Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(4):757-766. doi:10.7150/jca.22319 This issue Cite
Research Paper
Upregulation of HES1 Promotes Cell Proliferation and Invasion in Breast Cancer as a Prognosis Marker and Therapy Target via the AKT Pathway and EMT Process
Xiaoying Li1, Yu Cao1, Mu Li2, Feng Jin1 ![]()
1. Department of Breast Surgery, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China
2. Department of General Surgery, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China
Received 2017-8-10; Accepted 2017-12-3; Published 2018-2-11
Abstract
HES1 is a transcriptional repressor involved in cell differentiation and proliferation as well as in various cancer developments, but its expression pattern and biological roles in breast cancer have not been examined. In this study, we assessed HES1 expression in breast cancer tissues using immunohistochemistry and Western blot analyses and investigated HES1 function using MTT and Matrigel invasion assays. Significant relationships were observed between HES1 upregulation and advanced TNM stage (p=0.011), node metastasis (p=0.043), negative oestrogen receptor expression (p=0.001) and triple-negative status (p=0.001). HES1 overexpression was correlated with poor prognosis in breast cancer patients (p<0.05). The MTT and Matrigel invasion assays showed that silencing HES1 in MDA-MB-231 cells decreased cell proliferation and invasion, whereas overexpression of HES1 in MCF-7 cells enhanced its proliferation and invasion. Further analyses showed that silencing HES1 downregulated p-AKT and impeded epithelial-mesenchymal transition (EMT), whereas overexpression of HES1 upregulated AKT phosphorylation and induced EMT. Our study demonstrated that HES1 upregulation is a predictor of poor prognosis in human breast cancers and might be a critical contributor to the proliferation and invasion of breast cancer cells. Moreover, the proportion of cells with overexpression of HES1 in triple-negative breast cancer (TNBC) samples was significantly higher. Thus, HES1 might be a potential therapeutic target in the treatment of TNBC.
Keywords: HES1, triple-negative breast cancer (TNBC), epithelial-mesenchymal transition (EMT), AKT, phosphatase and tensin homologue (PTEN), prognosis
Introduction
Breast cancer is one of the most common malignancies, and it is the second leading cause of mortality in women [1]. Thus, new predictive biomarkers of tumour proliferation and targeted therapies are urgently needed [2,3]. Hairy and enhancer of split homolog-1 (HES1) is a basic helix-loop-helix (bHLH) transcriptional repressor. It is a mammalian counterpart of the Hairy and Enhancer of split protein and plays a critical role in many physiological processes and various morphogenetic events, including cellular differentiation, cell cycle arrest, apoptosis and self-renewal [4-7]. HES1 can be induced by both the canonical and non-canonical Notch pathways [8-11]. HES1 protein contains 3 conserved domains: the bHLH, Orange and WRPW domains. HES1 transcriptional repression is mediated through the bHLH domain, which binds to the N box (CACNAG) and Trp-Arg-Pro-Trp (WRPW) domain which interacts with the co-repressor transducin-like enhancer of split (TLE) [12-14].
Elevated HES1 expression is correlated with several neoplastic conditions, and its upregulation was found in multiple tumours, including non-small cell lung cancer, ovarian and colonic carcinomas and embryonal brain tumours [15-18]. In addition, HES1 has been shown to promote cancer cell proliferation, cell survival and cell metastasis by evading tumour cell differentiation or regulating various signalling pathways [5,19,20]. Based on these results, we hypothesized that HES1 may participate in breast cancer progression. To date, the expression pattern and biological roles of HES1 itself in human breast cancer are unknown. In the present study, we investigated the relationship between HES1 and breast cancers to determine whether HES1 is a contributor to malignancy and explored its underlying mechanism.
Materials and Methods
This study was approved by the Institutional Review Board of the China Medical University, according to the Declaration of Helsinki. All participants provided written informed consent.
Patients and specimens
We collected 150 breast cancer samples (32 triple-negative samples and 118 non-triple-negative samples) from patients who were diagnosed with primary breast cancer at the First Affiliated Hospital of the China Medical University between January 2009 and September 2011. The 150 patients enrolled in our study met the following criteria: (1) patients were diagnosed with invasive ductal breast cancer in clinical stages T1-T2, N0-N2 and M0; (2) none of the patients had received preoperational radiotherapy or chemotherapy; (3) all patients underwent complete surgical resection; and (4) all patients received routine standardized treatment after surgery according to the NCCN guidelines. Specimens from each individual were obtained through routine surgery dissections. All 150 patients were followed-up for a median of 77.15 months (range, 12-84 months), and during follow-up, there were 26 patients who relapsed or progressed to metastasis and 25 deaths due to relapse or distant metastases.
Immunohistochemical analysis
Tumour samples were fixed with 10% neutral formalin, embedded in paraffin, and sectioned into 4-µm-thick sections. Immunostaining was performed using the streptavidin-peroxidase immunohistochemical method. The sections were incubated overnight at 4ºC with a rabbit monoclonal HES1-specific antibody (1:300 dilution; ab108937, Abcam, Cambridge, UK). As a blank control, phosphate-buffered saline was used in place of the antibody. The sections were then incubated with biotin-labelled secondary antibodies (Ultrasensitive; MaiXin, Fuzhou, China) at 37°C for 30 min and were then developed with diaminobenzidine.
Positive nuclear staining was defined as positive results. We calculated the percentage of positively stained cells. The staining intensity was categorized as follows: 0=negative, 1=weak, 2=moderate and 3=strong. The percentage of positively stained cells was scored as follows: 0=no staining; 1, ≤10%; 2, 11-25%; 3, 26-50%; and 4, ≥51%. The scores of each tumour sample were multiplied to yield a final score of 0-8; tumour samples with 4-8 were defined as HES1 positive/overexpression.
Cell culture
MCF10A, MCF-7, ZR-75-1, ZR-75-30, SK-BR-3, MDA-MB-453, MDA-MB-231, HCC-1937, Hs578T cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM (Dulbecco's modified Eagle's medium) containing 10% foetal bovine serum (FBS) in a sterilized and humid incubator.
Plasmid transfection
The HES1 (NM_005524) plasmid was purchased from GeneChem. Attractene transfection reagent (Qiagen, Hilden, Germany) was used to transfect the HES1 plasmid according to the manufacturer's instructions. MCF-7 was transferred with the HES1 plasmid.
HES1 knockdown by shRNA
The HES1 shRNA sequences (shHES1 1#: HES1-RNAi (4387-1) GCCAGTTTGCTTTCCTCAT; shHES1 2#: HES1-RNAi (4388-1) AGATCAATGCCATGACCTA; shHES1 3#: HES1-RNAi (4389-1) GGACATTCTGGAAATGACA) were synthesized by GeneChem and transfected into cells using DharmaFECT1 (Qiagen, Hilden, Germany) according to the manufacturer's directions. MDA-MB-231 was transferred with the HES1 shRNA ((shHES1 1#, shHES1 2#, shHES1 3#). Forty-eight hours after transfection, the protein levels were assessed using Western blotting.
Western blot analysis
Total protein was extracted using the lysis buffer (Pierce, Rockford, IL, USA) and quantified through the Bradford method. Each 50-μg protein sample was separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), and the membranes were incubated with antibodies against HES1 (1:500 dilution, Abcam, ab108937, Cambridge, UK), PTEN, p-AKT, AKT, E-cadherin, N-cadherin, and vimentin (Cell Signalling Technology) and anti-β-actin (Abcam, Cambridge, UK) overnight at 4°C. After incubation, the bound proteins were determined by ECL (Pierce) using a BioImaging System (UVP Inc., Upland, CA, USA).
MTT assay
The transfected cells were seeded in 96-well plates at a concentration of 3000 cells per well and incubated for 5 days. Then, 20 μL of 5 mg/ml MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, thiazolyl blue) dye solution was added to each well, and the plates were cultured for 4 h at 37°C. The MTT solution was removed, and 150 μL of DMSO was used to solubilize the MTT formazan. The results were measured spectrophotometrically at a wavelength of 490 nm.
Transwell migration and Matrigel invasion assay
For the Transwell migration assay, 1x105 cells in 100 μL of DMEM without FBS were placed in the top chamber of Transwell migration chambers (8-μm BioCoat Control Inserts, Becton Dickinson Labware, Bedford, MA, USA), and 600 μL of DMEM with 10% FBS was added to the lower chamber to serve as the chemoattractant. After incubation for 24 h, the non-migrated cells on the top surface of the Transwell membrane were removed with a cotton swab, and the migrated cells on the lower membrane surface were fixed and stained. The stained cells in 5 randomly selected high-power fields were then counted.
The procedure used for the Matrigel invasion assay was similar to that used for the Transwell migration assay, with the exception that the Transwell membrane was precoated with 20 mL of Matrigel (1:3 dilution, BD Bioscience).
Statistical analysis
All statistical analyses were performed using SPSS version 20.0 (SPSS, Chicago, IL, USA). The correlations between HES1 and clinicopathological factors were analysed by Chi-square tests. Kaplan-Meier survival curves were prepared to evaluate the association between HES1 and patient survival, and the differences between subgroups of patients were compared through Cox regression analysis. p<0.05 was considered statistically significant.
Results
Expression and localization of HES1 in breast cancer tissues
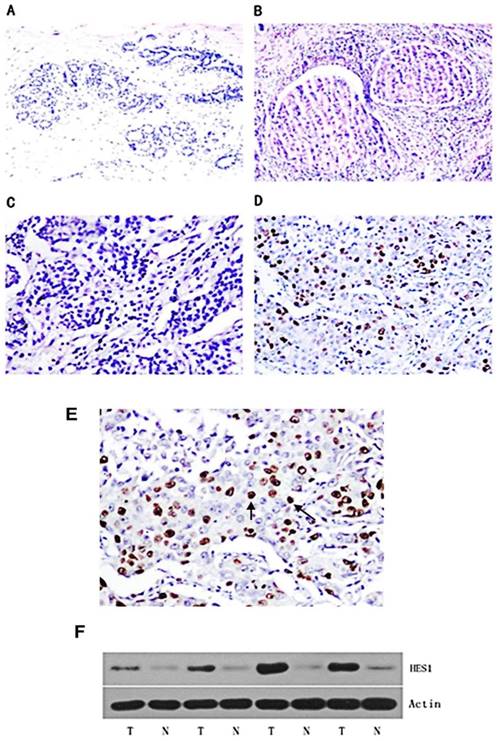
A panel of 150 primary breast cancer specimens and normal mammary gland tissues were analysed by immunohistochemistry. We found that HES1 protein was localized in the nuclear compartment. Negative staining was shown in the negative control using immunoglobulin. Negative expression of HES1 was found in normal breast samples (Figure 1A). Weak nuclear expression was detected in 5 cases of ductal carcinoma in situ (DCIS) (Figure 1B). We found that HES1 was upregulated in 53 of 150 (35.3%) invasive ductal carcinoma (IDC) specimens. Weak/negative HES1 staining was considered normal (Figure 1C). Strong/positive HES1 nuclear staining with a final score ≥4 was considered HES1 overexpression (Figure 1D).
Expression pattern of HES1 protein in breast cancer tissues. (A) Negative nuclear HES1 staining in the majority of normal breast tissues. (B) Negative/weak nuclear HES1 staining in ductal carcinoma in situ (DCIS). (C) Negative nuclear HES1 staining in invasive ductal carcinoma (IDC). (D) Strong nuclear staining of HES1 in invasive ductal carcinoma (IDC). (E) Magnification of HES1 positive staining in TNBC. (F) Western blot analysis of HES1 expression in 4 pairs of breast cancer and corresponding normal tissues. T, breast cancer tissue; N, matched adjacent noncancerous tissue.
We analysed the relationship between HES1 expression and the clinical parameters in order to evaluate the biological role of HES1 in breast cancer progression. As shown in Table 1, there were no correlations between HES1 overexpression and age (p=0.082), tumour size (p=0.097), progesterone receptor status (p=0.107), and ErbB2 status (p=0.407). However, significant correlations were observed between HES1 overexpression and advanced TNM stage (p=0.011), positive node metastasis (p=0.043) and negative oestrogen receptor status (p=0.001). The rate of HES1 overexpression was higher in stage II-III breast cancers (36/81) than that in stage I (17/69) breast cancers. Additionally, 24 of the 32 triple-negative breast cancer (ER-, PR-, and ErbB-2-negative expression, also as TNBC) samples exhibited HES1 overexpression, whereas only 11 of the 39 triple-positive breast cancer (ER-, PR-, and ErbB-2-positive expression) samples showed HES1 overexpression. These data demonstrated that the proportion of HES1-positive samples was significantly higher in TNBCs than that in other breast cancer subtypes (p=0.001), especially than that in triple-positive breast cancers (p<0.001).
Distribution of HES1 status in breast cancer on the basis of clinicopathological characteristics.
| Characteristics | Number of patients | HES1-negative | HES1-positive | P | ||||
|---|---|---|---|---|---|---|---|---|
| Age | 0.082 | |||||||
| <60 | 138 | 92 | 46 | |||||
| ≥60 | 12 | 5 | 7 | |||||
| TNM stage | ||||||||
| I | 69 | 52 | 17 | 0.011 | ||||
| II-III | 81 | 45 | 36 | |||||
| Tumour size | ||||||||
| <2 cm | 98 | 68 | 30 | 0.097 | ||||
| ≥2 cm | 52 | 29 | 23 | |||||
| Lymph node metastasis | ||||||||
| Absent | 90 | 64 | 26 | 0.043 | ||||
| Present | 60 | 33 | 27 | |||||
| Oestrogen receptor | ||||||||
| Absent | 48 | 19 | 29 | 0.001 | ||||
| Present | 102 | 78 | 24 | |||||
| Progesterone receptor | ||||||||
| Absent | 66 | 38 | 28 | 0.107 | ||||
| Present | 84 | 59 | 25 | |||||
| ErbB-2 | ||||||||
| Absent | 107 | 67 | 40 | 0.407 | ||||
| Present | 43 | 30 | 13 | |||||
| Triple-negative | ||||||||
| Absent | 118 | 89 | 29 | 0.001 | ||||
| Present | 32 | 8 | 24 | |||||
| Subtypes | ||||||||
| Triple-negative | 32 | 8 | 24 | <0.001 | ||||
| Triple-positive | 39 | 28 | 11 | |||||
We also investigated HES1 expression using Western blot analyses of freshly isolated breast cancer tissues. HES1 expression levels were significantly higher in the cancer samples compared with those of the corresponding normal samples (T vs N: 96.67±18.01 vs 19.69±3.94, respectively; p < 0.01) (Figure 1F).
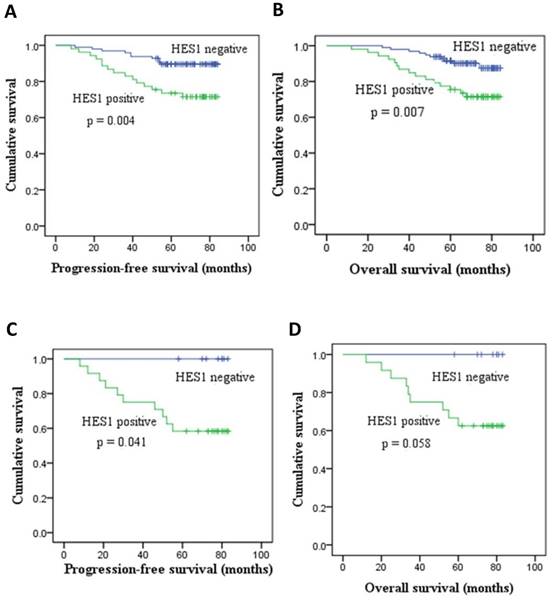
We found that patients with low HES1 levels had increased survival relative to patients with high HES1 levels. The rates of progression-free survival (PFS) and overall survival (OS) of HES1-negative patients were significantly higher than those of HES1-positive patients as determined by Kaplan-Meier survival analysis (p < 0.05) (Figure 2A, 2B). Moreover, the PFS rates of patients with HES1-negative TNBC were statistically higher than those of patients with HES1-positive TNBC as determined using a Kaplan-Meier survival analysis (p=0.041) (Figure 2C). Although there were no statistically significant differences in OS of patients with HES1-positive and HES1-negative TNBC (p=0.058) (Figure 2D), the OS rates of patients with HES1-positive TNBC were obviously lower than those in patients with HES1-negative TNBC. Thus, HES1 might be an important progressive and prognostic factor in breast cancers.
HES1 expression and its correlation with survival in breast cancer. (A-B) HES1 overexpression correlated with poor prognosis and lower PFS and OS of breast cancer patients. (C-D) HES1 overexpression was correlated with lower PFS and OS of patients with TNBC.
HES1 promoted breast cancer cell proliferation and invasion by regulating the AKT signalling pathway and EMT process
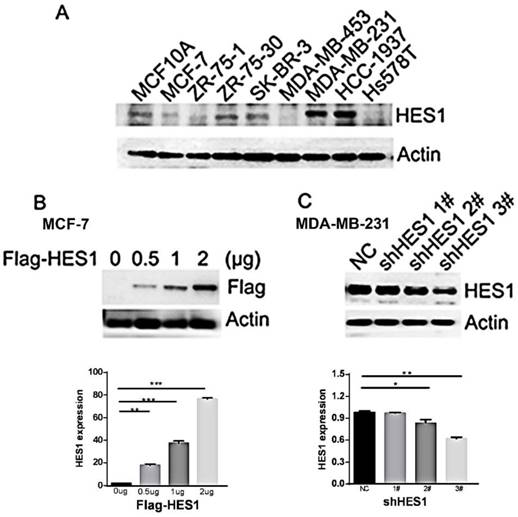
To determine the biological effect of HES1 in breast cancer cells, we detected HES1 expression in a normal breast cell line (MCF10A) and eight breast cancer cell lines using Western blot analyses. We observed low levels of HES1 expression in the MCF10A and MCF7 cell lines and high levels of HES1 expression in the MDA-MB-231 and HCC-1937 cell lines (MCF10A and MCF7 vs MDA-MB-231 and HCC-1937: 1.45±0.03 and 0.76±0.04 vs 2.21±0.05 and 2.38±0.05, respectively; p < 0.001) (Figure 3A). MDA-MB-231 and HCC-1937 cell lines were both derived from TNBC specimens. We depleted HES1 in MDA-MB-231 cells using siRNA and increased HES1 levels in MCF7 cells via HES1 plasmid transfection. Western blot analyses confirmed the HES1 transfection and knockdown efficiencies. HES1 plasmid transfection in MCF7 cells increased HES1 expression compared with cells transfected with a non-specific control (MCF7 con vs HES1 plasmid 2 μg: 1.83±0.05 vs 76.3±0.69, respectively; p < 0.001) (Figure 3B). MDA-MB-231 cells treated with either HES1 shRNA2# or HES1 shRNA3# exhibited decreased HES1 expression in cells compared with cells treated with non-specific control (231 con vs shRNA2#: 0.98±0.01 vs 0.84±0.2, respectively; 231 con vs shRNA3#: 0.98±0.01 vs 0.62±0.01, respectively; p < 0.01) (Figure 3C).
HES1 expression in breast cancer cell lines. (A) Western blot analysis of HES1 expression in the normal breast cell line MCF-10A and the breast cancer cell lines MCF-7, ZR-75-1, ZR-75-30, SK-BR-3, MDA-MB-453, MDA-MB-231, HCC-1937, and Hs578T. (B) Western blot analysis showed that transfection of the HES1 plasmid (0.5 μg, 1 μg, 2 μg) results in its ectopic expression in the MCF-7 cell line. (C) HES1 shRNA (shRNA1#, shRNA2#, shRNA3#) treatment decreases the HES1 protein levels in MDA-MB-231 cells. *p < 0.05, **p < 0.01, ***p < 0.001
MTT assays were performed to determine the effect of HES1 on breast cancer cell proliferation. We found that the HES1-specific shRNA treatment decreased cell survival compared with that of the control siRNA. Increased HES1 levels using HES1 plasmid transfection increased cell survival (Figures 4A and 4B). These results indicate that HES1 is a crucial regulator of cell survival and proliferation in breast cancers.
The biological role of HES1 in the proliferation and invasion of breast cancer cells. (A) MTT assays showed that HES1 upregulation promotes proliferation in MCF-7 cells. (B) MTT assays showed that HES1 depletion inhibits proliferation in MDA-MR-231 cells. (C) HES1 overexpression enhances migration and invasion in MCF-7 cells. (D) HES1 knockdown decreases migration and invasion in MDA-MR-231 cells. *p < 0.05, **p < 0.01, ***p < 0.001.
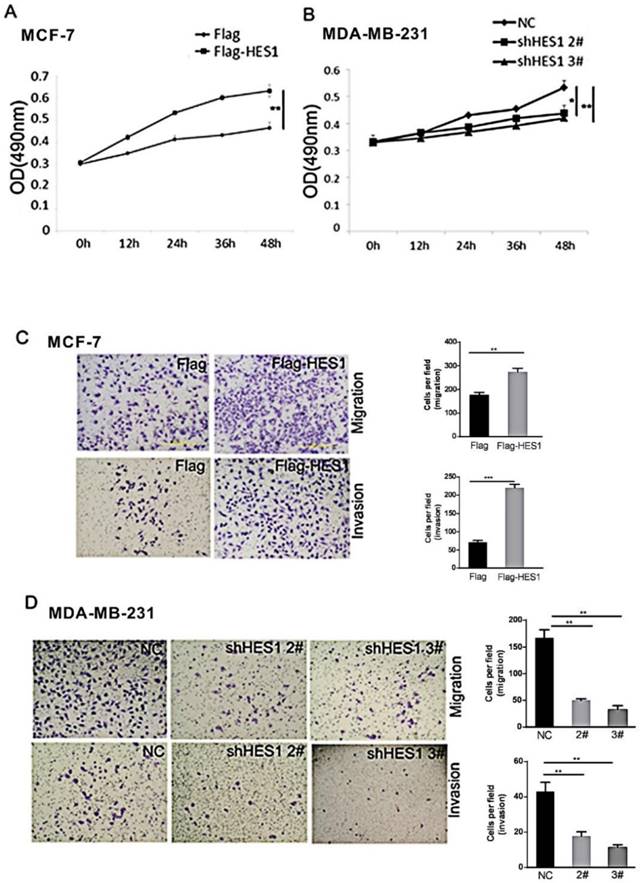
The role of HES1 in breast cancer cell migration and invasion was assessed using Transwell and Matrigel invasion assays. Our results showed that HES1 overexpression in MCF7 cells promoted migration and invasion, and a significant decrease in MDA-MB-231 cell migration and invasion was observed in cells with HES1 depletion compared with that of the scramble controls (Figures 4C and 4D). These results indicated that HES1 regulates proliferation and invasion in breast cancer cells.
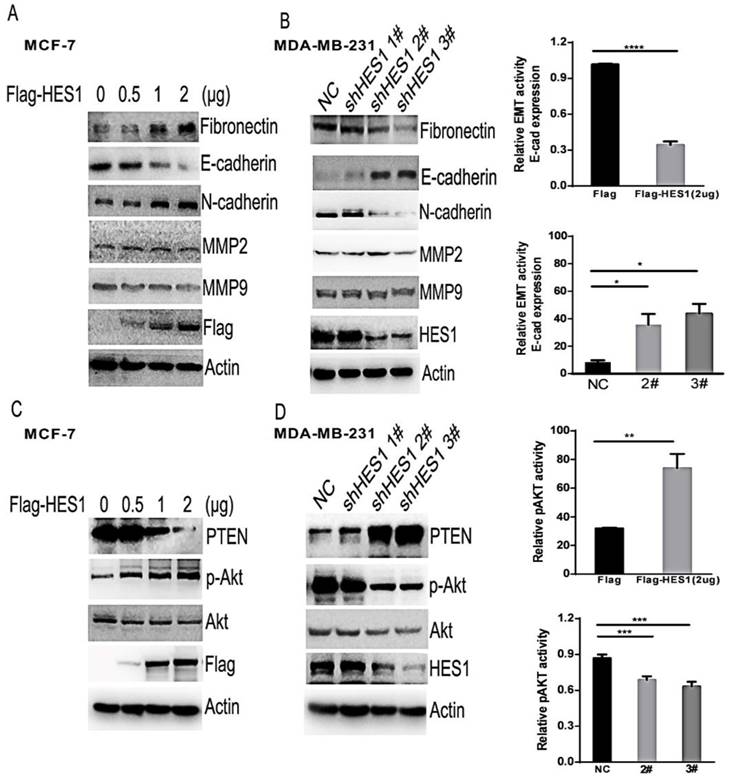
Epithelial-mesenchymal transition (EMT) is a key mechanism that contributes to invasion and metastasis of various cancers [21,22]. Additionally, the matrix metalloproteinase family members MMP2 and MMP9 facilitate cancer invasion by degrading the extracellular matrix (ECM) [23]. To further analyse the potential mechanisms of HES1 in breast cancer cells, we investigated the expression levels of proteins involved in EMT (eg. E-cadherin, N-cadherin or Fibronectin) and proteins belonging to the MMP family (Figures 5A and 5B). We found that there was no change in MMP2 and MMP9 expression levels following either upregulation of HES1 expression or silencing of HES1 expression. However, we observed a significant decrease in E-cadherin expression and a concurrent increase in N-cadherin and Fibronectin expression following HES1 overexpression. Conversely, depletion of HES1 using shRNA treatment resulted in increased E-cadherin expression and reduced N-cadherin and Fibronectin expression. These results indicated that HES1-induced migration and invasion in breast cancer cells is dependent on HES1-mediated EMT initiation. PTEN (phosphatase and tensin homologue) is well-known tumour suppressor that negatively regulates the AKT pathway, which is a critical regulator of cell survival, proliferation and migration [24,25]. Meanwhile, HES1 could also directly regulate PTEN expression by binding to the PTEN promoter [26]. Therefore, we investigated whether HES1-induced proliferation and invasion were mediated by inhibiting PTEN expression and activating the AKT pathway. Our results showed that upregulated HES1 expression in MCF7 cells significantly decreased PTEN expression and substantially increased p-AKT levels, whereas HES1 knockdown increased PTEN expression and reduced AKT phosphorylation (i.e., the p-AKT/AKT ratio) in MDA-MB-231 cells (Figures 5C and 5D). These results suggested that HES1 regulates cell survival, proliferation and invasion via AKT signalling pathway activation. Based on these results, we propose that HES1 promotes breast cancer cell proliferation and invasion by regulating the AKT signalling pathway and EMT process.
HES1 was correlated with EMT and the AKT signalling pathway. (A) Western blotting revealed that HES1 overexpression increases N-cadherin and vimentin and reduces E-cadherin, without significant changes in total AKT, MMP2, and MMP9. (B) Depletion of HES1 decreases the levels of N-cadherin and vimentin and increases E-cadherin, without significant changes in total AKT, MMP2, and MMP9. (C) Western blotting revealed that HES1 overexpression reduces the levels of PTEN and increases p-AKT. (D) Depletion of HES1 increases the levels of PTEN and decreases the levels of p-AKT and the p-AKT/AKT ratio. *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001.
Discussion
HES1 lies at the crossroads of multiple signalling pathways, and it is regarded as an excellent central target for treatment of multiple conditions. Although HES1 is usually considered a downstream targeted gene that is activated by the canonical Notch signalling pathway, HES1 expression is irrelevant to both activated and inactivated Notch signalling in ESFT cell lines [27]. Moreover, HES1 can also be enhanced via the hedgehog signalling pathway and other distinct pathways [9,10,28]. Therefore, HES1 levels are not always coupled with the activated Notch signalling pathway. The activation of these multiple signalling pathways in breast cancer can regulate cell proliferation and migration, indicating that upregulated HES1 may play an important role in increasing invasion and metastasis of breast cancer. However, the expression pattern of HES1 itself and its definite biological roles in breast cancer have not been examined. In our study, we demonstrated that HES1 levels in invasive breast cancer were significantly higher than those in the normal breast tissues. Furthermore, upregulated HES1 expression correlated with advanced TNM stage and lymph node metastasis. The proportion of HES1-positive samples in the TNBC samples was significantly higher than that in other subtypes of breast cancers, including triple-positive breast cancers. Similar outcomes were found in the breast cancer cell lines. TNBC is more aggressive and has decreased survival compared to that of other subtypes of breast cancers. These results not only revealed that high levels of HES1 were closely associated with TNBC and negative oestrogen receptor expression but also suggested that HES1 may be a critical contributor to TNBC. In addition, our study found that high HES1 expression was associated with unfavourable PFS and OS in breast cancer. Meanwhile, HES1 upregulation was correlated with lower PFS and OS in patients with TNBC. A recent study also revealed that HES1 upregulation predicts an unfavourable prognosis in TNBC [29]. To date, there is still no targeted and effective treatment strategy for TNBC due to deficiencies in therapeutic targets. Therefore, HES1 may be an independent prognostic factor for tumour progression and a potential therapeutic target in TNBC.
To analyse the potential mechanism by which upregulated HES1 promoted invasion of breast cancer cells, we tested the expression levels of invasion-related proteins and biological markers related to the EMT process. MMP2 and MMP9 are well-known cell migration-related proteins [23]. EMT is a process characterized by loss of epithelial cell polarization and cell adhesion and acquisition of mesenchymal characteristics and migration. These changes play a key role in morphogenesis during embryonic development and enhance epithelial-derived cancer invasion and metastasis and increase resistance to chemotherapy and radiotherapy [21,30]. EMT is initiated with repression of E-cadherin, an epithelial marker, and acquisition of N-cadherin and Fibronectin, mesenchymal markers [31]. In our study, we blocked HES1 expression using shRNA treatment in the MDA-MB-231 cell line, which has high endogenous HES1 expression. We found that HES1 depletion resulted in a significant decrease in the proliferation rate and migratory capacity. HES1 upregulation in the MCF-7 cell line enhanced its invasion and proliferation, which was consistent with our immunohistochemical data. Downregulated HES1 increased E-cadherin expression and decreased N-cadherin and Fibronectin expression. These results indicated that HES1 promotes invasion and migration of breast cancer cell via EMT induction.
PTEN is well-known tumour repressor and a negative regulator of the AKT pathway, which endows cancer cells with proliferative, invasive and migratory properties [24,25]. As a transcriptional repressor, HES1 could downregulate PTEN expression by binding to the PTEN promoter [26]. Our results showed that the knockdown of HES1 increased PTEN expression and decreased p-AKT protein levels and AKT activity, and overexpression of HES1 decreased PTEN expression and increased the p-AKT protein levels and AKT activity. These results revealed that HES1 suppressed PTEN expression and promoted activation of the AKT signalling pathway in breast cancer.
Our results also indicated that HES1 may be an important contributor to the malignant biological behaviour of breast cancer cell via the AKT pathway and EMT modulation. EMT is crucial for breast cancer migration and invasion and is achieved through a well-orchestrated transcriptional programme. There are three families of transcription factors that act as repressors and bind to the E-cadherin promoter directly to induce EMT; these proteins include Snail, Slug, ZEB1, ZEB2, E47 and Twist [32,33]. AKT plays a critical role in cell survival, proliferation and migration. AKT is a key mediator of EMT in human epithelial malignancies [34]. However, the mechanisms by which HES1 triggers EMT in breast cancer are currently unclear. Whether activating the AKT pathway mediates HES1-induced EMT indirectly or whether HES1 promotes EMT directly via suppression of E-cadherin by binding to its transcriptional promoter or activation of E-cadherin transcriptional repressors, such as Snail, Slug, ZEB1, ZEB2, E47 and Twist, should be elucidated in our future studies. HES1 was reported to enhance the phosphorylation of signal transducers and activators of transcription-3 (STAT3) by binding to STAT3 directly, and phosphorylated STAT3 can elevate MMP2 and MMP9 expression by directly binding to their promoters [35-37]. In our study, we observed that overexpression or depletion of HES1 had no effect on MMP2 and MMP9 expression.
In conclusion, the present study showed that increased HES1 was found in breast cancer tissues, and high levels of HES1 were positively correlated with advanced TNM stage and TNBC. HES1 played a crucial role in malignant cell proliferation and invasion via regulation of the AKT pathway and EMT process in breast cancer. These results suggested that HES1 may be an important regulator of breast cancer progression and a potential therapeutic target in TNBC treatment. Further studies should investigate the possibility of targeting HES1 in TNBC.
Compliance with ethical standards
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
This study was approved by the institutional review board of China Medical University. All procedures performed in this study were in accordance with the 1964 Declaration of Helsinki and its later amendments.
Informed consent
Written informed consent was obtained from all patients included in the study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81201886 and 81472599) to Feng Jin.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10-29 doi: 10.3322/caac.20138
2. Ahn S, Cho J, Sung J. et al. The prognostic significance of tumor-associated stroma in invasive breast carcinoma. Tumour Biol. 2012;33:1573-1580 doi: 10.1007/s13277-012-0411-6
3. Limame R, de Beeck KO, Van Laere S. et al. Expression profiling of migrated and invaded breast cancer cells predicts early metastatic relapse and reveals Krüppel-like factor 9 as a potential suppressor of invasive growth in breast cancer. Oncoscience. 2014;1:69-81 doi: 10.18632/oncoscience.10
4. Ishibashi M, Moriyoshi K, Sasai Y. et al. Persistent expression of helix-loop-helix factor HES-1 prevents mammalian neural differentiation in the central nervous system. EMBO J. 1994;13:1799-1805
5. Sun H, Ghaffari S, Taneja R. bHLH-Orange transcription factors in development and cancer. Transl Oncogenomics. 2007;2:107-120
6. Sang L, Roberts JM, Coller HA. Hijacking HES1: how tumors co-opt the anti-differentiation strategies of quiescent cells. Trends Mol Med. 2010;16:17-26 doi: 10.1016/j.molmed.2009.11.001
7. Tian C, Zheng G, Cao Z. et al. Hes1 mediates the different responses of hematopoietic stem and progenitor cells to T cell leukemic environment. Cell Cycle. 2013;12:322-331 doi: 10.4161/cc.23160
8. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237-255 doi: 10.1002/jcp.10208
9. Ingram WJ, McCue KI, Tran TH. et al. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene. 2008;27:1489-1500 doi: 10.1038/sj.onc.1210767
10. Stockhausen MT, Sjölund J, Axelson H. Regulation of the Notch target gene hes-1 by TGFalpha induced Ras/MAPK signaling in human neuroblastoma cells. Exp Cell Res. 2005;310:218-228 doi: 10.1016/j.yexcr.2005.07.011
11. Curry CL, Reed LL, Golde TE. et al. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene. 2005;24:6333-6344 doi: 10.1038/sj.onc.1208783
12. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007;134:1243-1251 doi: 10.1242/dev.000786
13. Davis RL, Turner DL. Vertebrate hairy and enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene. 2001;20:8342-8357 doi: 10.1038/sj.onc.1205094
14. Paroush Z, Finley RL Jr, Kidd T. et al. Groucho is required for drosophila neurogenesis, segmentation, and sex determination and interacts directly with hairy-related bHLH proteins. Cell. 1994;79:805-815 doi: 10.1016/0092-8674(94)90070-1
15. Konishi J, Kawaguchi KS, Vo H. et al. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007;67:8051-8057 doi: 10.1158/0008-5472.CAN-07-1022
16. Fre S, Huyghe M, Mourikis P. et al. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964-968 doi: 10.1038/nature03589
17. Wang X, Fu Y, Chen X. et al. The expressions of bHLH gene HES1 and HES5 in advanced ovarian serous adenocarcinomas and their prognostic significance: a retrospective clinical study. J Cancer Res Clin Oncol. 2010;136:989-996 doi: 10.1007/s00432-009-0744-8
18. Fan X, Mikolaenko I, Elhassan I. et al. Notch1 and Notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64:7787-7793 doi: 10.1158/0008-5472.CAN-04-1446
19. Maniati E, Bossard M, Cook N. et al. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits PPARγ expression and promotes pancreatic cancer progression in mice. J Clin Invest. 2011;121:4685-4699 doi: 10.1172/JCI45797
20. Wang SC, Lin XL, Wang HY. et al. HES1 triggers epithelial-mesenchymal transition (EMT)-like cellular marker alterations and promotes invasion and metastasis of nasopharyngeal carcinoma by activating the PTEN/AKT pathway. Oncotarget. 2015;6:36713-36730 doi: 10.18632/oncotarget.5457
21. Christofori G. New signals from the invasive front. Nature. 2006;441:444-450 doi: 10.1038/nature04872
22. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131-142 doi: 10.1038/nrm1835
23. Nelson AR, Fingleton B, Rothenberg ML. et al. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18:1135-1149 doi: 10.1200/JCO.2000.18.5.1135
24. Blanco-Aparicio C, Renner O, Leal JF. et al. PTEN, More than the AKT pathway. Carcinogenesis. 2007;28:1379-1386 doi: 10.1093/carcin/bgm052
25. Zhang J, Grindley JC, Yin T. et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518-522 doi: 10.1038/nature04747
26. Wong GW, Knowles GC, Mak TW. et al. HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRbeta-selected mouse thymocytes. Blood. 2012;120:1439-1448 doi: 10.1182/blood-2011-12-395319
27. Bennani-Baiti IM, Aryee DN, Ban J. et al. Notch signalling is off and is uncoupled from HES1 expression in Ewing's sarcoma. J Pathol. 2011;225:353-363 doi: 10.1002/path.2966
28. Wall DS, Mears AJ, McNeill B. et al. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J Cell Biol. 2009;184:101-112 doi: 10.1083/jcb.200805155
29. Orzechowska M, Jędroszka D, Bednarek AK. Common profiles of Notch signaling differentiate disease-free survival in luminal type A and triple negative breast cancer. Oncotarget. 2017;8:6013-6032 doi: 10.18632/oncotarget.13451
30. Parvani JG, Schiemann WP. Sox4, EMT programs, and the metastatic progression of breast cancers: mastering the masters of EMT. Breast Cancer Res. 2013;15:R72. doi: 10.1186/bcr3466
31. Fendrich V, Waldmann J, Feldmann G. et al. Unique expression pattern of the EMT markers snail, twist and E-cadherin in benign and malignant parathyroid neoplasia. Eur J Endocrinol. 2009;160:695-703 doi: 10.1530/EJE-08-0662
32. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415-428 doi: 10.1038/nrc2131
33. Yang J, Mani SA, Donaher JL. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-939 doi: 10.1016/j.cell.2004.06.006
34. Rafael D, Doktorovová S, Florindo HF. et al. EMT blockage strategies: targeting Akt dependent mechanisms for breast cancer metastatic behaviour modulation. Curr Gene Ther. 2015;15:300-312 doi: 10.2174/1566523215666150126123642
35. Kamakura S, Oishi K, Yoshimatsu T. et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547-554 doi: 10.1038/ncb1138
36. Xie TX, Wei D, Liu M. et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550-3560 doi: 10.1038/sj.onc.1207383
37. Song Y, Qian L, Song S. et al. Fra-1 and Stat3 synergistically regulate activation of human MMP-9 gene. Mol Immunol. 2008;45:137-143 doi: 10.1016/j.molimm.2007.04.031
Author contact
![]() Corresponding author: Prof Feng Jin, E-mail: cmu_13998866946com
Corresponding author: Prof Feng Jin, E-mail: cmu_13998866946com