Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(5):914-922. doi:10.7150/jca.22329 This issue Cite
Research Paper
Targeting hepatocarcinogenesis model in C56BL6 mice with pan-aurora kinase inhibitor Danusertib
Paschalis Gavriilidis1,8, Theofilos Poutahidis2, Alexander Giakoustidis5, Kali Makedou4, Katerina Angelopoulou3, Alexander Hardas2, Paola Andreani6, Argyro Zacharioudaki7, George Saridis2, Athanasios Gargavanis8, Eleni Louri5, Nikolaos Antoniadis8, Eleftheria Karampela7, Nikolaos Psychalakis7, Antonios Michalopoulos9, Apostolos Papalois7, Stavros Iliadis4, Satvinder Mudan5, Daniel Azoulay6, Dimitrios Giakoustidis8 ![]()
1. Department of Hepato-Pancreato-Biliary and Liver Transplant surgery, Queen Elizabeth University Hospitals Birmingham NHS Foundation Trust, B15 1NU, UK.
2. Laboratory of Pathology, School of Veterinary Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki.
3. Laboratory of Biochemistry and Toxicology, School of Veterinary Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki.
4. Laboratory of Biochemistry, School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki.
5. Academic Department of Surgery, The Royal Marsden Hospital, London, UK.
6. Service de Chirurgie Digestive et Hépatobiliaire, Hôpital Henri Mondor, Assistance Publique-Hôpitaux de Paris-Université Paris-Est, Créteil, France.
7. Experimental and Research Center ELPEN Pharmaceuticals, Athens, Greece.
8. Division of Transplant Surgery, Department of Surgery, School of Medicine, Faculty of Health Sciences, Aristotle University and Hippokration General Hospital, Thessaloniki, Greece.
9. Propaedeutic Division of Surgery, Department of Surgery School of Medicine, Faculty of Health Sciences, Aristotle University and AHEPA University Hospital, Thessaloniki, Greece.
Received 2017-8-11; Accepted 2018-1-29; Published 2018-2-27
Abstract
Background: To elucidate the expression of Aurora kinases (AURK) and the anticancer effects of pan-aurora kinase inhibitor Danusertib in hepatocarcinogenesis model in C56Bl6 mice.
Methods: Thirty mice C56Bl6 were randomly divided into Group A or control, Group B animals who underwent experimental hepatocarcinogenesis with diethylnitrosamine (DEN), and Group C animals with DEN-induced hepatocarcinogenenesis that treated with pan-aurora kinase inhibitor Danusertib. Primary antibodies for immunochistochemistry (IHC) included rabbit antibodies against Ki-67, DKK1, INCENP, cleaved caspase-3, NF-κB p65, c-Jun, β-catenin. Hepatocyte growth factor receptor (C-MET/HGFR) and Bcl-2 antagonist of cell death (BAD) serum levels were determined using a quantitative sandwich enzyme immunoassay technique.
Results: Inhibition of AURK reduced the number of DEN-induced liver tumours. Apoptosis and proliferation was very low in both DEN-induced and anti- AURK groups respectively. The hepatocellular adenoma cells of DEN-treated mice uniformly had ample nuclear INCENP whereas in anti- AURK markedly decreased. Expression of β-catenin, NF-kB and c-Jun did not differ in liver tumors of both AURK -depleted and non-depleted mice.
Conclusions: Depletion of AURK reduced the number of DEN-induced hepatic tumours. However, their size did not differ significantly between the groups.
Keywords: Hepatocarcinogenesis, Hepatocellular cancer, diethylnitrosamine, aurora kinases, danusertib
Introduction
Hepatocellular carcinoma (HCC) due to its increase of occurrence rate in western countries in the last decades represents a global health problem [1,2]. In mice, different models of hepaticocarcinogenesis have been developed (genetically engineered, xenograft and chemically induced models) and have been particularly useful in studying the molecular pathway of the disease [3,4]. Among them, is widely used and well-characterized the chemically induced model of Diethylnitrosamine (DEN) administration for HCC development. [3-5]
There are common molecular pathways of HCC pathogenesis that include phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), c-MET, H-Ras and vascular endothelial growth factor (VEGF)-mediated angiogenesis [6-8]. A significant number of proteins exhibit mitosis-specific phosphorylation, and a large number of kinases are implicated in executing signaling events. There are three members of Aurora kinases (AURK) family with differences in function, subcellular localization, and activation [8]. Aurora-A impacts centrosome maturation and spindle assembly and is referred as the 'polar kinase' and is essential for the correct centrosome cycle, aurora-B regulates cytokinesis and chromosome cohesion through spindle checkpoint. [9-12]. Aurora kinases have a paramount role during the cell division process, and over-expression was found to be associated with polyploid cell clones owing multiple centrosomes in the absence of p53 activity [13]. Aurora A overexpression was associated with the amplification of the coding AURKA gene which is located at 20q13. Gene amplification was first reported in breast cancer [14] followed by a wide range of epithelial tumours including colon, bladder, ovarian and pancreatic carcinoma.
Overexpression of AURK in solid and haematological cancers was linked with dismal prognosis [8]. Further research demonstrated that amplification and overexpression of AURK is associated with processes of carcinogenesis such as telomerase activity, migration and anchorage-independent growth, RAS/RAF/MEK/ERK/MAK kinase pathway. These findings triggered the interest of researchers to develop aurora kinase inhibitors [8,15]. Pan-Aurora kinase inhibition resulted in cellular changes similar to loss of Aurora B activity [16.] So far, the pan-aurora kinase inhibitor VE-465 showed antineoplastic effects in preclinical models of human HCC [17]. Danusertib a 3-aminopyrazol derivative is potent pan-aurora kinase inhibitor. It inhibits several cancers related to tyrosine kinases as well as Abl, Trk-a, FGFR-1 and Ret. Taken into account this feature of cross-reactivity of the danusertib researchers broaden the indications for further testing of its antineoplastic effects in several experimental and preclinical cancer models.
The aim of this study was to study the significance of AURK expression and the anticancer effects of pan-aurora kinase inhibitor danusertib in experimental model of DEN-induced hepatocarcinogenesis.
Materials and Methods
Animals
C56BL/6JOlaHsd male mice weighing 25-27 gr were purchased by the Hellenic Pasteur Institute. Mice were kept in stainless cages at constant 22 to 24oC temperature and allowed free access to food and water. All experimental procedures were according to the guide for care and use of laboratory animals [18] and were licensed (License reference number 4969) by the National Veterinary Administration Authorities according to Greek legislative Decree No 2015/92, 160/91 and European Communities Council directive no86/609/EEC.
Experimental design
A total of 30 male mice were used. At the age of 14 days, mice were injected with a single intraperitoneal (ip) injection of the carcinogen N-nitrosodiethylamine (DEN; 5mg/kg of BW) for the induction of hepatocellular carcinoma (n=20). Ten from the twenty carcinogen-injected mice were randomly picked and further treated at the age of 9 months with pan-aurora kinase inhibitor Danusertib 15mg/kg of BW twice a day intra-peritonealy for 1 week. Mice were killed with an overdose of ketamine and xylazine during anaesthesia at ten months of age. Blood was collected for ELISA and liver tissues were fixed in neutral-buffered formalin 10%.
Histopathology, immunohistochemistry and morphometry
Formalin-fixed livers were embedded in paraffin, cut at 5 μm, and stained with hematoxylin and eosin or immunohistochemistry (IHC). Primary antibodies for IHC included rabbit antibodies against Ki-67-Ab 16667, DKK1-Ab 61034, INCENP-Ab36453 (Abcam, Cambridge, UK), cleaved caspase-3-9661, NF-κB-D14E12XP p65, c-Jun-9165 (Cell Signaling, Beverly, MA), β-catenin-RB9035 (ThermoFisher Scientific/Lab Vision, Fremont, CA). Heat-induced antigen retrieval was performed with citrate buffer, pH 6, for c-Jun, cleaved caspase-3, NF-κB p65 and β-catenin or with EDTA buffer, pH 8, for Ki-67, DKK1 and INCENP. Rabbit primary antibody binding was detected with goat anti-rabbit polymer HRP (ZytoChem Plus, Berlin, Germany). Color was developed with Diaminobenzidine substrate-chromogen (ThermoFisher Scientific/Lab Vision) and tissues were counterstained with hematoxylin.
For quantitative histomorphometry, liver tumors in HE-stained sections were subscribed and their area was automatically measured in image pixels. The size of each tumor was recorded. The total tumor area per total liver area ratio was also calculated for each mouse liver section. IHC-positive cells or pixels were counted in hepatocellular adenoma images of x20 representative high power fields and results were recorded as number of cells or pixels per image as previously described. The ImageJ image processing and analysis program (NIH, Bethesda, MD) with cell counter, color threshold and histogram plug-ins was used for all histomorphometrical assessments.
Blood serum ELISA
Hepatocyte growth factor receptor (C-MET/HGFR) and Bcl-2 antagonist of cell death (BAD) serum levels were determined using a quantitative sandwich enzyme immunoassay technique of Cusabio Biotech Co, Ltd. Standards and serum samples diluted 1:2 in Sample Diluent and assayed in duplicate in a 96-well microplate, pre-coated with an antibody specific for C-MET/HGFR and BAD, respectively. A 2-hour incubation in 37°C was followed by the addition of the appropriate antibody, 1-hour incubation in 37°C, washing, the addition of avidin conjugated Horseradish Peroxidase (HRP), incubation of 1 hour in 37°C, washing and the addition of the appropriate substrate. Finally, the reaction was stopped and within 5 minutes the optical density was determined, using a microplate reader set to 450 nm. A standard curve was created and the concentration of the samples was calculated, taking into account the initial dilution of the samples. The inter-assay and intra-assay precision for both assays were <10% and <8%, respectively. The detection range for C-MET/HGFR was 0.078-5 ng/ml and for BAD was 31.2-2000pg/ml.
Statistical analyses
Histomorphometry and serum protein measurements data were compared between groups using Mann-Whitney U analysis. Statistical significance was set at P<0.05. All analyses were performed with the Graphpad Prism version 5.0 for windows, GraphPad software, San Diego, CA. Data representation was done with bar graphs depicting the mean and standard error of the parameter assessed for each experimental group.
Results
Inhibition of AURK suppresses DEN-induced liver tumours
The untreated control mice (n=10) had histologically normal livers. By contrast, DEN-challenged male mice (n=10) had several hepatic cell tumours that varied in size. Based on previously published histopathological criteria the tumours were diagnosed as hepatocellular adenomas [5]. They had the typical mouse DEN-induced adenoma histomorphology (Fig. 1A), which has been described previously in detail [5]. Aurora kinase (AURK) are emerging promising targets in the treatment of various neoplasms including hepatocellular cancer [24, 25]. For that, we used the DEN mouse model of chemical liver carcinogenesis to test whether the inhibition of AURK affects hepatocellular tumours. We examined DEN-challenged mice at the age of 10 months and find that those treated with anti- AURK antibodies for ten consecutive days at the age of 9 months, had significantly fewer liver tumours compared to their untreated counterparts (Fig. 1B). However, the liver tumours of AURK-inhibited mice although fewer did not differ significantly in size from those of untreated mice (Fig. 1C).
(A) High-power magnification demonstrating the typical histomorphology of DEN-induced hepatocellular adenomas. There is increased cellular pleomorphism and atypia and spherical, variably-sized eosinophilic hyaline inclusion bodies. (B) Depletion of Aurora kinases worked to reduce the number of hepatocellular adenomas in DEN-treated mice at statistically significant levels. (C) DEN-induced liver tumors (arrows) appearing as sharply demarcated, hypercellular, basophilic hepatic cell nodules. Despite reduction in number, hepatocellular adenomas of anti-Aks-treated mice had comparable sizes with tumors of untreated controls. A and C. Hematoxylin and Eosin. Scale bars: 25 μm (A) and 250 μm (C). B and bar graph in C. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameter assessed; NS p>0.05; **p<0.01
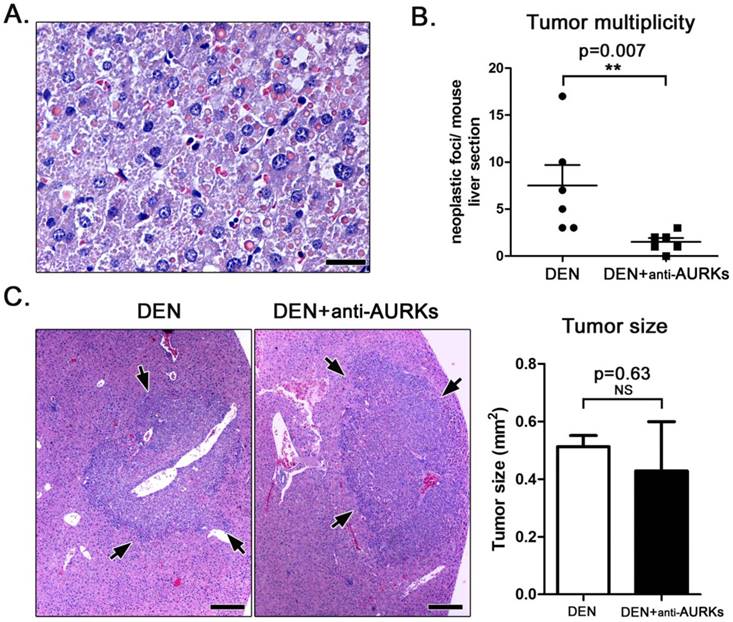
Effects of AURK inhibition on proliferation and apoptosis of liver tumour cells
Besides its major role of AURK in aneuploidy induction, several studies suggest that AURK influences cell cycle progression and apoptosis of tumor cells [19]. For that we next probed the effect of AURK neutralization in the proliferation and apoptosis of mouse hepatocellular adenomas. Proliferation in tumour cells was assessed in liver sections by means of Ki-67-specific immunohistochemistry. The morphometric counts of ki-67-positive tumour cells showed that the treatment with anti- AURK antibodies did not affect the proliferation of hepatocellular adenoma cells at statistically significant levels (Fig 2). To label apoptotic tumour cells, we next applied caspase-3-specific immunohistochemistry. We found that the numbers of apoptotic cells in DEN-induced liver adenomas was low and the effect of AURK depletion was not statistically significant (Fig 2).
Effects of Aks depletion on the immunohistochemical expression of tumor-associated factors. DEN-induced adenomas had relatively low numbers of proliferating ki-67-positive cells and occasional apoptotic Caspase-3-positive cells (arrows). The depletion of Aks had no effect on proliferation and apoptosis of neoplastic cells. Likewise, it did not alter the expression of tumor-associated proteins NFκ-B p65 and c-Jun. NFκ-B p65 expression in tumor cells remains cytoplasmic, whereas non-parenchymal cells morphologically consistent with kupffer cells show nuclear expression. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 50 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. NS p>0.05
Effects of AURK inhibition on selected tumour markers
The NFκ-B and c-Jun signaling pathways have been shown to correlate with increased malignancy in hepatocellular tumors of both humans and mouse models [20]. Using immunohistochemistry, we found that tumour cells in both anti-AURK-treated and non-treated control mice had multi-focally diffuse cytoplasmic NFκ-B p65 expression in a background of NFκ-B p65-negative non-tumoral liver tissue. Morphometric counts of positively stained image pixels showed that hepatocellular adenomas in both groups did not differ in terms of NFκ-B p65 expression (Fig. 2). The positive c-Jun signal in the liver sections restricted to tumor areas with several neoplastic cells in hepatocellular adenomas showing typical c-Jun nuclear expression. The morphometric counts, however, suggested that the liver adenomas in both anti-AURK treated and non-treated groups had comparable amounts of c-Jun-positive tumour cells (Fig. 2)
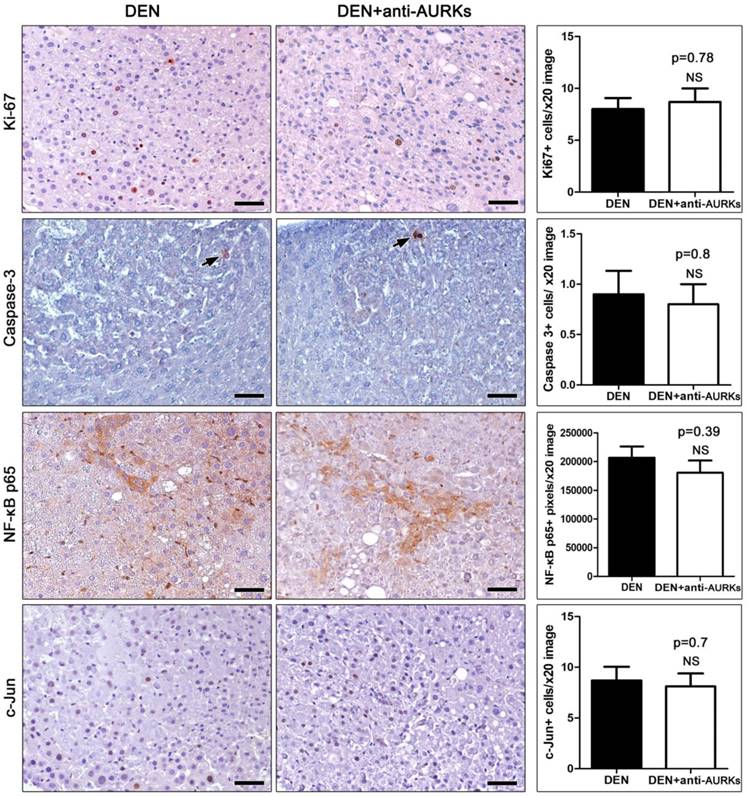
β-Catenin and the WNT signaling pathway inhibitor Dickkopf-related protein 1 (DKK1) are overexpressed in liver cancer. Upon immunohistochemistry, mouse hepatocellular adenomas stood out from non-tumoral liver tissue due to denser β-catenin membrane staining (Fig. 3). The tumour cells, however, did not have aberrant nuclear or cytoplasmic accumulation of β-catenin (Fig. 3). Counts of immunohistochemically-positive signal image pixels revealed that the expression of β-catenin did not differ significantly between liver tumours of AURK-inhibited and non-inhibited mice. Likewise, the tumours of both experimental groups had comparable levels of DKK1 expression (Fig. 3).
Effects of Aks depletion on the immunohistochemical expression of neoplastic cell Wnt/β-catenin signaling. Hepatocellular adenomas show increased β-catenin immunohistochemical signal. However, β-catenin remains localized in cell membranes with only few cells showing cytoplasmic stabilization and none nuclear translocation of β-catenin. The tumor cells also show prominent cytoplasmic DKK1 expression. Anti-Aks therapy affects neither β-catenin, nor DKK1 expression in liver tumors. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 50 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. NS p>0.05
Inhibition of AURK diminishes INCENP expression
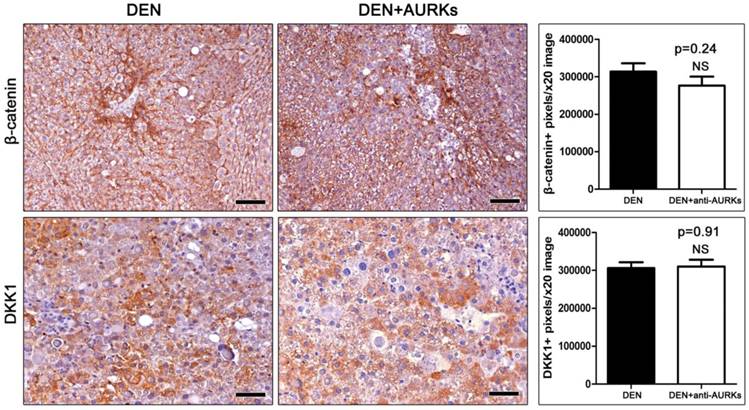
Hepatocarcinogens induce cytomegaly, which contributes to increased cellular pleomorphism and increased ploidy [21]. For that, we next sought to detect the expression of inner centromere protein (INCENP), which is a regulator of AURK and integral for accurate chromosome segregation and completion of cytokinesis during mitosis [19]. By using INCENP-specific immunohistochemistry we found that while non-tumoral liver tissue had no positivity, the hepatocellular adenoma cells of DEN-treated mice uniformly had ample nuclear INCENP. By contrast, neoplastic liver cells in hepatocellular tumors of AURK-inhibited mice had absent INCENP expression (Fig. 4A).
Effects of Aks depletion on INCENP expression and on blood serum tumor markers. (A) Hepatocellular adenomas show ample nuclear INCENP. In the tumors of anti-Aks-treated mice, however, INCENP expression is practically absent. (B) Mice with DEN-induced hepatocellular adenomas have significantly less Bcl-2 in their blood serum by comparison with tumor-free controls. The levels of Bcl-2 increase significantly after anti-Aks treatment. (C) Serum c-met is significantly increased after the neutralization of Aks. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 25 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. NS p>0.05, *p<0.05, **p<0.01, ***p<0.001.
Impact of AURK inhibition in serum Bcl-2 and cMet
We next assessed Bcl-2 levels in the serum of mice using ELISA. We found that mice bearing liver tumors had significantly decreased serum levels of Bcl-2 compared to cancer-free controls. Treatment with anti- AURK had a statistically significant effect in increasing circulating levels of Bcl-2 (Fig. 4B). This result suggests that anti- AURK blockade increases Bcl-2-associated anti-apoptotic signals in mice with DEN-induced liver tumors.
By using ELISA, we also measured c-Met serum protein levels in the mice of our study. c-Met, also called hepatocyte growth factor receptor (HGFR), is a tyrosine-protein kinase activator that is abnormally upregulated in liver cancer. We found that serum c-Met is slightly upregulated in mice with DEN-induced liver tumors by comparison with cancer-free controls, with the difference not reaching statistical significance. The group treated with of anti- AURK showed a statistical significance increase of c-Met levels when compared to DEN-exposed mice that received no further treatment (Fig. 4C).
Discussion
Aurora Kinases consists a family of serine/threonine mitotic kinases and play a key role in accurate cell cycle and genomic stability. It is shown that genomic and chromosomal instability are correlated with hepatocarcinogenesis and differential status in human HCC [22,23] Overexpression of AURK in solid and haematological cancers was linked with dismal prognosis [8]. Further research demonstrated that amplification and overexpression of AURK is associated with processes of carcinogenesis such as telomerase activity, migration and anchorage-independent growth, and RAS/RAF/MEK/ERK/MAK kinase pathway [8].
Tanaka et al. [24] found aurora-B gene expression in resected HCCs as the most significant predictor for aggressive tumor recurrence or genetic instability. Lin et al [25] have reported an Aurora B mRNA over-expression in 61 % of HCC cases by RT-PCR. They showed that Aurora B increase was associated with aggressive tumour phenotype and advanced tumor stage and grade, and finally poor overall survival.
Tang et al 2017 [19] described the major role of AURKB in aneuploidy, cell cycle progression and apoptosis.
In our study, we have used a validated model of experimental hepatocarcinogenenis in order to examine the impact of blocking Aurora kinases with an anti-Pan-Aurora kinases inhibitor. DEN-administered male mice had several hepatic cell tumors that varied in size compared to untreated controls. Based on previously published histopathological criteria the tumors were diagnosed as hepatocellular adenomas [5]. In DEN-challenged mice we found that those treated with anti-Pan-Aurora specific antibodies for seven consecutive days had significantly fewer liver tumors compared to their untreated counterparts. Interestingly, inhibition of the AURK provoked reduction in the numbers of tumours without to reduce significantly the size of the residual tumors. Benten et al [26] in a in a xenograft tumor model showed that treatment with PHA-739358 at a dosage of 2 × 15 mg/kg per day significantly decreased tumor growth of both, rapidly proliferating Huh-7 tumors and moderately growing HepG2 xenografts. The mean absolute tumor volume was reduced by 80.4%, the mean percent tumor growth was reduced by 84.4%, and the mean tumor weight on necropsy was reduced by 71.8%, respectively, compared with vehicle-treated controls. Antiproliferative efficacy was even more pronounced in HepG2 tumors at the same dosage level. The difference in the results could be attributed to the different hepaticocarcinogenesis protocols and possible upregulation of different molecular pathways.
Hepatocarcinogens induce cytomegaly, which contributes to increased cellular pleomorphism and increased ploidy [21]. AURK plays a key role as essential regulators of the mitotic spindle and protein phosphorylation in mitotic control [27-29]. They interact with other proteins to control chromosome assembly and segregation. Aurora kinase B, inhibition rapidly results in catastrophic mitosis with senescence [30,31]. During mitotic karyokinesis under downregulation of Aurora kinases, a process termed micronucleation occurs in the cancer cells. Thus, unable to maintain G2 arrest, they enter mitosis and after being arrested for several hours at metaphase, they eventually die without successfully completing mitosis. This process is known as p53-independent cell death or mitotic catastrophe [31]. INCENP plays a central role in the activation of AURKB, via a positive-feedback loop mediated by phosphorylation of the Thr-Ser-Ser (TSS) motif within the INbox near the C-terminus of INCENP [32-33]. This functional interaction is involved in the subcellular localization of the CPC itself and the regulation of mitotic checkpoints such as the spindle assembly checkpoint (SAC), which monitors and corrects errors in kinetochore-microtubule attachment. In addition, depletion of endogenous INCENP or expression of inactive INCENP mutants (i.e., alleles harboring non-functional mutations of the TSS motif or deletion of the C-terminal region) causes severe cytokinesis defects [34,35]. From our results, we could see that while non-tumoral liver tissue had no positivity, the hepatocellular adenoma cells of DEN-treated mice uniformly had ample nuclear INCENP. By contrast, neoplastic liver cells in hepatocellular tumors of AURK-treated mice had absent INCENP expression. This finding could be attributed to catastrophic mitosis with senescence and provide a possible explanation of our observation concerning the disappearances of a number of tumors in the treated animals and the negative impact in size in the remaining tumors. Our results show that a variable degree of INCENP expression is a consistent feature of DEN-induced carcinogenesis. Also, that the liver tumors of AURK-inhibited mice universally lack INCENP expression, which indicates that the drug had a suppressive effect on the expression of this molecule. Our study suggests that the efficacy of AURK inhibition of DEN-induced liver is not universal for all tumors. It is highly probable that it depends on their size that may in turn reflect their evolutional stage. Whether this phenomenon directly relates or rather co-exists with AURK-inhibited associated loss of INCENP cannot be addressed in the current study. The discrepancy regarding the regression of tumors after AURK-inhibition treatment we have observed, highlights the need for additional elaborate and more targeted studies for explaining the role of INCENP in hepatocellular cancer evolution. Also, for identifying additional molecules and mechanisms that determine the traits of AURK-inhibited hepatocellular tumor treatment efficacy.
It is not very easy and accurate to compare different studies using DEN-induced hepatocarcinogenesis in mice using different strains of mice, and the application of divergent experimental designs and morphometrical approaches for accessing proliferation and apoptosis.
Qi et al [36] studied the expression of AURKB and its correlation with cell proliferation in 40 oral squamous cell carcinoma cases. They showed the elevation of AURKB and Ki67 expressing cell fractions in carcinomas compared to normal epithelial tissue, with a linear correlation between the two datasets. They also found a prominent kinase expression in cases with lymph node metastasis.
In our present study, the hepatocellular adenomas that were developed had low neoplastic cell proliferation and apoptosis levels, a finding however that has been reported also previously [37]. Lee et al [38] reported that DEN-induced hepatocellular proliferating lesions including adenomas have only 11 PCNA-positive proliferating hepatocytes in every 100 abnormal hepatocytes counted; the relatively low proliferating index, which is a typical feature of human hepatocellular adenoma as well [32], probably reflects the low proliferation rate of hepatocytes that in normal conditions rest in the G0 phase of the cell cycle. It also explains the slow growth of hepatocellular tumors in DEN-treated rodent models, that, as in the present study, are not further manipulated to promote tumor initiation and growth. In our study, we have found that the numbers of apoptotic cells in DEN-induced liver adenomas was low and the effect of AURK inhibition was not statistically significant. Similarly, to proliferation, apoptosis in hepatocellular adenomas of mice has also been reported to be as low as 0.02 to 0.44% regardless of mouse strain and induction of carcinogenesis protocol [39]. The results of caspase-3 specific IHC performed in the present study further confirm that apoptosis in mouse hepatocellular adenomas is rather rare.
Matsunaga et al [40] proposed that Bcl-2 proteins control a checkpoint of cellular survival during polyploidization and its major function is to modulate mitochondrial membrane permeability directly. Moreover, that regulates the release of pro-apoptotic factors via the intermembrane space into the cytoplasm. The Bcl-2 family is associated with the mitotic catastrophe after polyploidization by the activation of mitochondrial membrane permeabilization [41].
The HGF/c-MET pathway has been involved in HCC progression demonstrated to play a key role in growth, epithelial-mesenchymal transition, angiogenesis, invasiveness and metastasis in HCC cells [42]. c-MET has also been reported to be essential for the metastatic potential of HCC cells and represents as a powerful prognostic indicator for the early stage of cancer invasion and metastasis [42].
In our study, we have evaluated the serum levels of BCL-2 and c-MET. Interestingly BCL-2 was systematically decreased in DEN-induced hepaticocarcinogenesis mice and it was increased with the anti-Aurora treatment despite the very low local apoptotic rate. In the case of c-MET control and DEN-induced mice levels were not significantly different although pan-Aurora inhibitors dramatically increased the serum levels. The interpretation of above results is not easy and need further investigation.
Inhibiting AURK in our study did not affected NF-kB and c-Jun expression. The NF-κB and c-Jun signaling pathways have been shown to correlate with increased malignancy in hepatocellular tumors of both humans and mouse models [43]. In our experiment, hepatocellular adenomas in both groups did not differ in terms of NFκ-B p65 expression. The positive c-Jun signal in the liver sections restricted to tumor areas with several neoplastic cells in hepatocellular adenomas showing typical c-Jun nuclear expression. The liver adenomas in both anti-AURK treated and non-treated groups had comparable amounts of c-Jun-positive tumor cells.
We have also examined the possible impact of Aurora kinase inhibitor in the expressing of β-catenin and the Dickkopf-related protein 1 (DKK1) which are usually over-expressed in liver cancer [43]. Hepatocellular adenomas stood out from non-tumoral liver tissue due to denser β-catenin membrane staining whilst the tumor cells did not have aberrant nuclear or cytoplasmic accumulation of β-catenin. At the end the expression of β-catenin did not differ significantly between liver tumors of AURK-inhibited and non-inhibited mice. Likewise, the tumors of both experimental groups had comparable levels of DKK1 expression. Taken together those results we could see that inhibiting Aurora kinases pathway has a minimal impact on NF-kB, c-Jun and b-catenin pathways and the antineoplastic effect comes from another pathway as we mentioned earlier.
In conclusion pan anti- AURK inhibition is likely to have a significant impact in DEN-induced hepatocarcinogenesis in mice. Treated animals exhibited fewer tumors compared with the non-treated, despite the low apoptotic and proliferative index and the minimal impact in traditional hepatocellular carcinoma pathways.
Acknowledgements
This study was funded as Research Scholarship by the Experimental - Research Center ELPEN. Also, we would like to express our sincere thanks to Mr E. Gerakis and Mr S. Gerakis for their assistance in the implementation of the experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907-17
2. Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat Med. 2012;4:19-37
3. Santos NP, Colaço AA, Oliveira PA. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumor Biol. 2017;39:1010428317695923
4. Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367-86
5. Goldfarb S, Pugh TD, Koen H, He YZ. Preneoplastic and neoplastic progression during hepatocarcinogenesis in mice injected with diethylnitrosamine in infancy. Environ Health Perspect. 1983;50:149-61
6. Kim Y, Sills RC, Houle CD. Overview of the molecular biology of hepatocellular neoplasms and hepatoblastomas of the mouse liver. Toxicol Pathol. 2005;33:175-80
7. Wilkens L, Flemming P, Gebel M, Bleck J, Terkamp C, Wingen L. et al. Induction of aneuploidy by increasing chromosomal instability during differentiation of hepatocellular carcinoma. Proc Natl Acad Sci USA. 2004;101:1309-14
8. Gavriilidis P, Giakoustidis A, Giakoustidis D Aurora kinases, potential medical applications of aurora kinases inhibitors. A review. J Clin Med Res. 2015;7:742-751
9. Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639-1648
10. Salaun P, Rannou Y, Prigent C. Cdk1, Plks, Auroras, and Neks: the mitotic bodyguards. Adv Exp Med Biol. 2008;617:41-56
11. Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5:42-50
12. Lukasiewicz KB, Lingle WL. Aurora A, centrosome structure, and the centrosome cycle. Environ Mol Mutagen. 2009;50:602-619
13. Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21:483-492
14. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643-649
15. Carpinelli O, Ceruti R, Giorgini ML, Cappela P, Gianellini L, Croci V, Degrassi A. et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158-68
16. Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S. Molecular basis of drug resistance in aurora kinases. Chem Biol. 2008;15:552-562
17. Lin ZZ, Hsu HC, Hsu CH, Yeh PY, Huang CY, Huang YF. et al. The aurora kinase inhibitor VE-465 has anticancer effects in preclinical studies of human hepatocellular carcinoma. J Hepatol. 2009;50:518-527
18. Committee. Guide for the Care and Use of Laboratory Animals. 8th Edition. 2011:118
19. Tang M, Chen J. 53BP1: keep an eye on merometry. Oncotarget. 2017;25:48527-48528
20. Maeda S, Kamata H, Luo JL, Leffert H, Kavin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977-90
21. Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T. et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 2010;38:5s-81s
22. Kim GJ, Cho SJ, Won NH, Sung JM, Kim H, Chun YH, Park SH. Genomic imbalances in Korean hepatocellular carcinoma. Cancer Genet Cytogenet. 2003;142:129-133
23. Wilkens L, Flemming P, Gebel M, Bleck J, Terkamp C, Wingen L. et al. Induction of aneuploidy by increasing chromosomal instability during differentiation of hepatocellular carcinoma. Proc Natl Acad Sci USA. 2004;101:1309-14
24. Tanaka S, Arii S, Yasen M, Mogushi K, Su NT, Zhao C, Imoto I, Eishi Y, Inazawa J, Miki Y. et al. Aurora kinase B is a predictive factor for the aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg. 2008;95:611-619
25. Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL, Lee PH, Cheng AL, Hsu HC. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer. 2010;10:461
26. Benten D, Keller G, Quaas A, Schrader J, Gontarewicz A, Balabanov S, Braig M, Wege H, Moll J, Lohse AW, Brummendorf TH. Aurora kinase inhibitor PHA-739358 suppresses growth of hepatocellular carcinoma in vitro and in a xenograft mouse model. Neoplasia. 2009;11:934-44
27. Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer. 2010;10:825-841
28. Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60-72
29. Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927-936
30. Girdler F, Gascoigne KE, Eyers PA. et al. Validating Aurora B as an anticancer drug target. J Cell Sci. 2006;119:3664-75
31. Vakifahmetoglu V, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153-62
32. Adams RR, Wheatley SP, Gouldsworthy AM, Kandels-Lewis SE, Carmena M, Smythe C. et al. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol. 2000;10:1075-8
33. Bishop JD Schumacher JM. Phosphorylation of the carboxyl terminus of inner centromere protein (INCEP) by the Aurora B kinase stimulates Aurora B kinase activity. J Biol Chem. 2002;277:27577-80
34. Honda R, Körner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP and survivin in mitosis. Mol Biol Cell. 2003;14:3325-41
35. Xu Z, Vagnarelli P, Ogawa H, Samejima K, Earnshaw WC. Gradient of increasing Aurora B kinase activity is required for cells to execute mitosis. J Biol Chem. 2010;285:40163-70
36. Qi G, Ogawa I, Kudo Y, Miyauchi M, Siriwardena BS, Shimamoto F, Tatsuka M, Takata T. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007;450:297-302
37. Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA. et al. Beta-catenin signaling in murine liver zonation and regeneration: A Wnt-Wnt situation!. Hepatology. 2014;60:964-76
38. Lee GH, Ooasa T, Osanai M. Mechanism of the paradoxical, inhibitory effect of phenobarbital on hepatocarcinogenesis initiated in infant B6C3F1 mice with diethylnitrosamine. Cancer Res. 1998;58:1665-9
39. Bursch W, Chabicovsky M, Wastl U, Grasl-Kraupp B, Bukowska K, Taper H. et al. Apoptosis in stages of mouse hepatocarcinogenesis: Failure to counterbalance cell proliferation and to account for strain differences in tumor susceptibility. Toxicol Sci. 2005;85:515-29
40. Matsunaga H. et al. A Novel Therapeutic Combination Sequentially Targeting Aurora B and Bcl-xL in Hepatocellular Carcinoma. Ann Surg Oncol. 2015;22:3079-3086
41. Casredo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. et al. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825-37
42. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989-5005
43. Giakoustidis A, Giakoustidis D, Mudan S, Sklavos A, Williams R. Molecular signaling in hepatocellular carcinoma: Role of and crosstalk among WNT/ß-catenin, Sonic Hedgehog, Notch and Dickkopf-1. Can J Gastroenterol Hepatol. 2015;29:209-17
Author contact
![]() Corresponding author: Dimitrios Giakoustidis MD, PhD, Ass. Professor of Surgery and Transplantation, Division of Transplant Surgery, Department of Surgery, School of Medicine, Faculty of Health Sciences, Aristotle University and Hippokration General Hospital, Konstantinoupoleos 49, 54642 Thessaloniki, GREECE. Tel: +302310992875; Fax: +302310237914; Email: dgiakgr
Corresponding author: Dimitrios Giakoustidis MD, PhD, Ass. Professor of Surgery and Transplantation, Division of Transplant Surgery, Department of Surgery, School of Medicine, Faculty of Health Sciences, Aristotle University and Hippokration General Hospital, Konstantinoupoleos 49, 54642 Thessaloniki, GREECE. Tel: +302310992875; Fax: +302310237914; Email: dgiakgr