Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(5):880-889. doi:10.7150/jca.20869 This issue Cite
Research Paper
Large Intergenic Non-coding RNA-RoR Inhibits Aerobic Glycolysis of Glioblastoma Cells via Akt Pathway
Yong Li1,2, Zhi-Cheng He1,2, Qing Liu1,2, Kai Zhou1,2, Yu Shi1,2, Xiao-Hong Yao1,2, Xia Zhang1,2, Hsiang-Fu Kung1,2 ![]() , Yi-Fang Ping1,2
, Yi-Fang Ping1,2 ![]() , Xiu-Wu Bian1,2
, Xiu-Wu Bian1,2 ![]()
1. Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University
2. Key Laboratory of Tumor Immunopathology, Ministry of Education of China, Chongqing 400038, China
Received 2017-5-4; Accepted 2018-1-29; Published 2018-2-23
Abstract
Reprogramming energy metabolism is a hallmark of malignant tumors, including glioblastoma (GBM). Aerobic glycolysis is often utilized by tumor cells to maintain survival and proliferation. However, the underlying mechanisms of aerobic glycolysis in GBM remain elusive. Herein, we demonstrated that large intergenic non-coding RNA-RoR (LincRNA-RoR) functioned as a critical suppressor to inhibit the aerobic glycolysis and viability of GBM cells. We found that LincRNA-RoR was markedly reduced in GBM tissues compared with adjacent non-tumor tissues from 10 cases of GBM patients. Consistently, LincRNA-RoR expression in GBM cells was significantly lower than that in normal glial cells. The aerobic glycolysis of GBM cells, as determined by the measurement of glucose uptake and lactate production, was impaired by LincRNA-RoR overexpression. Mechanistically, LincRNA-RoR inhibited the expression of Rictor, the key component of mTORC2 (mammalian target of rapamycin complex 2), to suppress the activity of Akt pathway and impair the expression of glycolytic effectors, including Glut1, HK2, PKM2 and LDHA. Finally, enforced expression of LincRNA-RoR reduced the proliferation of GBM cells in vitro, restrained tumor growth in vivo, and repressed the expression of glycolytic molecules in GBM xenografts. Collectively, our results underscore LincRNA-RoR as a new suppressor of GBM aerobic glycolysis with therapeutic potential.
Keywords: Glioblastoma, Glycolysis, LincRNA-RoR, Proliferation
Introduction
Cancer cells prefer to obtain energy from aerobic glycolysis [1-3], which produces more lactate but less ATP, a phenomenon called “Warburg effect”. The conversion from glucose to lactate in the presence of oxygen facilitates proliferation, migration and invasion of cancer cells [2, 4, 5]. Consequently, the amount of lactate produced in the microenvironment is closely correlated with tumor malignancy, and lactate promotes the survival of tumor cells by decreasing the oxidative stress via consuming the reactive oxygen species [4, 6, 7].
Glioblastoma (GBM) is the most common and aggressive primary human brain tumor with high mortality [8]. The survival of GBM patients has not been significantly prolonged despite of significant progress in therapeutic strategies, such as surgical practices, new radiotherapy techniques and molecule-targeted therapies [9, 10]. Although GBM is highly-vascularized [11], bevacizumab, an antibody targeting vascular endothelial growth factor (VEGF), failed to improve the survival of GBM patients partially due to the induction of metabolic switch toward glycolysis in GBM cells [12]. In addition, several key glycolytic effectors, such as glucose transporter 1 (Glut1), hexokinase 2 (HK2), pyruvate kinase 2 (PKM2) and lactate dehydrogenase A (LDHA), are aberrantly expressed in glioma cells and play vital roles in tumorigenesis [5,13]. Thus, clarification of the regulatory molecules of glycolysis is important for understanding the mechanism of GBM progression and exploring new targeted therapeutic strategies for GBM.
The long non-coding RNAs are RNA molecules over 200 nucleotides in length [14], and involved in multiple physiological and pathological processes, including carcinogenesis [15, 16]. Large intergenic non-coding RNA-RoR (LincRNA-RoR) was firstly identified and characterized in induced pluripotent stem cells and human embryonic stem cell by microarray screening and functional validation [17, 18]. Altered expression of LincRNA-RoR has also been shown to mediate various cellular processes in cancer cells, including proliferation, metastasis, invasion and apoptosis [19]. However, the regulation of glucose metabolism by LincRNA-RoR in cancer cells has not been explored.
In this study, we detect the expression of LincRNA-RoR in GBM tissues and the matched non-tumor tissues, investigate the effect and corresponding mechanism of LincRNA-RoR on aerobic glycolysis, as well as explore the therapeutic significance of LincRNA-RoR on GBM xenografts.
Materials and Methods
Cell culture
The normal human glial cell line, HEB, was generously provided by Professor Guang-mei Yan (Department of Pharmacology, Sun Yat-sen University, Guangzhou, China). The human U87-MG, T98G and LN229 glioblastoma cell lines from ATCC was authenticated by short tandem repeat profiling (Beijing Microread Genetics, Beijing, China) and passaged for less than 6 months. Human primary GBM1 cells, primary astrocytes (sw16-N) and its matched GBM cells (sw16-T) were established and maintained in our laboratory [20, 21]. HEB, U87-MG, T98G, LN229, sw16-N, sw16-T and GBM1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Grad Island, NY), 100 μg/ml penicillin and 100 U/ml streptomycin. All the cells were incubated at 37℃ in a humidified incubator with 5% CO2.
Patients and tissue specimens
Ten fresh GBM tumor specimens and the matching non-tumor tissues were collected by surgery from Department of Neurosurgery, Southwest Hospital, Third Military Medical University (TMMU). Written informed consents were obtained from the patients or their guardians. All patients had not received radiotherapy or chemotherapy before surgery. All the experimental procedures regarding human subjects had been approved by the ethics committees of the Third Military Medical University.
Lentiviral vectors transfection procedures
The human LincRNA-RoR overexpression lentiviral vector and its negative control vector were purchased from GenePharma (Shanghai, China). Cells were infected with lentivirus in the presence of 8 μg/ml polybrene.
Glucose uptake, Lactate production and ATP Assay
QuantiChromTM Glucose Assay Kit (DIGL-100) and EnzyChromTM L-Lactate Assay Kit (ECLC-100) were obtained from BioAssay Systems (Hayward, CA). ATP Assay Kit (S0026) was purchased from Beyotime (Shanghai, China). Briefly, cells were plated in 12-well culture dishes, and the culture medium was replaced with FBS-free DMEM 24 hrs later. After cultured for additional 24 hrs, glucose and lactate assays were performed with culture media collected from each sample according to the manufacturer's instructions. ATP assay was conducted on the cultured cells according to the manufacturer's protocol.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Real-time qRT-PCR was performed as previously described [22]. The sequences of PCR primers were listed as follow: β-actin, F 5'-GACCTGACTGACTACCTCATG AAGAT-3', R 5-'GTCACACTTCATGATGGAGTTGAAGG-3'; LincRNA-RoR, F 5'-CTTGATGGCATTGTCGCTAA3'-3', R 5'-TCCAGTGGCTGTGCTAGATG-3'.
Western Blot
Western blotting was conducted as previously described [22]. Commercial primary antibodies included β-actin (1:1000) (AA132, Beyotime), Glut1(1:1000) (ab115730, Abcam, Cambridge, UK), glycolysis antibody sample kit (1:1000) (#8337, Cell Signal Technology), Rictor (1:1000) (#2114, Cell Signal Technology), p-Akt (1:1000) (#4060, Cell Signal Technology) and Akt (1:1000) (#4685, Cell Signal Technology), p-mTOR(1:1000) (#5536P, Cell Signal Technology) and mTOR (1:1000) (#2983P, Cell Signal Technology), S6 (#2317s, Cell Signal Technology) and p-S6 (#4857s, Cell Signal Technology).
Cell proliferation assays
The cell proliferation was assessed by using Cell Counting Kit (CCK-8) (C0038, Beyotime). Briefly, cells were seeded in a 96-well plate at a density of 1.5×103 cells/well. CCK8 was added to each well every 24 hrs for up to 4 days. The light absorbance was then measured at a wavelength of 450 nm.
In vivo xenograft experiments
For evaluation of tumor growth in vivo, 1×106 U87 cells were injected subcutaneously into 5-week-old male BALB/c nude mice. Four weeks later, tumor volume was calculated by the formula V= ab2/2 (a: tumor length, b: tumor width). The animal experiments were approved by the Institutional Animal Care and Use Committee of Southwest Hospital, Third Military Medical University in accordance with the Guide for the Care and Use of Laboratory Animals.
HE and immunohistochemistry staining
HE and immunohistochemistry staining of xenograft tumors were performed as previously described [20]. Commercial antibodies were mouse anti-human Ki67 (Kit-0005, Maixin, Fuzhou, China), rabbit anti-human Glut1 (1:200) (ab115730, Abcam), rabbit anti-human HK2 (1:200) (#8337, Cell Signal Technology), rabbit anti-human PKM2 (1:200) (#8337, Cell Signal Technology), rabbit anti-human LDHA (1:200) (#8337, Cell Signal Technology), rabbit anti-human Rictor (1:100) (#2114, Cell Signal Technology), rabbit anti-human p-Akt (1:200) (#4060, Cell Signal Technology). Mean density of immunohistochemistry staining was calculated by Image Pro-plus software (Media Cybernetics, Silver Spring, MD).
Statistical analysis
All experiments were performed at least in triplicates and representative data were shown. The unpaired two-group comparison and multiple comparisons were made with Student's t test or one-way ANOVA, respectively. Data were presented as the mean ± SD. P < 0.05 (*) and P < 0.01 (**) were considered statistically significant.
Results
Expression of LincRNA-RoR is reduced in human GBM tissues and GBM cells
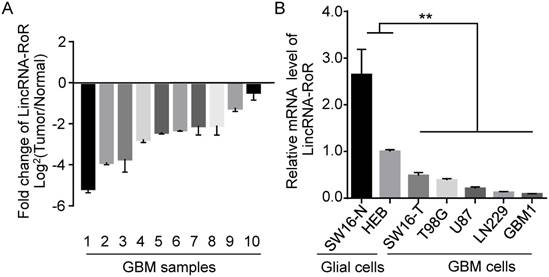
To determine the expression of LincRNA-RoR in human GBMs, we investigated 10 cases of GBM specimens and found that LincRNA-RoR expression in tumor tissues was significantly lower than that in adjacent non-tumor tissues (Figure 1A). We further examined the endogenous level of LincRNA-RoR in a panel of GBM cells, including U87, T98G, LN229, primary GBM1 and sw16-T, as well as normal glial cells (human primary astrocytes sw16-N and HEB). The level of LincRNA-RoR was significantly lower in GBM cells (T98G, LN229, U87 GBM1 and sw16-T) compared to HEB and sw16-N cells (Figure 1B). These results indicate that the expression of LincRNA-RoR is reduced in GBM.
Expression of LincRNA-RoR in GBM tissues and GBM cells by qRT-PCR. (A) LincRNA-RoR expression was lower in GBM tissues compared with the adjacent non-tumor tissues from the same patient. (B) LincRNA-RoR expression was lower in GBM cells (T98G, U87, LN229 and primary GBM1, sw16-T) than that in glial cells (HEB and sw16-N). **, P<0.01.
LincRNA-RoR inhibits the aerobic glycolysis of GBM cells
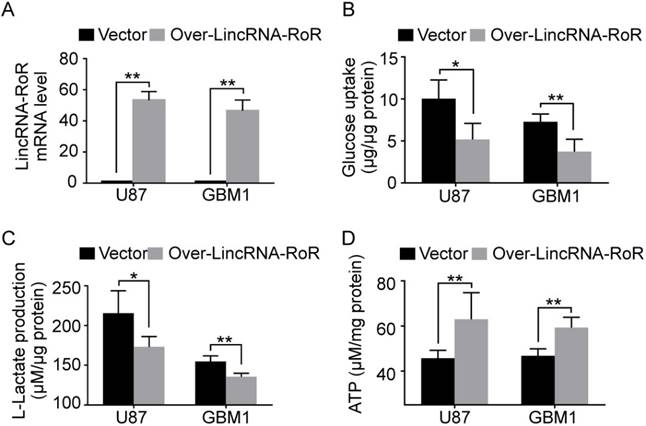
To explore the role of LincRNA-RoR in aerobic glycolysis, U87 and GBM1 cells were used to generate LincRNA-RoR-overexpression cells (Figure 2A). We found that LincRNA-RoR overexpression significantly impeded glucose uptake (Figure 2B) and L-lactate production (Figure 2C). Accordingly, GBM cells with LincRNA-RoR-overexpression produced more ATP than control cells (Figure 2D). These data indicate that LincRNA-RoR inhibits aerobic glycolysis of GBM cells.
LincRNA-RoR inhibits the aerobic glycolysis of GBM cells in vitro. (A) LincRNA-RoR is significantly elevated in LincRNA-RoR-lentivirus infected GBM cells. (B) Glucose uptake and (C) L-lactate production were significantly inhibited in LincRNA-RoR-overexpression GBM cells, while the total ATP (D) was significantly increased in LincRNA-RoR-overexpression GBM cells as compared with control cells. *P<0.05, **P<0.01.
LincRNA-RoR inhibits the expression of Rictor, p-Akt and glycolytic molecules to suppress the proliferation of GBM cells
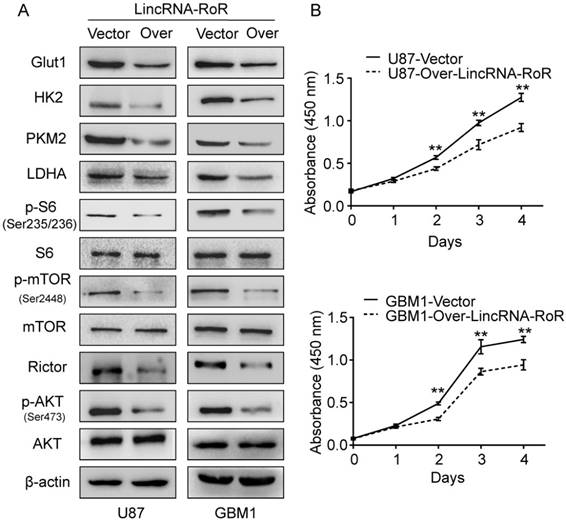
The molecular mechanisms on the regulation of glucose metabolism by LincRNA-RoR were also investigated. mTORC2 has been reported as an important regulator of Akt activation and plays pivotal role in cancer metabolism [23, 24]. Rictor is the key component of mTORC2 [25]. Of note, mTORC2 activity is significantly elevated and exerts oncogenic function in glioma cells [26]. We thus investigated whether LincRNA-RoR could regulate mTORC2. Our results revealed that LincRNA-RoR overexpression reduced the level of Rictor and attenuated the phosphorylation of Akt (Ser473) (Figure 3A). The important downstream molecules of Akt pathway, p-mTOR (Ser2448) and p-S6 (Ser235/236), were also decreased upon LincRNA-RoR overexpression (Figure 3A). Moreover, the expression of glycolytic effectors, including Glut1, HK2, PKM2 and LDHA, was remarkably decreased in LincRNA-RoR-overexpressed GBM cells in comparison with their control counterparts (Figure 3A). Functionally, LincRNA-RoR overexpression suppressed the proliferation (Figure 3B) of GBM cells in vitro. These results indicate that LincRNA-RoR inhibits proliferation of GBM cells through Rictor/Akt pathway.
LincRNA-RoR inhibits Rictor expression, Akt phosphorylation and suppresses the expression of glycolytic molecules. (A) Over-expression of LincRNA-RoR in GBM cells inhibited the expression of Rictor, the phosphorylations of Akt, mTOR and S6, and down-regulate expression of Glut1, HK2, PKM2 and LDHA. (B) LincRNA-RoR suppressed the proliferation of GBM cells in vitro. *P<0.05, **P<0.01.
LincRNA-RoR restrains tumor growth in vivo and inhibits the expression of glycolytic molecules in GBM xenografts
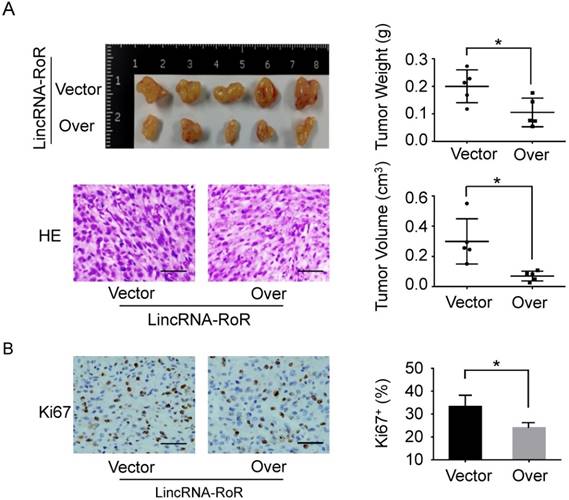
Furthermore, we investigated the effect of LincRNA-RoR on tumorigenesis of GBM cells. As shown in Figure 4A, the weight and volume of the xenografts with LincRNA-RoR overexpression were dramatically reduced compared with control xenografts. Meanwhile, the percentage of Ki67 positive cells was significantly decreased in LincRNA-RoR overexpression xenografts (Figure 4B). Additionally, down-regulation of Glut1, HK2, PKM2, LHDA, Rictor, p-Akt (Ser473), p-mTOR (Ser2448) and p-S6 (Ser235/236) was observed in the xenografts derived from LincRNA-RoR-overexpressed cells as compared with that derived from control cells (Figure 5A, B, C and Figure 6A, B), demonstrating that LincRNA-RoR might inhibit tumor growth via suppressing tumor glycolysis.
LincRNA-RoR restrains GBM tumor growth in vivo. (A) The gross and HE staining images of xenografts derived from LincRNA-RoR-overexpression U87 cells and its control counterparts. Tumor volumes were calculated by ab2/2. a = length. b = width. Both tumor volumes and tumor weight were significantly decreased in LincRNA-RoR-overexpression groups. (B) The expression of Ki67 (Brown staining) was analyzed by immunohistochemistry. Scale bar represents 100 μm. Data are represented as means ± SD of each group. *P<0.05.
LincRNA-RoR inhibited the expression of glycolytic effectors in xenograft tumors by western blot. (A) The mRNA level of LincRNA-RoR is higher in over-LincRNA-RoR-tumor xenografts as compared with that in control xenografts. (B) The expression of Glut1, HK2, PKM2 and LDHA are significantly impaired in over-LincRNA-RoR-tumor xenografts as compared with that in control xenografts. (C) LincRNA-RoR over-expression significantly inhibits Rictor expression, as well as the phosphorylations of Akt, mTOR and S6 in tumor xenografts.
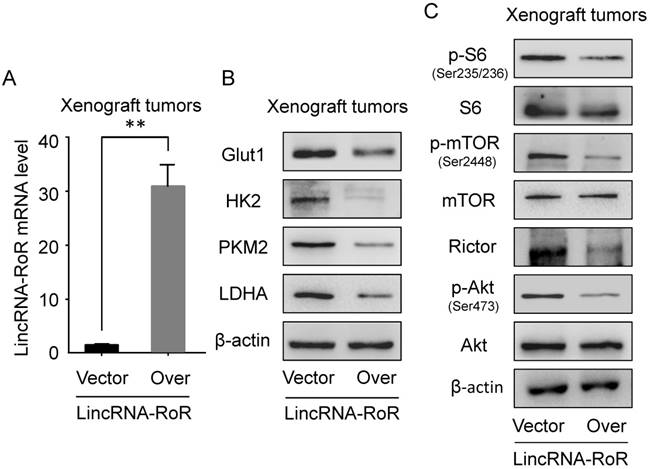
Immunostaining of Glut1, HK2, PKM2, LDHA, p-Akt and Rictor in xenograft tumors in presence of LincRNA-RoR. (A) The expression of Glut1, HK2, PKM2, LDHA as well as (B) p-Akt and Rictor were analyzed by immunohistochemistry. Mean density was calculated by Image Pro-plus software. Scale bar represents 100 μm. Data are represented as means ± SD of each group. *P <0.05, **P <0.01.
Discussion
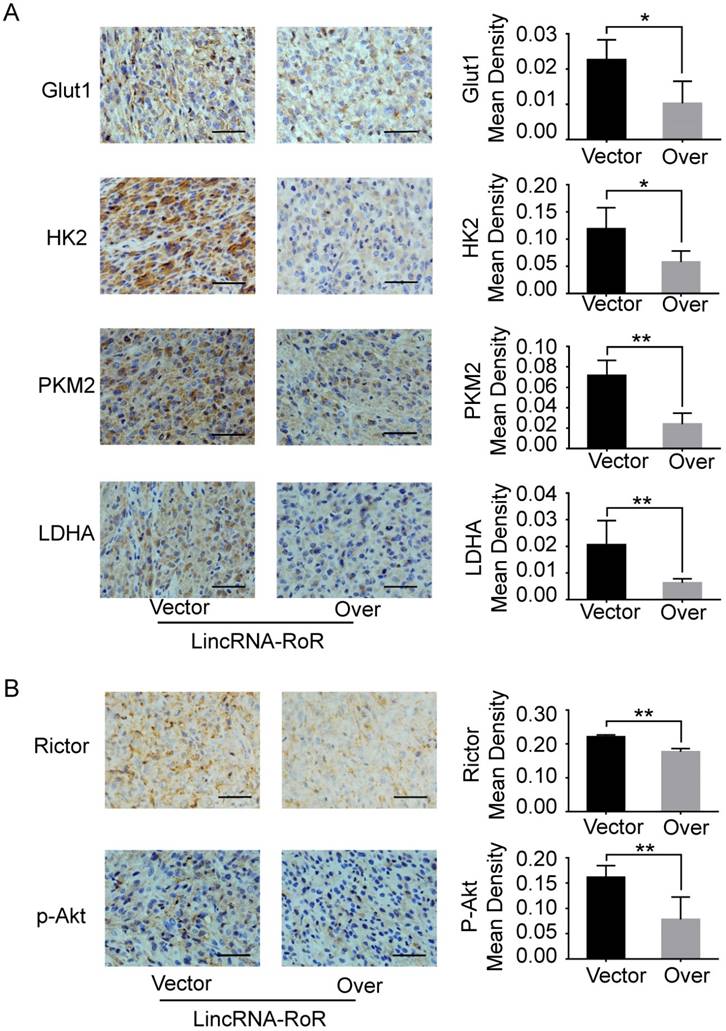
GBM cells prefer to acquire energy from glycolysis instead of oxidative phosphorylation. Targeting glycolysis is proposed as a promising therapeutic strategy for GBM treatment [13, 27, 28]. Our results for the first time showed that LincRNA-RoR inhibited the expression of Rictor, reduced the activity of Akt, and impaired the expression of glycolytic molecules, by which the proliferation of GBM cells was restrained (Figure 7B). These data revealed LincRNA-RoR as a new suppressor of GBM aerobic glycolysis with therapeutic potential.
Schematic presentation shows the effect of LincRNA-RoR on aerobic glycolysis of GBM cells.
In glioma, Akt activation has been proved to be tightly correlated with tumor malignancy, including drug-resistant and tumor progression [29-32]. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells [33], implying that targeting key regulators controlling Akt signaling activity might disrupt the malignant behaviors of gliomas. Previous studies have demonstrated that Akt activity is tightly regulated by mTORC2, which responds to growth factor and functions as a major activator of Akt [24]. However, the mechanisms on mTORC2 regulation in gliomas are still poorly understood. In this study, we identified LincRNA-RoR as an important negative regulator of mTORC2-Akt signaling. The data showed that LincRNA-RoR impaired the expression of Rictor, an important component of mTORC2 complex, and thus interrupted mTORC2 function followed by inhibition of Akt activity, which functionally disrupted aerobic glycolysis in GBM cells. Our finding is in consistent with previous studies showing that upregulated Rictor expression and mTORC2 activity in glioma promotes cell growth and motility [26], underscoring the important role of this signaling pathway in glioma progression.
Akt stimulates aerobic glycolysis in cancer cells [34]. Several glycolytic effectors, such as Glut1, HK2, PKM2 and LDHA, are aberrantly expressed in glioma, and play vital roles in tumorigenesis [5, 35]. Glut1 has been identified as one of the proteins that are up-regulated in GBM to enhance the “Warburg effect”, especially in hypoxic microenvironments [36]. HK2 is the first key enzyme of glycolysis. Depletion of HK2 in GBM cells can restore oxidative glucose metabolism and increase sensitivity to cell death inducers, such as radiation and temozolomide [26]. Pyruvate kinase is an important enzyme of glycolysis by transferring the phosphoryl group of phosphoenolpyruvate (PEP) onto ADP to form ATP and pyruvate.
In glioma, PKM2 expression is up-regulated in a grade-specific manner and loss of PKM2 results in cell death [37, 38]. Meanwhile, PKM2 dephosphorylation by Cdc25A promotes the “Warburg effect” and tumorigenesis [39]. LDHA catalyzes the conversion of pyruvate to lactate and is considered to be a key checkpoint of anaerobic glycolysis [3]. Our results show that LincRNA-RoR overexpression inhibits the growth of GBM xenografts with the decreased expression of Glut1, HK2, PKM2 and LDHA, suggesting that LincRNA-RoR could be used as a potential therapeutic target for GBM.
We also determined the inhibition of aerobic glycolysis in GBM cells by LincRNA-RoR through measuring glucose and L-lactate in the medium, total ATP, and a panel of glycolytic effectors in the cells, which are known parameters in the literature reflect the status of cell glycometabolism [7, 28]. However, we wished to mention that the amount of glycolysis related-metabolites and the glucose flux might provide another set of evidence to confirm the inhibitory effects of LincRNA-RoR on glycolysis.
Our results also showed that the expression of LincRNA-RoR was significantly lower in GBM tissue as compared with their adjacent non-tumor tissue. However, the expression of LincRNA-RoR exhibits heterogeneity in different type of malignancy. For example, LincRNA-RoR is up-regulated in epithelial malignant tumors, including breast cancer, hepatocellular cancer, colorectal cancer, pancreatic cancer and endometrial cancer [19]. In consideration of the critical role of LincRNA-RoR in regulating cell metabolism and other oncogenic phenotypes [19], it should be interesting to clarify the regulatory mechanism on LincRNA-RoR expression. Zhang et al. found that NRF2 could bind to the promoter region of LincRNA-RoR to suppress the expression of LincRNA-RoR [40]. Since NRF2 had been reported to serve as an oncogenic protein in GBM [41-43], we supposed that high expression of NRF2 might inhibit the expression of LincRNA-RoR in GBM cells, which needed to be further investigated.
Conclusion
Taken together, our study reveals a novel function of LincRNA-RoR in GBM. LincRNA-RoR is significantly down-regulated in GBM and enforced expression of LincRNA-RoR inhibits glycolysis and the growth of GBM xenografts. Therefore, LincRNA-RoR may be a promising therapeutic target towards GBM glycolysis.
Abbreviations
Akt: protein kinase B;
GBM: glioblastoma;
Glut1: glucose transporter 1;
HK2: hexokinase 2;
LDHA: lactate dehydrogenase A;
LincRNA-RoR: long intergenic non-protein coding RNA, regulator of reprogramming;
mTOR: mammalian target of rapamycin;
mTORC2: mammalian target of rapamycin complex 2;
PKM2: pyruvate kinase 2;
qRT-PCR: quantitative reverse transcriptase-PCR;
S6: S6 Ribosomal Protein.
Acknowledgements
This research was supported by grants from China National Science and Technology Major Project (2016YFA0101203).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269-70
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
3. Miao P, Sheng S, Sun X, Liu J, Huang G. Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. Iubmb Life. 2013;65:904-10
4. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27-47
5. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11-31
6. Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118:3835-7
7. Lee KM, Nam K, Oh S, Lim J, Lee T, Shin I. ECM1 promotes the Warburg effect through EGF-mediated activation of PKM2. Cell Signal. 2015;27:228-35
8. Filbin MG, Suva ML. Gliomas genomics and epigenomics: arriving at the start and knowing it for the first time. Annu Rev Pathol. 2016;11:497-521
9. Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma stem cells: signaling, microenvironment, and therapy. Stem Cells Int. 2016:7849890
10. Sanzey M, Abdul Rahim SA, Oudin A, Dirkse A, Kaoma T, Vallar L. et al. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PloS One. 2015;10:e0123544
11. Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181:1126-41
12. Fack F, Espedal H, Keunen O, Golebiewska A, Obad N, Harter PN. et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2015;129:115-31
13. Seyfried TN, Flores R, Poff AM, D'Agostino DP, Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett. 2015;356:289-300
14. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47-62
15. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452-63
16. Yu C, Xue J, Zhu W, Jiao Y, Zhang S, Cao J. Warburg meets non-coding RNAs: the emerging role of ncRNA in regulating the glucose metabolism of cancer cells. Tumor Biol. 2015;36:81-94
17. Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH. et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113-7
18. Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L. et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69-80
19. Pan Y, Li C, Chen J, Zhang K, Chu X, Wang R. et al. The emerging roles of long noncoding RNA ROR (lincRNA-ROR) and its possible mechanisms in human cancers. Cell Physiol Biochem. 2016;40:219-229
20. Shi Y, Chen C, Yu SZ, Liu Q, Rao J, Zhang HR. et al. miR-663 suppresses oncogenic function of CXCR4 in glioblastoma. Clin Cancer Res. 2015;21:4004-13
21. Gao L, Guan W, Wang M, Wang H, Yu J, Liu Q. et al. Direct generation of human neuronal cells from adult astrocytes by small molecules. Stem Cell Rep. 2017;8:538-547
22. Shi Y, Chen C, Zhang X, Liu Q, Xu JL, Zhang HR. et al. Primate-specific miR-663 functions as a tumor suppressor by targeting PIK3CD and predicts the prognosis of human glioblastoma. Clin Cancer Res. 2014;20:1803-13
23. Masui K, Cavenee WK, Mischel PS. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab. 2014;25:364-73
24. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101
25. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
26. Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712-20
27. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R. et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208:313-26
28. Zhao S, Liu H, Liu Y, Wu J, Wang C, Hou X. et al. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013;333:253-60
29. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT. et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226-35
30. Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T. et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81
31. Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NF kappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941-51
32. Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001;61:6674-8
33. Fujiwara K, Iwado E, Mills GB, Sawaya R, Kondo S, Kondo Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int J Oncol. 2007;31:753-60
34. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR. et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892-9
35. Nakano I. Therapeutic potential of targeting glucose metabolism in glioma stem cells. Expert Opin Ther Targets. 2014;18:1233-6
36. Labak CM, Wang PY, Arora R, Guda MR, Asuthkar S, Tsung AJ. et al. Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am J Cancer Res. 2016;6:1599-608
37. Han D, Wei W, Chen X, Zhang Y, Wang Y, Zhang J. et al. NF-kappaB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget. 2015;6:26119-28
38. Liu VM, Vander HM. The role of Pyruvate Kinase M2 in cancer metabolism. Brain Pathol. 2015;25:781-3
39. Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X. et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431
40. Zhang Y, Xia J, Li Q, Yao Y, Eades G, Gernapudi R. et al. NRF2/long noncoding RNA ROR signaling regulates mammary stem cell expansion and protects against estrogen genotoxicity. J Biol Chem. 2014;289:31310-8
41. Tsai WC, Hueng DY, Lin CR, Yang TC, Gao HW. Nrf2 Expressions Correlate with WHO Grades in Gliomas and Meningiomas. Int J Mol Sci. 2016:17 pii: E722
42. Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W. et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int J Cancer. 2014;135:574-84
43. Zhu J, Wang H, Sun Q, Ji X, Zhu L, Cong Z. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer. 2013;13:380
Author contact
![]() Corresponding author: Dr. Hsiang-Fu Kung, Dr. Yi-Fang Ping and Dr. Xiu-Wu Bian, Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing 400038, China. Tel: +86 23 68754431; Fax: +86 23 65397004; E-mail: Hsiang-Fu Kung: hkungedu.hk; Yi-Fang Ping: pingyifangedu.cn; Xiu-Wu Bian: bianxiuwuedu.cn; bianxiuwunet
Corresponding author: Dr. Hsiang-Fu Kung, Dr. Yi-Fang Ping and Dr. Xiu-Wu Bian, Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing 400038, China. Tel: +86 23 68754431; Fax: +86 23 65397004; E-mail: Hsiang-Fu Kung: hkungedu.hk; Yi-Fang Ping: pingyifangedu.cn; Xiu-Wu Bian: bianxiuwuedu.cn; bianxiuwunet