting cells, such as activated macrophages and dendritic cells [7]. GPNMB is overexpressed in various cancer tissues, including melanoma, glioma, breast cancer (BC), and GC [8-11]. The physiological function of GPNMB promotes cell invasion and motility, leading to tumor metastasis in the case of cancer [7,10,12-14]. Furthermore, high levels of GPNMB expression in cancer cells results in poor prognoses, suggesting that GPNMB might be an oncogene [7,10,12-14]. GPNMB is also reportedly involved in GC-cell apoptosis. Wei et al [11] reported that GPNMB expression is decreased in BGC-823 cells after treatment with trichostatin A (TSA) according to microarray analysis, speculating that TSA might increase acetylated histone H4 and lead to inhibited apoptosis, given its role as a potent and specific inhibitor of histone acetyltransferase. Our group previously reported that serum GPNMB levels were significantly higher in BC patients, particularly in HER2-positive cases. Moreover, GPNMB expression is significantly elevated after treatment with trastuzumab. From these results, we concluded that GPNMB might exhibit cross-talk with HER2-signaling pathways, perhaps via AKT pathway in BC [15].
Recently, a GPNMB-targeting antibody conjugated to a cytotoxic tubulin destabilizer, monomethylauristatin E (MMAE), was developed. This drug, named CDX-011 (formerly CR011-vcMMAE) and also known as glembatumumab, is now under investigation in clinical trials for the treatment of metastatic melanoma and BC [10,16-19]. However, there are no reports on interactions between HER-family proteins and GPNMB in GI cancer. In this study, we investigated the cross-talk between GPNMB and HER-family proteins in GC and CRC.
Materials and Methods
Cell Lines and Culture
Human GC cell lines (MKN1, MKN45, MKN74, and TMK1), CRC cell lines (Caco-2, SW48, LS174T, and HCT116), and BC cell lines (SK-BR-3 and BT-474) used in this study were all obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in media consisting of Roswell Park Memorial Institute medium 1640 (MKN1, MKN45, MKN74, TMK1, SW48, and LS174T), Dulbecco's modified Eagle medium (SK-BR-3, BT-474, and HCT116) or minimal essential medium (MEM) (Caco-2) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 2% L-glutamine (MP Biomedicals, Eschwege, Hesse, Germany), 2% pyruvate (Sigma-Aldrich), 1% MEM non-essential amino acid solution (Sigma-Aldrich), 1% antibiotic/antimyotic solution (Sigma-Aldrich), and 0.1% tylosin solution (Sigma-Aldrich). Cells were incubated at 37°C in an atmosphere containing 5% CO2.
Western Blot
Cells were harvested and lysed in radioimmunoprecipitation assay buffer (Sigma-Aldrich) containing phosphatase inhibitors (Sigma-Aldrich) and protease inhibitors (Sigma-Aldrich) for 15 min on ice. Cell lysates (15-20 mg/mL) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis using Super Sep (Wako, Osaka, Japan) and transferred onto polyvinylidene difluoride membranes, which were blocked for 1 h and incubated with the respective primary antibody overnight at 4°C. The antibody against GPNMB was from R&D Systems (Minneapolis, MN, USA), antibodies against HER2 and phosphorylated HER2 (p-HER2) were purchased from Abcam (Cambridge, UK), and all other antibodies [EGFR, p-EGFR, p42/44 MAPK (ERK), p-p42/44 MAPK (ERK), v-AKT murine thymoma viral oncogene homolog (AKT), p-AKT, and β-actin] were obtained from Cell Signaling Technology (Danvers, MA, USA). The membranes were then washed and incubated with the appropriate secondary antibodies, and immunoreactive proteins were visualized and captured by an LAS-4000 camera system (FUJIFILM, Tokyo, Japan).
Small-interfering RNA (siRNA) against GPNMB
Two GC cell lines (MKN1 and MKN74) and three CRC cell lines (Caco-2, SW48, and LS174T) were seeded 24 h and 48 h before transfection at ~80% confluence. Cells were transiently transfected with stealth siRNA (Invitrogen, Carlsbad, CA, USA) using Lipofectamine RNAiMAX (Invitrogen) at a final concentration of 10 nM in Opti-MEM (Invitrogen). The nucleotide sequences were as follows for GPNMB siRNA (GPNMBsi): 5′-CCAGCCCUCGUGGGCUCAAAUAUAA-3′ (sense) and 5′-UUAUAUUUGAGCCCACGAGGGCUGG-3′ (antisense) for GPNMBsi1; and 5'-GGCCUGUUUGUUUCCACCAAUCAUA-3' (sense) and 5'-UAUGAUUGGUGGAAACAAACAGGCC-3' (antisense) for GPNMBsi2. Stealth siRNA negative control (Invitrogen) was used as a control. Knockdown efficiencies of siRNA were confirmed by western blot [15].
Drugs
Cetuximab (CTX) was purchased from Merck (Darmstadt, Germany), the ERK inhibitor FR180204 was purchased from EMD Millipore (Billerica, MA, USA), and the AKT inhibitor MK-2206 was from Merck Oncology (Whitehouse Station, NJ, USA).
Cell-proliferation Assay
Cell proliferation was evaluated by 3-[4,5-dimethylthiazol-2- yl]-2, 5-diphenyltetrazoliumbromide (MTT; Sigma-Aldrich) assay. Briefly, cells were seeded in 96-well plates at a concentration of 2500 to 5000 cells/well and allowed to adhere overnight in a humidified atmosphere of 5% CO2. The medium was then removed and replaced by fresh culture medium containing CTX (100-200 μg/mL). After incubation for 48 h, 96 h, or 120 h, MTT solution (5 mg/mL) was added to each well, and the cells were incubated for another 4 h at 37°C. The formazan product of MTT was measured by absorbance at 570 nm using an Envision 2104 multilabel reader (Perkin-Elmer, Waltham, MA, USA).
Samples and Immunohistochemistry (IHC)
Clinical samples for IHC were obtained from CRC patients who received treatment at Gifu University (Gifu, Japan) from January 2010 to January 2016. Seven sets of naive primary tumors and remaining liver metastatic tumors after chemotherapy regimens, such as folinic acid, fluorouracil, and oxaliplatin (FOLFOX) containing anti-EGFR therapy, were used for this analysis [2,3]. Informed consent was obtained from each patient.
IHC for GPNMB was performed according to standard procedures using the same antibody described here and a biotin-conjugated donkey anti-goat secondary antibody (Jackson Laboratories, West Grove, PA, USA) [19]. Goat IgG, a polyclonal isotype control (Abcam), was used as a negative control. For preliminary experiments, we created cell blocks containing three cell lines (GPNMB expression levels: high, SK-BR-3; moderate, MKN1; and low, LS174T). We used PROTEASE 1 (Ventana, Tuscon, AZ, USA) for antigen retrieval and determined the best conditions for GPNMB IHC (Fig. S1). Changes in GPNMB-staining intensity between each primary tumor and metastasis were determined by an experienced pathologist. This study was approved by the Central Ethics Committee of Gifu University.
Statistical Analysis
The statistical significance of the results was calculated using Student's t test. All statistical tests were two-sided, and a p < 0.05 was considered statistically significant.
Results
GPNMB Expression Levels in GC and CRC Cell Lines
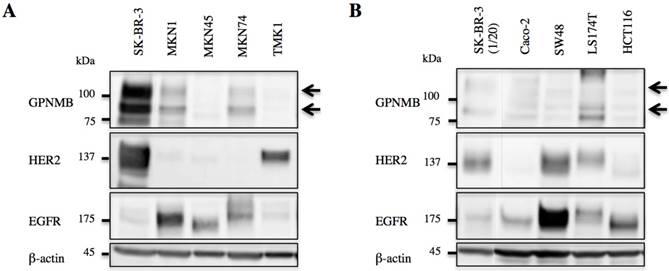
GPNMB expression levels were evaluated by western blot, revealing expression in both GC (MKN1 and MKN74) and CRC cells (Caco-2, SW48, and LS174T). However, GPNMB expression levels in GC and CRC cells were lower than those in BC cells used as positive controls (SK-BR-3). All of these cells also expressed either EGFR or HER2 (Fig. 1).
GPNMB expression in GI cancer cells. A. GC and B. CRC cell lines. Arrows indicate GPNMB expression. SK-BR-3 cells were used as a positive control. Although the expression of GPNMB was lower than that of the positive control, it was expressed in two GC cell lines (MKN1 and MKN74) and three CRC cell lines (Caco-2, SW48, and LS174T), all of which also showed overexpression of HER2 or/and EGFR.
Relationship between GPNMB and HER-family Proteins
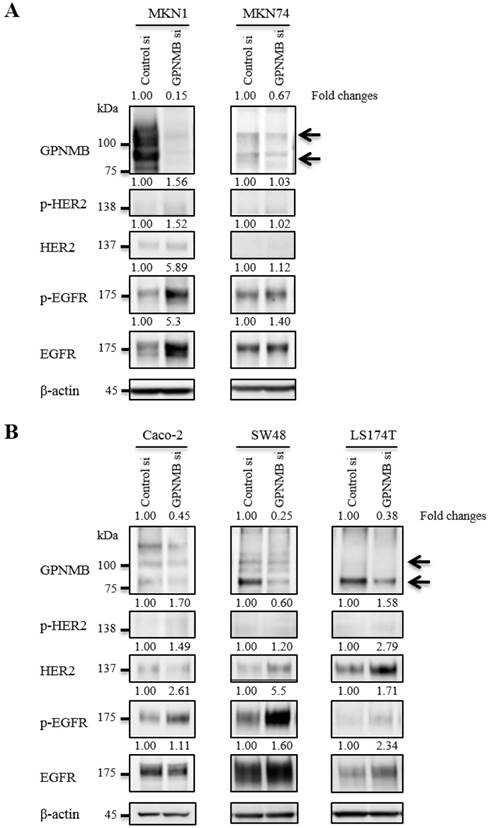
To verify relationships between GPNMB and HER-family proteins, we used the following cell lines that expressed relatively high levels of GPNMB among the cells tested: MKN1 and MKN74 for GC; and Caco-2, SW48, and LS174T for CRC. At 48 h after transfection of GPNMBsi, expression levels of depleted GPNMB, HER2, and EGFR were evaluated in all cells (Fig. 2). HER2 and/or p-HER2 levels increased slightly in CRC cell lines, but not in GC cell lines, and EGFR and/or p-EGFR increased in all cell lines examined. Similar results were observed upon use of a different GPNMB-specific siRNA (GPNMBsi1 and GPNMBsi2) (Fig. S2A).
Changes in HER2/EGFR expression and phosphorylation after GPNMB knockdown. A. GC cell lines MKN1 and MKN74 and B. CRC cell lines Caco-2, SW48, and LS174T. Relative ratios of protein expression are indicated, respectively. EGFR or p-EGFR expression levels were elevated in all GC and CRC cells; however, HER2 expression was only slightly elevated in two CRC cell lines (SW48 and LS174T), but not in GC cell lines.
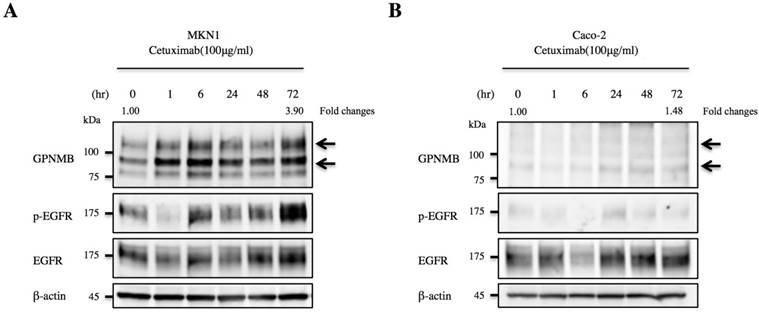
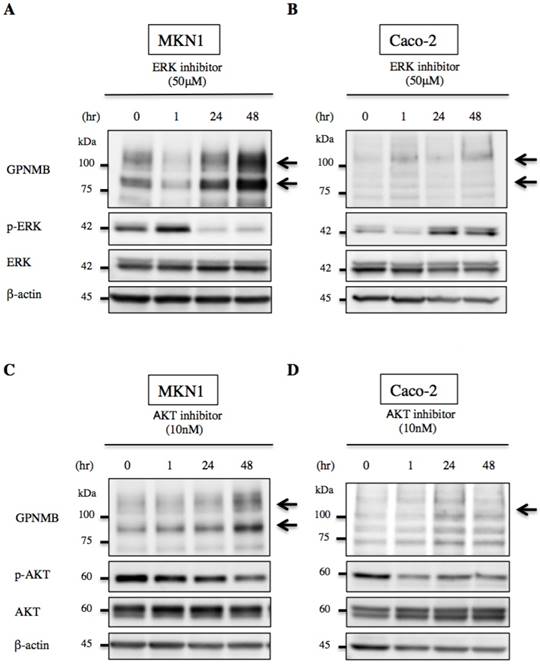
We then treated MKN1 and Caco-2 cells with CTX to investigate changes in GPNMB expression. Blockage of EGFR increased expression of GPNMB in both GC and CRC cell lines after 1 h, 6 h, 24 h, 48 h, and 72 h (Fig. 3). Furthermore, we also inhibited the ERK- or AKT-signaling pathways, followed by evaluation of GPNMB expression. In MKN1 cells, treatment with both ERK and AKT inhibitors increased GPNMB expression (Fig. 4A and C), whereas in Caco-2 cells, only AKT inhibition increased GPNMB expression (Fig. 4B and D). These results suggested that GI cells might exhibit cross-talk between GPNMB and EGFR through the AKT- and/or MAPK-signaling pathways.
Association between GPNMB and HER-family proteins. Changes in GPNMB expression after treatment with CTX in A. MKN1 and B. Caco-2 cells. GPNMB expression increased in both GC (MKN1) and CRC (Caco-2) cell lines at 1, 6, 24, 48, and 72 h after EGFR blockage by CTX treatment. Expression of EGFR/p-EGFR also increased after anti-EGFR therapy. Relative ratios of protein expression are indicated, respectively.
Changes in GPNMB expression after signaling inhibition. Treatment with ERK inhibitor (FR180204, 50 μM) or AKT inhibitor (MK2206, 10 nM) of A., C. MKN1 and B., D. Caco-2 cells. In MKN1 cells, GPNMB expression increased in a time-dependent manner following inhibition of either ERK or AKT signaling. However, GPNMB expression was increased only by AKT inhibition in Caco-2 cells.
Growth Inhibition by Double Blockade of GPNMB and EGFR
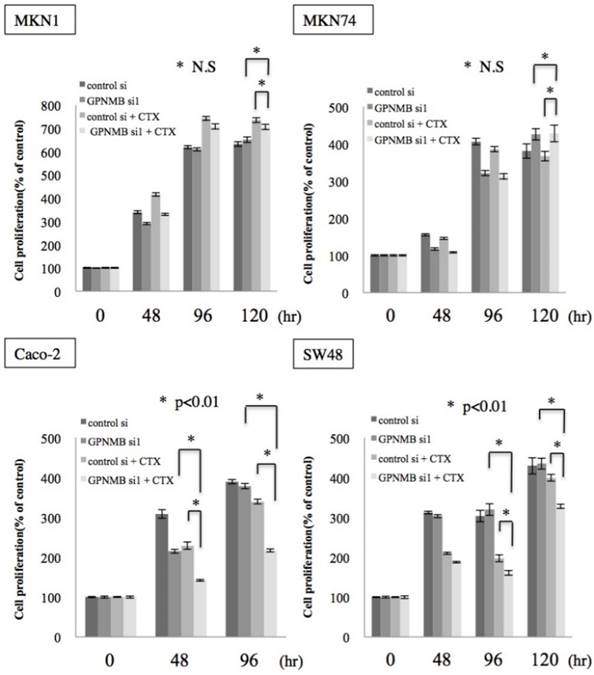
To investigate whether this cross-talk is clinically significant, we examined the effect of GPNMB depletion alone and the synergistic effect of GPNMB depletion and anti-EGFR therapy using CTX. No significant change was observed in either GC or CRC cells transfected with GPNMBsi. However, a synergistic effect was observed with a combination of GPNMBsi and CTX (double blockage) treatment in KRAS wild-type CRC cells (Caco-2 and SW48) (Fig. 5 and Fig. S2B). However, there was no synergistic effect observed in GC cells (MKN1 and MKN74), a KRAS-mutant CRC cell line (LS174T), or a BRAF-mutant CRC cell line (SW1417) (Fig. 5 and Fig. S3). These results indicated that GPNMB depletion enhanced CTX-induced growth suppression.
Cell-proliferation assay. GC cell lines (MKN1 and MKN74) and CRC cell lines (Caco-2 and SW48) were treated with cetuximab (100 μg/mL) after GPNMB depletion, and cell growth was measured by MTT assay. Although there was no significant change in either GC or CRC cells transfected with GPNMBsi1, a synergistic effect was observed with a combination of GPNMBsi1 and CTX treatment in KRAS wild-type CRC cells (Caco-2 and SW48). This phenomenon was not observed in GC cells (MKN1 and MKN74).
GPNMB Expression In Vivo
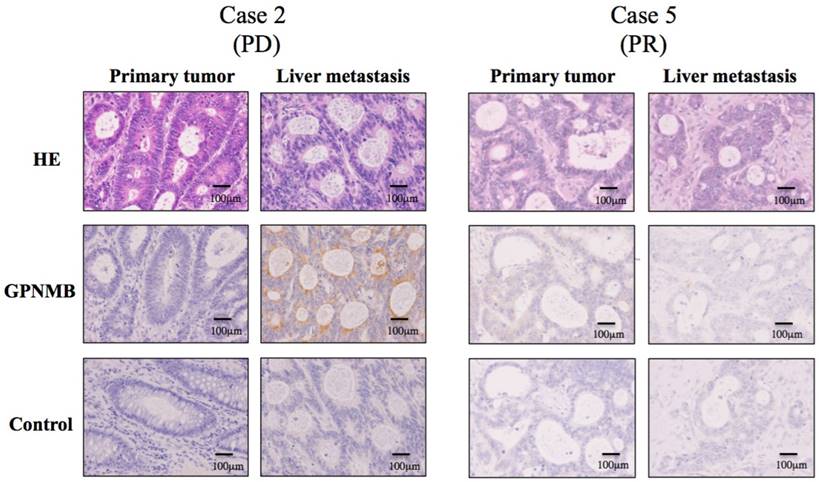
Based on the results in Figure 3, we speculated that GPNMB might be induced by CTX treatment in vivo, suggesting a possible association between GPNMB and resistance to anti-EGFR therapy. To test this hypothesis, we performed IHC using surgical specimens treated with FOLFOX along with anti-EGFR antibody, such as CTX or panitumumab, and compared GPNMB expression levels in primary tumors with those in residual liver metastatic tumors following treatment with anti-EGFR therapies. Patient profiles are shown in Table S1, with six patients showing partial response (PR) and one progressive disease (PD). Among seven CRC patients, only one (case 2) patient with PD demonstrated stronger GPNMB expression in a liver metastatic tumor as compared with that observed in a naive primary tumor. Conversely, in case 5 (PR), no GPNMB signal was observed in residual liver metastatic tumors, suggesting that GPNMB expression might be induced as a method of resistance to anti-EGFR therapy (Fig. 6).
IHC staining for GPNMB in CRC patients (200×). GPNMB expression levels according to IHC were compared between primary tumors and those in residual liver metastatic tumors after treatment with anti-EGFR therapies. Among seven CRC patients, only one (Case 2), who showed PD after combination therapy with FOLFOX and panitumumab, demonstrated stronger GPNMB expression in a liver metastatic tumor than in a naive primary tumor (Left). However, in case 5, who resulted in PR, GPNMB was expressed in neither the primary tumor nor in the liver metastatic tumor (Right). This suggested that GPNMB expression might promote resistance to anti-EGFR therapy.
Discussion
Rapid progress in the elucidation of molecular mechanisms in cancer has accelerated the development of molecular-targeted drugs and clinical applications for the treatment of various types of cancer. First, HER2, a representative membrane receptor of the HER family, was discovered as an BC oncogene [20-22], and anti-HER2 therapy has been widely used to treat HER2-overexpressing BC [23,24]. HER2 overexpression was also detected in certain GCs, leading to expanded use of anti-HER2 antibodies for treating GC and BC [5,25]. Second, EGFR was demonstrated to be overexpressed in various types of cancer, including CRC, lung, and head and neck cancer [26,27]. Anti-EGFR therapy is now widely used in clinical practice, and anti-EGFR antibodies are effective against KRAS wild-type CRC [2,3].
Further elucidation of cancer-specific molecular mechanisms will enable the development of new techniques to overcome resistance to molecular-targeted therapies. For BC, a previous study reported cross-talk between the estrogen receptor and HER-family proteins [28]. Successful treatment for such forms of BC was subsequently reported involving a combination of an aromatase inhibitor with lapatinib or a mammalian target of rapamycin inhibitor (everolimus) [29-31]. Additionally, activation of the c-Met pathway reportedly plays an important role in the acquisition of HER2 resistance in BC and resistance to an EGFR inhibitor in non-small-cell lung cancer [32-34], for which combination therapy using anti-EGFR and anti-c-Met drugs was effective by downregulating the Erb-B2 receptor tyrosine kinase-3/phosphoinositide 3-kinase/AKT-signaling pathway [33,34]. However, in GI cancer, there are few reports of cross-talk between growth factors, such as HER2, c-Met, insulin-like growth factor receptor (IGFR), and EGFR [35]. Therefore, we sought to clarify the mechanism of resistance to molecular-targeted therapies in GI cancer.
Our interests in investigating GPNMB as a BC oncogene are as follows: 1) GPNMB is highly expressed and might be a potential molecular target in BC; 2) the extracellular domain of GBPMB shed by a disintegrin and metalloprotease 10 might be useful as a prognostic marker for HER2-positive BC; and 3) GPNMB might be associated with resistance to anti-HER2 therapy [15]. These data prompted us to investigate the role of GPNMB in GI cancer. Here, we found lower GPNMB expression levels in GC and CRC cells than those observed in BC cells, which agreed with a previous report by Qian et al [36]. This phenomenon was also supported by a recent report by Mokarram et al [37], indicating that GPNMB is a representative methylated gene in CRC. By contrast, we speculated that GPNMB might play an important role in GI cancer and particularly in CRC cancer.
Despite low levels of GPNMB expression in GI cancer cells, its depletion increased the expression and phosphorylation of membrane receptors, such as EGFR and HER2. Moreover, anti-EGFR therapy accelerated GPNMB stimulation. These findings suggested that there is cross-talk between GPNMB and EGFR in GC and CRC cells, similar to that with HER2 in BC cells. Signaling cross-talk between GPNMB and HER-family proteins is not likely to be specific to BC, but appears to be a common phenomenon among various types of cancer, including GI cancer.
A remarkable finding in this study was that GPNMB depletion enhanced EGFR expression and phosphorylation, and that additional treatment by CTX suppressed cell growth, despite the low baseline levels of GPNMB in CRC cells. This suggested that the cross-talk between GPNMB and EGFR might be clinically important. By contrast, despite the detection of cross-talk with EGFR, no add-on effect of double blockage was shown in GC cells. Although HER2-targeted therapy is clinically used for HER2-positive GC, expression of HER2 is heterogeneous in GC and differs from both HER2 expression in BC and EGFR expression in CRC [38,39]. Additionally, expression of various receptor tyrosine kinases, including EGFR, HER2, c-Met, and IGFR, is also reportedly involved in GC [40].
From the standpoint of downstream signaling, it is likely that GPNMB interacts with HER-family proteins via both the ERK- and AKT-signaling pathways, as the administration of ERK and AKT inhibitors led to enhanced GPNMB expression. Here, Caco-2 and SW48 cells harboring wild-type KRAS and BRAF, exhibited growth suppression following double blockage of GPNMB and EGFR; however, no growth suppression was observed in LS174T cells harboring mutant KRAS or in SW1417 cells harboring mutant BRAF (Fig. S3).
Moreover, we were able to detect GPNMB in residual liver metastatic tumors after anti-EGFR therapy. GPNMB expression levels in residual tumors was higher than that in primary tumors resected before anti-EGFR therapy. However, it is unclear from where GPNMB-expressing cells originate. There are two possibilities: 1) that resistant cells spontaneously express GPNMB to encourage cross-talk between growth factors, and 2) that a very small population of cells originally expressing GPNMB survived and proliferated during anti-EGFR therapy. However, we suggest that these possibilities are acceptable for understanding resistance to anti-EGFR therapy, and our results strongly suggest the possible involvement of GPNMB in mechanisms facilitating metastasis and resistance to EGFR inhibitors. Currently, clinical trials of new drugs targeting GPNMB are ongoing and involve patients with BC or melanoma. The results of this study indicated that a particular combination of a GPNMB-targeting drug with an EGFR-targeting drug would be more effective against not only BC but also GI cancers. However, determination of the optimal timing for anti-GPNMB therapy currently remains difficult. Nevertheless, this therapy might be appropriate for patients harboring metastatic tumors or recurrent BC after confirmation of GPNMB expression by biopsy, as GPNMB-expressing metastatic BC is sensitive to glembatumumab [19].
We previously reported that the addition of zoledronic acid to anti-EGFR therapy results in tumor-growth suppression, even in KRAS-mutated CRC cells [41]. This strategy can correct both the AKT- and MAPK-signaling pathways and enhance the effect of anti-EGFR therapy, and it is possible that this additional treatment might help to enhance the effects of anti-GPNMB and anti-EGFR/HER2 therapy.
With advances in molecular biology, the development of molecular-targeted therapies has progressed rapidly, prolonging survival of patients with various cancers. However, drug resistance remains a significant problem with such treatments; therefore, double blockage of targeted molecules to overcome drug resistance might represent a useful strategy, similar to that of co-treatment with trastuzumab and pertuzumab, which is currently effective against metastatic HER2-positive BC [42]. The precise roles of GPNMB in cancer progression remain unclear; therefore, further investigation is required to clarify its biological function prior to possible future clinical use.
Conclusion
In this study, we demonstrated the existence of cross-talk between GPNMB and EGFR in GC and CRC, revealing that double blockage of GPNMB and EGFR resulted in a synergistic effect against CRC. These results suggested that GPNMB might participate in tolerance to molecular-targeted drugs, such as anti-EGFR therapy, and might be a potential candidate for new cancer treatments.
Abbreviations
BC: Breast cancer; CRC: Colorectal cancer; CTX: Cetuximab; EGFR: Epidermal growth factor receptor; FOLFOX: Folinic acid, fluorouracil, and oxaliplatin; GC: Gastric cancer; GI: Gastrointestinal; GPNMB: Glycoprotein non-metastatic B; GPNMBsi: GPNMB siRNA; HER2: Human epidermal growth factor receptor 2; IGFR: Insulin-like growth factor receptor; IHC: Immunohistochemistry; MAPK: Mitogen-activated protein kinase; MEM: Minimum essential medium; AKT: AKT8 virus oncogene cellular homolog; MMAE: Monomethylauristatin E; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide; PD: Progressive disease; PR: Partial response; siRNA: Small-interfering RNA; TSA: Trichostatin A.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank K. Shizu for technical support and K. Enya, M. Hirata, A. Iwata, K. Mori, and K. Takano for administrative assistance. We would like to thank Editage (www.editage.jp) for English-language editing.
Competing Interests
KY has received grants and personal fees from Taiho Pharmaceutical Co., Ltd., Pfizer Inc., Chugai Pharmaceutical Co., Ltd., and Yakult Honsha Co., Ltd., grants from Bristol-Myers Squibb and Kyowa Hakko Kirin Co., Ltd., honoraria from Taiho Pharmaceutical Co., Ltd., Pfizer Inc., Chugai Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., and Yakult Honsha Co., Ltd., and had a consultant or advisory relationship with Taiho Pharmaceutical Co., Ltd. and La Roche, Ltd. All remaining authors declare that they have no conflicts of interest.
References
1. Torre LA, Bray F, Siegel RL. et al. Global Cancer Statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Alberts SR, Sargent DJ, Nair S. et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383-93
3. Ocvirk J, Brodowicz T, Wrba F. et al. Cetuximab plus FOLFOX6 or FOLFIRI in metastatic colorectal cancer: CECOG trial. World J Gastroenterol. 2010;16:3133-43
4. Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523-9
5. Bang YJ, Van Cutsem E, Feyereislova A. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomized controlled trial. Lancet. 2010;376:687-97
6. Van Emburgh BO, Bianchi AS, Di Nicolantonio F. et al. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol Oncol. 2014;8:1084-94
7. Maric G, Rose AAN, Annis MG. et al. Glycoprotein non-metastatic b (GPNMB): a metastatic mediator and emerging therapeutic target in cancer. Onco Targets Ther. 2013;6:839-52
8. Weterman MA, Ajubi N, Van Dinter IM. et al. Nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int J Cancer. 1995;60:73-81
9. Kuan CT, Wakiya K, Dowell JM. et al. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin Cancer Res. 2006;12:1970-82
10. Rose AAN, Pepin F, Russo C. et al. Osteoactivin promotes breast cancer metastasis to bone. Mol Cancer Res. 2007;5:1001-14
11. Ruan WM, Li YL, Nie G. et al. Differential expression of glycoprotein non-metastatic melanoma protein B (GPNMB) involved in trichostatin A-induced apoptosis in gastric cancer. Int J Clin Exp Med. 2014;7:4857-66
12. Onaga M, Ido A, Hasuike S. et al. Osteoactivin expressed during cirrhosis development in rats fed a choline-deficient, L-amino acid defined diet, accelerates motility of hepatoma cells. J Hepatol. 2003;39:779-85
13. Rich JN, Shi Q, Hjelmeland M. et al. Bone-related genes expressed in advanced malignancies induce invasion and metastasis in a genetically defined human cancer model. J Biol Chem. 2003;278:15951-7
14. Oyewumi MO, Manickavasagam D, Novak K. et al. Osteoadtivin (GPNMB) ectodomain protein promotes growth and invasive behavior of human lung cancer cells. Oncotarget. 2016;7:13932-44
15. Kanematsu M, Futamura M, Takata M. et al. Clinical significance of glycoprotein nonmetastatic B and its association with HER2 in breast cancer. Cancer Med. 2015;4:1344-55
16. Tse KF, Jeffers M, Pollack VA. et al. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin Cancer Res. 2006;12:1373-82
17. Pollack VA, Alvarez E, Tse KF. et al. Treatment parameters modulating regression of human melanoma xenografts by an antibody-drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother Pharmacol. 2007;60:423-35
18. Ott PA, Hamid O, Pavlick AC. et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patient with advanced melanoma. J Clin Oncol. 2014;32:3659-66
19. Yardley DA, Weaver R, Melisko ME. et al. EMERGE: a randomized phase II study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein NMB-expressing breast cancer. J Clin Oncol. 2015;33:1609-19
20. Coussens L, Yang-Feng TL, Liao YC. et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230:1132-9
21. King CR, Kraus MH, Aaronson SA. Amplification of novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229:974-6
22. Slamon DJ, Clark GM, Wong SG. et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-82
23. Slamon DJ, Jones BL, Shak S. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783-92
24. Marty M, Cognetti F, Maraninchi D. et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23:4265-74
25. Jaehne J, Urmacher C, Thaler HT. et al. Expression of Her2/neu oncogene product p185 in correlation to clinicopathological and prognostic factors of gastric carcinoma. J Cancer Res Clin. 1992;118:474-9
26. Spano JP, Lagorce C, Atlan D. et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16:102-8
27. Rusch V, Baselga J, Cardo CC. et al. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993;53:2379-85
28. Johnston SR. Enhancing the efficacy of hormonal agents with selected targeted agents. Clin Breast Cancer. 2009;9:S28-36
29. Arpino G, Wiechmann L, Osborne CK. et al. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29:217-33
30. Johnston S, Pippen Jr J, Pivot X. et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27:5538-46
31. Baselga J, Campone M, Piccart M. et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-9
32. Shattuck DL, Miller JK, Carraway III KL. et al. Met receptor contributes to trastuzumab resistance of her2-overexpressing breast cancer cells. Cancer Res. 2008;68:1471-7
33. Engelman JA, Zejnullahu K, Mitsudomi T. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-43
34. Yano S, Wang W, Li Q. et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479-87
35. Hofmann TB, Sun MY, Chen CT. et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther. 2008;7:3499-3508
36. Qian X, Mills E, Torgov M. et al. Pharmacologically enhanced expression of GPNMB increases the sensitivity of melanoma cells to the CR011-vcMMAE antibody-drug conjugate. Mol Oncol. 2008;2:81-93
37. Mokarram P, Kumar K, Brim H. et al. Distinct high-profile methylated genes in colorectal cancer. PLoS ONE. 2009;4:e7012
38. Grillo F, Fassan M, Sarocchi F. et al. HER2 heterogeneity in gastric/gastroesophageal cancers: From benchside to practice. World J Gastroenterol. 2016;22:5879-87
39. Grabsch H, Sivakumar S, Gray S. et al. HER2 expression in gastric cancer: Rare, heterogeneous and of no prognostic value - conclusions from 924 cases of two independent series. Cell Oncol. 2010;32:57-65
40. Nagatsuma AK, Aizawa M, Kuwata T. et al. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer. 2015;18:227-38
41. Kato J, Futamura M, Kanematsu M. et al. Combination therapy with zoledronic acid and cetuximab effectively suppresses growth of colorectal cancer cells regardless of KRAS status. Int J Cancer. 2016;138:1516-27
42. Baselga J, Cortés J, Kim SB. et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109-19
Author contact
![]() Corresponding author: Tel: +81-58-230-6231; Fax: +81-58-230-6236; E-mail: mfutamurac.jp
Corresponding author: Tel: +81-58-230-6231; Fax: +81-58-230-6236; E-mail: mfutamurac.jp