Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(3):521-527. doi:10.7150/jca.22355 This issue Cite
Review
Overexpression of MICAL2, a novel tumor-promoting factor, accelerates tumor progression through regulating cell proliferation and EMT
Yongqiang Cai, Jinping Lu, Faqing Tang ![]()
Clinical Laboratory and Medical Research Center, Zhuhai Hospital, Jinan University, Zhuhai 519000, Guangdong, China
Received 2017-8-13; Accepted 2017-10-20; Published 2018-1-1
Abstract
Molecule interacting with CasL 2 (MICAL2), a microtubule associated monooxygenase, is involved in cell growth, axon guidance, vesicle trafficking and apoptosis. Recent studies have demonstrated that MICAL2 is highly expressed in tumor and accelerates tumor progression and it is deemed to be a novel tumor-promoting factor. MICAL2 overexpression increases cell proliferation to accelerate tumor growth, and MICAL2 also promotes epithelial-mesenchymal transition (EMT)-related proteins to increase cancer cell metastasis. On mechanism, MICAL2 induces EMT by regulating SRF (serum response factor)/MRTF-A (myocardin related transcription factor A) signaling, Semaphorin/Plexin pathway and inducing ROS (Reactive oxygen species) production. In the present review, we introduced MICAL family, expatiated the structure and functions of MICALs, and summarized the mechanisms of MICAL2 involving tumor progression. The challenges and perspectives for MICAL2 in tumor are also discussed.
Keywords: MICAL2, cancer, epithelial-mesenchymal transition, metastasis.
Introduction
MICAL (Molecule interacting with CasL) 2 is a member of MICAL family. MICALs are an evolutionary conserved family involved in cell growth, axon guidance, vesicle trafficking and apoptosis. MICAL was found by Suzuki et al in 2002 [1]. It was reported that there are one MICAL and MICAL-like (MICAL-L) protein in Drosophila, three MICALs (MICAL-1, MICAL-2, and MICAL-3) and two MICAL-L (MICAL-L1 and MICAL-L2) in homo sapiens, and eight MICALs including two paralogs for MICAL2, MICAL3, and MICAL-L2 in zebra fish [2]. MICALs are multidomain proteins containing N-terminal flavor protein monooxygenase (MO) domain, calponin homology (CH) domain, LIM domain, and C-terminal domain. But MICAL-like proteins lack of flavorprotein monooxygenase domain. The functions of MICAL family are to participate in the control of cytoskeleton dynamics and the related fundamental biological processes, such as cell-cell contacts, cell differentiation and migration, axon growth, dendrite pruning, formation of neuromuscular junctions, angiogenesis, vesicle trafficking and gene transcription [3].
In the recent, people further found MICALs' importance in the diseases, particular in tumors. MICAL2 prostate cancer variants (MICAL2-PV) overexpresses in prostate cancer cells with high Gleason score, and MICAL2-PV expression is correlated with c-Met expression [4]. c-Met can be activated by binding of semaphorin (Sema) 4D to plexin B1, and involve in metastasis and invasion of cancer cells [5]. Loria et al also demonstrated that MICAL1 and Sema6A are highly expressed in melanoma [6]. MICAL family regulates survival and migration of cancer cell by the Sema/plexin/mical pathway [5, 6]. In additional, MICAL1 overexpression induces reactive oxygen species (ROS) generation and increases Akt phosphorylation in breast cancer cell [7]. ROS can induce the metastasis and invasion of cancer cells by epithelial-mesenchymal transition (EMT)[8-10]. MICAL-L2 protein is upregulated in ovarian cancer by Wnt/β-catenin pathway, and the silencing of MICAL-L2 is correlated with the decrease of SNAIL, TWIST, ZEB1 and ZEB2 [11], which are the transcription factors of EMT [12]. MICAL2 knockdown results in ROS decrease, viability reduction, and loss of cancer motility and invasion [13]. In this review, we summarized the main functions of MICALs, and analyzed the mechanism of MICAL2 accelerating tumor, including SRF/MRTF-A pathway, Sema/Plexin pathway and ROS production.
The structures of MICALs
MICAL protein is composed of a flavorprotein monooxygenase (MO) domain, a calponin homology domain (CH), a LIM (Lin1-1, Isl-1and Mec-3), and C-terminal domain (CTD) (Figure 1). The CTD includes a proline-rich region, a glutamic acid-repeat, and a coiled-coil motif [1]. However, MICAL2 doesn't contain CTD, and MICAL-like (MICAL-L) shares a same structure with MICALs except for N-terminal Redox enzymatic region (Figure 1). In human cells, MICAL-1 was mainly found in the cytosol, and MICAL-3 has also been found in the cytosol, while MICAL-2 was enriched in the nuclear [14].
A diagram of MICALs structure. MICAL1 is composed of monooxygenase (MO), calponinhomology (CH), LIM domain (LIM), and C-terminal domains (CTD). MICAL2 is composed of MO, CH, and LIM domains. MICAL3 is composed of MO, CH, LIM and CTD domains. MICAL-L1, 2 are composed of CH, LIM, and CTD domains.
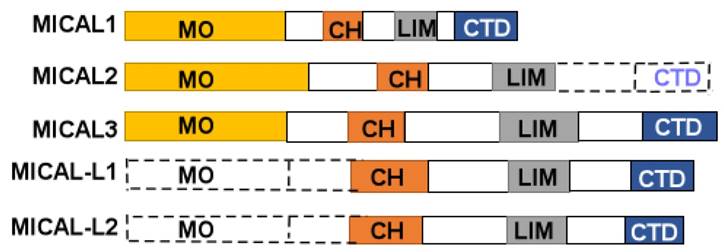
The MO domain is located at the N-terminal part of MICAL protein, and covers about 500 amino acids. Within the MICAL MO domain, three separate conversed motifs can be discemed that defined the flavin adenine dinucleotide (FAD) binding domain (FBD) present in flavoprotein monooxygenases [3]. The main function of MO domain is NADPH-dependent methionine sulfoxidase activity. MICAL selectively modifies F-actin's methionine (M) 44 and 47 residues by adding two oxygens through MO domain [15]. This MO domain can produce ROS [16]. Collapsin response mediator proteins (CRMP) may serve a substrate binding to MO domain, and activate Sema1A axon guidance [17]. In the Drosophila test, MO domain can lead to a loss of axon repulsion induced by Sema [18]. And the monooxygenase inhibitor, epigallocatechin gallate (EGCG) can reduce the repulsive effects of Sema3A and Sema3F in vitro [19]. Zhou et al reported that MICAL1 can use its MO domain to suppress independently apoptosis [20].
The CH domain follows MO domain within MICALs. The main function of CH domain is as an actin-binding protein to involve in muscle contractility and non-muscle cell motility. There are three types of CH domain, type1, type2 and type3. The type1 and type2 domains are coordinated one by one in cytoskeletal proteins, and type2 can accelerate type1 to bind to F-actin [21]. The type 3 domain involves in regulating muscle contraction and signal proteins [22]. The recent studies showed that CH domain can cause an increase of KNADPH, improve catalytic efficiency of monooxygenase reaction, but don't bind directly to F-actin [23]. CH domain advances recognition of F-actin and NADPH, and accelerates NADPH oxidation of MO domain activity in the presence of F-actin [24]. So CH domain is important to MO domain functions.
The LIM domain is composed of small cysteine-rich zinc-finger structures, and connected to CH domains by a variable region in all MICAL proteins. The zinc-finger of LIM domain serves mainly as a docking site for kinases, transcription factors, and cytoskelatal proteins, which involves in various cellular functions such as cytoarchitecture, cell adhesion and motility, and signal transduction [25]. For example, LIM domain of MICAL-L2 interacts with F-actin, and promotes F-actin cross linking [26].
The CTD domain is existed in C-terminal domain, and it is composed of aproline-rich region, glutamic acid-repeat, and coiled-coil motif. In the proline-rich region, there is a PPKPP (Pro-Pro-Lys-Pro-Pro) motif that interacts with SH3 domain of Cas family members, Cas, and CasL [1]. The glutamic acid-repeat region in MICAL-1 is poorly understood its function [19]. In the coiled-coil motif, there exists a piexin-interacting region that is similar to the alpha-helical region [18]. Furthermore, CTD domain may autoinhibit the activity of MO domain [17], likewise, CTD domain also interacts with vimentin, a protein components of intermediate filaments in mesenchymal tissues [1]. And CTD domain interacts with Rad small GTPases, regulating vesicle fusion at the plasma membrane [25].
MICAL family and its functions
MICALs have been found in brain, skeletal muscle, heart, fibroblasts, lung, kidney, bone marrow, thymus, spleen, liver, and testis - as well as in various cell lines and cancer cell lines [27]. MICAL proteins participate in various cellular functions. The main functions of MICALs in cell are redox regulation of cytoskeleton, regulation of axon guidance, dendritic pruning, and synaptic bouton distribution [3].
The important role of MICALs is as actin disassembly factors to directly regulate actin cytoskeleton and intracellular trafficking [28]. MICAL1 serves as a cytoskeletal regulator to connect CasL to intermediate filaments and interact with vimentin through COOH-terminal region, leading to cytoskeletal rearrangement [1]. In the drosophila experiment, Hung et al demonstrated that MICALs alter the instability of F-actin and reorganize parallel F-actin networks into meshwork arrays [29]. All MICALs can mediate vesicle trafficking between endoplasmic reticulum and Golgi complex by binding Rab1A/B [3]. MICAL3 can link Rab8A and ELKS (a Rab6-interacting) to promote vesicle docking and fusion [25]. MICAL-L1 interacting with Rab8, Rab10, Rab13, Rab15, Rab35 or Rab36, can promote endocytic recycling of transferrin and β1 integrin receptors through binding to EH domain-containing protein 1 (EHD1) on recycling endosomes and regulating trafficking [30]. The main function of EHD1 is interacting with Arf6 and Rabs to control membrane and receptor trafficking [31]. Likewise, Rab13 is mainly interacting with MICAL-L2 to mediate endocytic recycling of occluding [32]. In a recent work, Sun et al found that MICAL-L2 and Rab13 formed an insulin-dependent complex in vesicle traffic, which increasing Glucose transporter type (GLUT) 4 on plasma membrane [33].
NDR (nuclear Dbf2-related) kinase is a number of the protein kinases that regulate cellular growth and apoptosis by catalyzing their relevant substrate. MammalianSTE20-like 1 (MST1) is a tumor suppressor protein regulating cell proliferation and apoptosis [34]. NDR1/2 kinase mediates apoptosis through MST1 downstream. MICAL1 can compete with MST1 for NDR binding to downregulate MST1-NDR apoptotic signaling pathway. MICAL1 combines with hydrophobic motif of NDR1 and colocalizes with NDR1 in cells, and ectopic expression of MICAL1 downregulates NDR kinase activation and activity [20]. Kanda et al reported that MICAL-L2 is involved in epithelial cell scattering. MICAL-L2 knockdown can inhibit Madin-Dar by canine kidney (MDCK) cells scattering, but re-expression of MICAL-L2 can rescue cells scattering. Rab13 and MICAL-L2 may decrease cell-cell adhesion and increase cell motility to break the balance between cell-cell adhesion and cell motility [35]. MICAL-L2 can bind to GTP-bind form of Rab13 by its C -terminus to mediate the endocytic recycling of occludin and the formation of functional tight junctions [32]. Rab13 is activated by its effector MICAL-L2 to recruit actin-binding proteins to promote migrating cell. Moreover, Rad13 can control the recycle of integrins and play a key role in cancer metastasis. Actinin-4 links MICAL-L2 to F-actin, and recruits MICAL-L2 to cell-cell junctions, and forms functional tight junction, which are associated with cell motility and cancer invasion [36]. Throughout epithelial junctional development from establishment to maturation of cell-cell adhesion, Rab13 and MICAL-L2 may regulate the reorganization of actin cytoskeleton [37]. Rab13 and MICAL-L2 regulate the transport of claudin-1 and occludin to plasma membrane. And Rab8 and MICAL-L2 also mediate the recycling of E-cadherin to plasma membrane by using a Ca2+ switch [38].
MICALs high expresses in tumors and promotes tumor progress
In Drosophila, Mical gene mutations were found, and gene mutation mediated abnormal MICALs high expression [39]. Xu Y, et al reported that Mical gene copy number was associated with MICAL expression, high gene number had high expression of MICAL2 [40]. In cellular experiments, low-serum condition induces MICAL2 overexpression [14]. MICALs expression is linked to various diseases including cancers [41]. MICAL2-PV overexpresses in prostate cancer cells, and MICAL2 may activate c-Met to increase invasive ability of prostate cancer cells [4]. Additionally, Sema6A and MICAL1 are highly expressed in melanoma, and MICAL1 as an effector of Sema/Plexin axis regulates Sema6A. Sema6A complex with plexins regulates melanoma cell growth, motility, and invasion [6]. MICAL1 overexpresses in breast cancer cell, and promotes cell migration and invasion by activating ROS/Akt signaling [7]. Likewise, MICAL-L2 protein is up-regulated in ovarian cancer, and MICAL-L2 silencing suppresses ovarian cancer proliferation, migration and invasion ability [11]. It was also found that MICAL-L2 silencing induces ovarian cancer cells MET [11]. In a recent study, it is also found that MICAL2 overexpresses in gastric cancer (GC), non small cell lung cancer (NSCLC) and renal carcinoma, and MICAL2 mRNA expression is higher in GC diffuse histotype than intestinal histotype, is higher in NSCLC adenocarcinoma than squamous cell carcinoma. In additional, MICAL2 expression is high in aggressive primary cancer and metastatic emboli, but undetected in cancer cells at metastatic site [13].
MICAL2 participates in cell proliferation
In the renal carcinoma, Mical2 gene expression has a relevance with differentiation and proliferation [13]. MICAL2 is involved in the proliferation of prostate cancer through mediating NADPH oxidation [42]. Mical2 gene expression is correlated with the expression of MKI67 in bladder cancer, and MICAL2 may involve in bladder cancer cell proliferation [43]. Ectopic expression of MICAL2 in breast cancer cell augments epidermal growth factor receptor (EGFR) expressions, further its mechanism is that MICAL2 triggers a delay in EGFR degradation. MICAL2 may promote cell proliferation and migration [44].
MICAL2 as a regulator of EMT in cancer metastasis
The gene expression analysis of cancer metastasis had proved MICAL2 as an important gene in EMT regulation [45]. EMT plays an important role in cell growth, tissue repair, stem cell transformation, and promoting migratory and invasive properties [12, 46]. EMT process is a loss of cell-cell adhesion and cell polarity, and acquisition of migratory and invasive properties. Furthermore, EMT induces stem cell properties, prevents apoptosis and senescence, and contributes to immune suppression [47, 48]. EMT process involves the loss of epithelial cell junctional proteins such as E-cadherin, claudins and ZO-1, and the increasing in mesenchymal markers such as N-cadherin, vimentin and fibronectin and cytoskeletal reorganization [49]. MICAL2 could oxygenate M44 and 47 amino acid residues on F-actin, resulting in F-actin disassembly and cytoskeleton dynamics [15, 42]. When F-actin depolimerization and cytoskeleton dynamics change, the epithelial cell has ability of EMT.
1) MICAL2 regulates cell EMT through SRF/MRTF-A signaling pathway
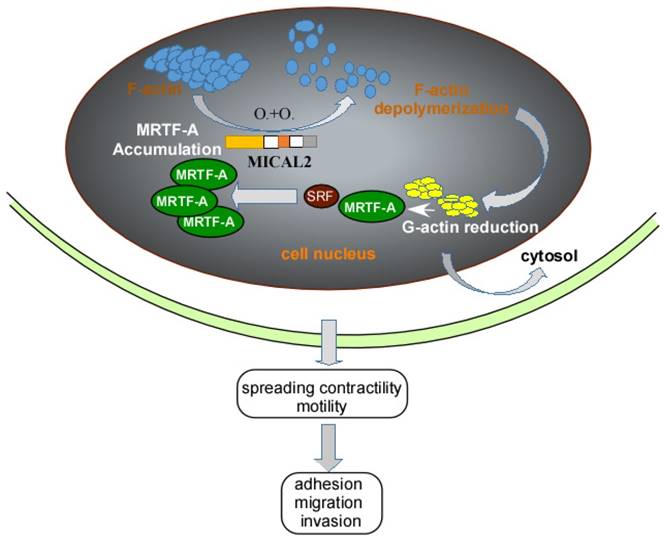
Myocardin-related transcription factor (MRTF)-A, a cofactor of serum response factor (SRF) participates in EMT progress [50]. MRTF and SRF activities are dependent on Rho-actin signaling, Rho GTPases regulates G-actin and F-actin to influence the activation of SRF/MRTF-A-dependent gene transcription [51]. MICAL2 can induce F-actin depolymerization by Redox modification. F-actin depolymerization reduces G-actin, causing the interaction of SRF and MRTF-A, finally resulting in MRTF-A accumulation in nucleus (Figure 2). MRTF-A accumulation activates SRF/MRTF-A-dependent gene transcription [14]. And activation of SRF/MRTF-A-dependent gene transcription increases the capacity of adhesion, migration and invasion of cancer cells, and inhibits migration of adherent non-invasive cells [51, 52] (Figure 2). Additionally, MRTF-A is a downstream of transforming growth factor (TGF) β in the regulation of TGFβ1-induced EMT [53, 54]. MRTF-A could also stimulate transcriptional activation of actin cytoskeletal genes to change the organization of actin cytoskeleton, and downregulate E-cadherin and upregulate ZEB1, vimentin and slug expression in cancer cell [50, 55].
Schemastic illustration of MICAL2 functions. MICAL2 induces F-actin depolymerization by redox modification. The depolymerization of F-actin reduces G-actin and accumulates MRTF-A through clustering. The accumulation of MRTF-A in nucleus actives SRF/MRTF-A-dependent gene transcription, and finally promotes cancer cells migration and invasion.
2) MICAL2 involves in cell EMT via Sema/Plexin pathway
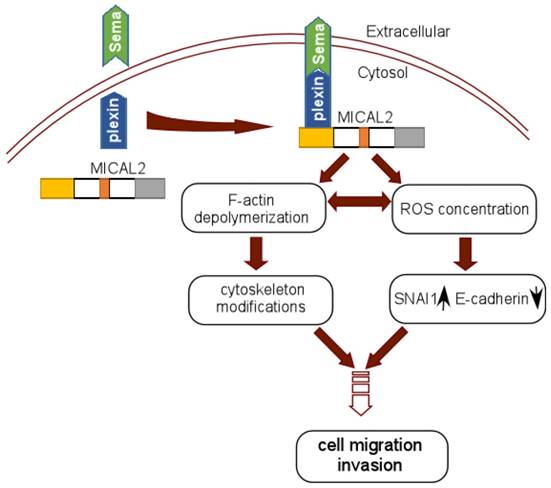
In Sema/Plexin pathway, MICAL2 serves as a platform to recruit Sema to the site of plexin activation, which enhances MICAL2's monoxygenase activity or local intracellular concentration of ROS [56]. MICAL2 may also link Sema and plexin to depolymerize F-actin and induce actin rearrangement [29] (Figure 3). Termam et al reported that MICALs interact with neuronal plexin A receptor [18], so Sema, plexin, and MICALs constitute a Sema/plexin/mical pathway. The Sema/plexin/mical pathway involves in repulsive guidance, regulating cell morphology and migratory functions. Downregulation of E-cadherin and upregulation of ß-catenin are the key step to EMT, Sema also affects the expressions of adhesion protein E-cadherin and ß-catenin [57], and regulates tumor cell EMT [58]. MICAL2-PV overexpresses in prostate cancer cell, its expression is strongly concordant with classification of prostatitis, lymphatic metastasis, vascular invasion and Gleason scores of prostatic cancer. Likewise, it was found that MICAL2-PV expression is correlated with c-Met level, MICAL2 may upregulate c-Met to promote tumor cell invasive [4].
Schematic reprentation of MICAL2 as Sema platform. Sema interacts with plexin and activates plexin, and the activated plexin enhances MICAL2 monoxygenase activity and ROS intracellular concentration. MICAL2 accelerates F-actin depolymerization and increases cancer cell migration and invasion. ROS concentration induces EMT to promote migration and invasion.
The functions of MICALs
| MICAL | associated protein | Function | Reference |
|---|---|---|---|
| Mical-1 | vimentin and CasL | cytoskeletal rearrangement | [1] |
| CRMP-2 | Axon guidance | [28] | |
| PlexA-1 | Axon guidance | [17] | |
| NDR1 | apoptosis | [20] | |
| Mical-2 | F-actin | cytoskeletal rearrangement | [14] |
| Rab1 | vesicle trafficking | [29] | |
| Mical-3 | Rab8A and ELKS | vesicle docking and fusion | [25] |
| F-actin | cytoskeletal rearrangement | [14] | |
| Mical-L1 | Rab8,10, 13,15,35 and36 | recycling endosomes and regulating trafficking | [30] |
| EHD1 | Focal adhesion and endocytic function | [31, 32] | |
| Mical-L2 | Rab13 | neurite outgrowth and exocytosis | [33, 34] |
| F-actin | cytoskeletal rearrangement | [35] |
3) MICAL2 mediates cell EMT through inducing ROS production
MICAL2 induces constitutive F-actin depolymenzation by increasing the generation of ROS [26]. Some experiments confirmed that ROS increase promotes EMT in cytoskeleton dynamics and microenvironment [10] (Figure 3). MICAL2 uses NADPH to produce ROS through its MO domain binding to FAD [13]. This progress produces H2O2, and then H2O2 affects actin filaments, subsequently causes actin remodeling [16]. On the one hand, ROS generation mediated by MICAL2 is significant for actin-regulatory function. On the other hand, the high levels of ROS can increase metabolic activity, mitochondrial dysfunction and peroxisome activity, and deregulates cellular receptor signaling, oncogene activity and enhances activity of COXs, LOXs and thymidine phosphorylase [59]. ROS can promote EMT process by stimulating overexpression of RTKs, EGFR and c-Met/HGFR, and hyper activation of Rac-1 GTPase, hypoxia and Ras [10]. ROS also induces the repression of E-cadherin and the increase stability of SNAL1, and cell-cell disengagement [10].
Conclusions
In summary, MO domain is a critical structure of MICALs, and its pivotal function is redox regulation to the cytoskeleton. MICAL2 induces cancer cell invasion and proliferation, its mechanism is to induce F-actin depolymerization by redox modification. F-actin depolymerization interacts with MRTF-A and results in MRTF-A accumulation. MRTF-A accumulation in nucleus actives SRF/MRTF-A-dependent gene transcription, promoting cancer cells migration and invasion. In addition, MICAL2 also mediates ROS production, activates wnt/β-catenin signaling and Sema/Plexin pathway, and promotes tumor cell EMT. However, some mechanisms of MICALs involving cancer progress are still undefined.
Acknowledgements
We thank the members in Clinical Laboratory of Zhuhai Hospital for contributions and helpful discussion.
Author contributions
Cai Y wrote the paper. Tang F and Lu J designed and revised the paper.
Funding
This work was supported in part by the National Natural Science Foundation of China (81372282, 81402368, 81402265, 81502346), Program for New Century Excellent Talents in University, NCET (NCET-06-0685), Guangdong Natural Science Foundation (S2013010013360), China Postdoctoral Science Foundation funded project (2016M592580).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Suzuki T, Nakamoto T, Ogawa S, Seo S, Matsumura T, Tachibana K. et al. MICAL, a novel CasL interacting molecule, associates with vimentin. J Biol Chem. 2002;277:14933-41
2. Xue Y, Kuok C, Xiao A, Zhu Z, Lin S, Zhang B. Identification and expression analysis of mical family genes in zebrafish. J Genet Genomics. 2010;37:685-93
3. Zhou Y, Gunput RA, Adolfs Y, Pasterkamp RJ. MICALs in control of the cytoskeleton, exocytosis, and cell death. Cell Mol Life Sci. 2011;68:4033-44
4. Ashida S, Furihata M, Katagiri T, Tamura K, Anazawa Y, Yoshioka H. et al. Expression of novel molecules, MICAL2-PV (MICAL2 prostate cancer variants), increases with high Gleason score and prostate cancer progression. Clin Cancer Res. 2006;12:2767-73
5. Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D. et al. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol. 2002;4:720-4
6. Loria R, Bon G, Perotti V, Gallo E, Bersani I, Baldassari P. et al. Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget. 2015;6:2779-93
7. Deng W, Wang Y, Gu L, Duan B, Cui J, Zhang Y. et al. MICAL1 controls cell invasive phenotype via regulating oxidative stress in breast cancer cells. BMC cancer. 2016;16:489
8. Qin X, Kuang H, Chen L, Wei S, Yu D, Liang F. Coexpression of growth differentiation factor 11 and reactive oxygen species in metastatic oral cancer and its role in inducing the epithelial to mesenchymal transition. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:697-706
9. Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011;1:2045-3701
10. Giannoni E, Parri M, Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid Redox Signal. 2012;16:1248-63
11. Zhu LY, Zhang WM, Yang XM, Cui L, Li J, Zhang YL. et al. Silencing of MICAL-L2 suppresses malignancy of ovarian cancer by inducing mesenchymal-epithelial transition. Cancer Lett. 2015;363:71-82
12. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
13. Mariotti S, Barravecchia I, Vindigni C, Pucci A, Balsamo M, Libro R. et al. MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget. 2016;7:1808-25
14. Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR. et al. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 2014;156:563-76
15. Hung RJ, Pak CW, Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science (New York, NY). 2011;334:1710-3
16. Giridharan SS, Caplan S. MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid Redox Signal. 2014;20:2059-73
17. Schmidt EF, Shim SO, Strittmatter SM. Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J Neurosci. 2008;28:2287-97
18. Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002;109:887-900
19. Pasterkamp RJ, Dai HN, Terman JR, Wahlin KJ, Kim B, Bregman BS. et al. MICAL flavoprotein monooxygenases: expression during neural development and following spinal cord injuries in the rat. Mol Cell Neurosci. 2006;31:52-69
20. Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, Li KW. et al. MICAL-1 is a negative regulator of MST-NDR kinase signaling and apoptosis. Mol Cell Biol. 2011;31:3603-15
21. Gimona M, Djinovic-Carugo K, Kranewitter WJ, Winder SJ. Functional plasticity of CH domains. FEBS Lett. 2002;513:98-106
22. Gimona M, Mital R. The single CH domain of calponin is neither sufficient nor necessary for F-actin binding. J Cell Sci. 1998;111:1813-21
23. Vitali T, Maffioli E, Tedeschi G, Vanoni MA. Properties and catalytic activities of MICAL1, the flavoenzyme involved in cytoskeleton dynamics, and modulation by its CH, LIM and C-terminal domains. Arch Biochem Biophys. 2016;593:24-37
24. Alqassim SS, Urquiza M, Borgnia E, Nagib M, Amzel LM, Bianchet MA. Modulation of MICAL Monooxygenase Activity by its Calponin Homology Domain: Structural and Mechanistic Insights. Sci Rep. 2016;6:22176
25. Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E. et al. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol. 2011;21:967-74
26. Giridharan SS, Rohn JL, Naslavsky N, Caplan S. Differential regulation of actin microfilaments by human MICAL proteins. J Cell Sci. 2012;125:614-24
27. Hung RJ, Terman JR. Extracellular inhibitors, repellents, and semaphorin/plexin/MICAL-mediated actin filament disassembly. Cytoskeleton (Hoboken, NJ). 2011;68:415-33
28. Wu H, Hung RJ, Terman JR. A simple and efficient method for generating high-quality recombinant Mical enzyme for in vitro assays. Protein Expr Purif. 2016;127:116-24
29. Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N. et al. Mical links semaphorins to F-actin disassembly. Nature. 2010;463:823-7
30. Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, Caplan S. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol Biol Cell. 2009;20:5181-94
31. Giridharan SS, Cai B, Naslavsky N, Caplan S. Trafficking cascades mediated by Rab35 and its membrane hub effector, MICAL-L1. Commun Integr Biol. 2012;5:384-7
32. Terai T, Nishimura N, Kanda I, Yasui N, Sasaki T. JRAB/MICAL-L2 is a junctional Rab13-binding protein mediating the endocytic recycling of occludin. Mol Biol Cell. 2006;17:2465-75
33. Sun Y, Jaldin-Fincati J, Liu Z, Bilan PJ, Klip A. A complex of Rab13 with MICAL-L2 and alpha-actinin-4 is essential for insulin-dependent GLUT4 exocytosis. Mol Biol Cell. 2016;27:75-89
34. Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet. 2010;11:60-74
35. Kanda I, Nishimura N, Nakatsuji H, Yamamura R, Nakanishi H, Sasaki T. Involvement of Rab13 and JRAB/MICAL-L2 in epithelial cell scattering. Oncogene. 2008;27:1687-95
36. Nakatsuji H, Nishimura N, Yamamura R, Kanayama HO, Sasaki T. Involvement of actinin-4 in the recruitment of JRAB/MICAL-L2 to cell-cell junctions and the formation of functional tight junctions. Mol Cell Biol. 2008;28:3324-35
37. Sakane A, Abdallah AA, Nakano K, Honda K, Ikeda W, Nishikawa Y. et al. Rab13 small G protein and junctional Rab13-binding protein (JRAB) orchestrate actin cytoskeletal organization during epithelial junctional development. J Biol Chem. 2012;287:42455-68
38. Yamamura R, Nishimura N, Nakatsuji H, Arase S, Sasaki T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol Biol Cell. 2008;19:971-83
39. Beuchle D, Schwarz H, Langegger M, Koch I, Aberle H. Drosophila MICAL regulates myofilament organization and synaptic structure. Mech Dev. 2007;124:390-406
40. Xu Y, Zhang L, Shi T, Zhou Y, Cai H, Lan X. et al. Copy number variations of MICAL-L2 shaping gene expression contribute to different phenotypes of cattle. Mamm Genome. 2013;24:508-16
41. Wilson C, Terman JR, Gonzalez-Billault C, Ahmed G. Actin filaments-A target for redox regulation. Cytoskeleton (Hoboken, NJ). 2016;73:577-95
42. McDonald CA, Liu YY, Palfey BA. Actin stimulates reduction of the MICAL-2 monooxygenase domain. Biochemistry. 2013;52:6076-84
43. Ho JR, Chapeaublanc E, Kirkwood L, Nicolle R, Benhamou S, Lebret T. et al. Deregulation of Rab and Rab effector genes in bladder cancer. PloS one. 2012;7:e39469
44. Wang Y, Deng W, Zhang Y, Sun S, Zhao S, Chen Y. et al. MICAL2 promotes breast cancer cell migration by maintaining epidermal growth factor receptor (EGFR) stability and EGFR/P38 signalling activation. Acta physiologica (Oxford, England). 2017
45. Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P. et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158:1094-109
46. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995;154:8-20
47. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
48. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
49. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548-58
50. Morita T, Mayanagi T, Sobue K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J Cell Biol. 2007;179:1027-42
51. Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257-68
52. Leitner L, Shaposhnikov D, Mengel A, Descot A, Julien S, Hoffmann R. et al. MAL/MRTF-A controls migration of non-invasive cells by upregulation of cytoskeleton-associated proteins. J Cell Sci. 2011;124:4318-31
53. O'Connor JW, Gomez EW. Cell adhesion and shape regulate TGF-beta1-induced epithelial-myofibroblast transition via MRTF-A signaling. PloS one. 2013;8:e83188
54. Gupta M, Korol A, West-Mays JA. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor beta-induced epithelial to mesenchymal transition of lens epithelial cells. Mol Vis. 2013;19:1017-28
55. Song Z, Liu Z, Sun J, Sun FL, Li CZ, Sun JZ. et al. The MRTF-A/B function as oncogenes in pancreatic cancer. Oncol Rep. 2016;35:127-38
56. Ventura A, Pelicci PG. Semaphorins: green light for redox signaling? Sci STKE. 2002;2002:pe44
57. Herman JG, Meadows GG. Increased class 3 semaphorin expression modulates the invasive and adhesive properties of prostate cancer cells. Int J Oncol. 2007;30:1231-8
58. Capparuccia L, Tamagnone L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment-two sides of a coin. J Cell Sci. 2009;122:1723-36
59. Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881-96
Author contact
![]() Corresponding author: Dr. Faqing Tang, Clinical Laboratory, Hunan Cancer Hospital & The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, 410008, Changsha, China. E-mail: tangfaqing33com; Tel.: 86-756-2157645; Fax: 86-756-2157634
Corresponding author: Dr. Faqing Tang, Clinical Laboratory, Hunan Cancer Hospital & The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, 410008, Changsha, China. E-mail: tangfaqing33com; Tel.: 86-756-2157645; Fax: 86-756-2157634