Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(5):923-928. doi:10.7150/jca.22802 This issue Cite
Research Paper
Genome-wide Association Study (GWAS) of Germline Copy Number Variations (CNVs) Reveal Genetic Risks of Prostate Cancer in Chinese population
Yishuo Wu1,2,3*, Haitao Chen4*, Guangliang Jiang1,2*, Zengnan Mo5, Dingwei Ye6, Meilin Wang7, Jun Qi8, Xiaoling Lin2, S. Lilly Zheng3, Ning Zhang1,2, Rong Na2,9 ![]() , Qiang Ding1,2
, Qiang Ding1,2 ![]() , Jianfeng Xu2,3
, Jianfeng Xu2,3 ![]() , Yinghao Sun10
, Yinghao Sun10
1. Department of Urology, Huashan Hospital, Fudan University, Shanghai, PR China
2. Fudan Institute of Urology, Huashan Hospital, Fudan University, Shanghai, PR China
3. Program for Personalized Cancer Care, NorthShore University HealthSystem, Evanston, Illinois, USA
4. State Key Laboratory of Organ Failure Research, Guangdong Key Laboratory of Viral Hepatitis Research, Department of Infectious Diseases and Hepatology Unit, Nanfang Hospital, Southern Medical University, GuangZhou, China.
5. Department of Urology and Nephrology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China.
6. Department of Urology, Fudan University Shanghai Cancer Center, Shanghai Medical College, Fudan University, Shanghai, China.
7. Department of Molecular and Genetic Toxicology, The Key Laboratory of Modern Toxicology of the Ministry of Education, School of Public Health, Nanjing Medical University, Nanjing, China.
8. Department of Urology, Xinhua Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China.
9. Department of Urology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
10. Department of Urology, Shanghai Changhai Hospital, Second Military Medical University, Shanghai, China.
*These authors contributed equally to this study.
Received 2017-9-12; Accepted 2018-1-29; Published 2018-2-27
Abstract
Introduction: The associations between Prostate cancer (PCa) and germline copy number variations (CNVs) in genome-wide level based on Chinese population are unknown. The objective of this study was to identify possible PCa-risk associated CNV regions in Chinese population.
Materials and Methods: We performed a genome-wide association study for CNV in 1,417 PCa cases and 1,008 controls in Chinese population.
Results: 7 risk-associated CNVs were identified for PCa after association analyses (P <7.2×10-6). Another 34 CNVs were found to be potentially risk-associated CNVs (P<0.05). Among the total 41 CNVs, 27 CNVs were risk variations and the other 14 were found to be protective of PCa. 25 of the CNVs (19 duplications and 6 deletions) were located in gene regions while 16 CNVs (9 duplications and 7 deletions) were located in intergenic regions. We identified a higher burden of gaining PCa-risk CNVs and a lower frequency of protective CNVs in cases than controls. Bioinformatics analyses suggested that genes related to PCa risk-associated CNVs were significantly enriched in some biological processes, cellular components and molecular functions.
Conclusion: These results provided additional information of genetic risks for PCa. Several CNV regions involved actionable genes that might be potential gene for target therapy. Additional validation and functional studies are warranted for these results.
Keywords: copy number variation, genome-wide association study, prostate cancer, China.
Introduction
Prostate cancer (PCa) represents the second most common cancer and the fifth most common cause of cancer-related death in men, with more than 900,000 new cases diagnosed each year [1]. In China, the incidence has increased by nearly six-fold during last 30 years [2]. Genetic risk is one of the most important factors causing PCa, which can explain ~42% of the disease carcinogenesis [3]. Till now, more than 100 PCa risk-associated single nucleotide polymorphisms (SNPs) have been found through genome-wide association studies (GWAS) [4]. However, a study showed these risk-associated SNPs could only explain about 33% of the familial risk for PCa [5]. In addition, most of these SNPs are located in intergenic regions [4]. Thus, it is hard to understand the possible functional effects of these SNPs on pathogenesis.
Copy-number variations (CNVs) are considered another major fact of genetic diversity, which could directly modulate cellular biological functions [6]. In addition, studies showed that CNVs cover ~35% of the human genome, while SNPs only account for less than 1% [7]. Several studies indicated that germline CNVs were associated with PCa in Caucasian and African-American populations [8-12]. However, the association between PCa and CNVs in a genome-wide level has not been reported in Chinese population.
In the present study, we performed a genome-wide CNV association study with PCa in a Chinese population by using the published GWAS data from our previous study [13]. Our objective was to identify possible germline PCa-risk CNV regions, which might provide additional insight to the inherited risk of PCa.
Materials and Methods
Study population
The study population is a part of Chinese Consortium for Prostate Cancer Genetics (ChinaPCa). The demographic characteristics and clinical features of the study were reported in our previous study [13]. All the subjects are male Han Chinese from the southeastern region of China. A total of 1,417 PCa cases and 1,008 controls were recruited. All of the cases were pathologically diagnosed with primary PCa. Controls were recruited from the community population. Written informed consent was obtained from each participant. The study was approved by the institutional review board of each medical center that participated in ChinaPCa.
Genotyping
DNA was extracted from blood sample of each subject. Illumina Human OmniExpress BeadChips (Illumina, San Diego, California) were used to genotype the samples. Finally, a total of 731,458 SNPs were genotyped. Genotyping was performed at State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, Shanghai, China.
CNV Calling
Raw signal intensity files were first generated by the export function provided in Illumina GenomeStudio. Then we used UCSC Genome Browser's liftover tool to map SNPs to the newer reference genome assembly (http://genome.ucsc.edu/cgi-bin/hgLiftOver). By choosing proper population frequency of B allele (PFB) and GCmodel files, we then used PennCNV to identify CNVs and generate a quality control summary for each sample [14].
Quality Controls
During SNP genotyping, samples were removed if they (i) had an overall genotyping rate of <95%; or (ii) were duplicates or showed familial relationships (PI_HAT > 0.025). SNPs were excluded if they had (i) a call rate of <95%; (ii) a minor allele frequency (MAF) of <0.01; or (iii) P < 1 × 10-3 in a Hardy-Weinberg equilibrium test among controls. During individual quality control analysis, samples with highly variable signal intensity was removed, including (i) LRR_SD (standard deviation of Log R Ratio) >0.3; (ii) BAF_drift (measuring departure of the B Allele Frequency from the expected values) > 0.01; (iii) WF (waviness factor, the amount of dispersion in signal intensity) >0.05 or WF<-0.05; (iv) Number of CNVs >50 (number of called-CNVs). After individual quality control, a total of 753 cases and 982 controls with 51788 CNVs remained.
During CNV calling, CNVs were removed if (i) number of SNPs spanning less than 10; (ii) length of CNV >50 kb. After call-level quality control, 8352 CNVs remained.
Then we merged adjacent CNV calls using a threshold of gap<20% of total length during which 91 paired CNV were merged.
We also removed the CNVs calls located in certain regions, including HLA regions, immunoglobulin regions, telomere regions, centromere regions. After quality control analysis, a total of 6932 CNV regions were able to be used for further analyses.
Statistical Analysis
Predicted CNVs from the sample genomic locus can have various start and end points across individuals. To make the analysis simpler, we divided the genome into copy number polymorphic regions (CNPRs). Each individuals is then assigned a copy number (CN) state for each CNPR according to the CNV predicted in that region, with CN = 2 if no CNV is predicted.[14] In order to discriminate the CNV with a single duplication of one allele and single deletion of the other (CN=2) from non-CNV status (CN=2), we further encoded CN state with deletion and duplication variables.[14] P-link 1.09 was used to perform association analysis. Logistic regression analysis was used to adjust for age. Fisher exact test was used if 25% of cells with expected count less than 5. Two-tail Bonferroni corrected p value of 7.2×10-6 (0.05/6932) was considered statistical significant.
Bioinformatics analyses
The CNVs found in this study were compared with the data based on normal populations in the Datablse of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home). Gene annotation in the CNV regions was performed by using UCSC gene browser. DAVID was used to perform functional annotation of affected genes located within CNVs [15].
Results
After quality control, a total of 753 cases (PCa patients) and 982 controls were available for further analyses. The characteristics of the study population are shown in Table 1. The mean age of case group was significantly higher than that of control group. The difference of age was adjusted in the further association analyses.
Characteristics of the study population
| Variables | Case (N=753) | Control (N=982) | P |
|---|---|---|---|
| Age (year, mean±SD)* | 71.4±8.04 | 61.5±9.5 | <1.0×10-4 |
| PSA (ng/mL, median, IQR)* | 26.6(13.2-79.1) | 0.6(0.3-1.2) | <1.0×10-4 |
| Gleason Score (No., %) | |||
| <7 | 189(25.1) | - | |
| ≥7 | 500(66.4) | - | |
| Missing | 64(8.5) | - |
*Age and PSA at diagnosis for cases or at recruitment for controls.
Seven risk-associated CNVs were identified for PCa after association analyses (P value in bold<7.2×10-6). Another 34 CNVs were found to be potentially risk-associated CNVs (P<0.05) (Table 2). After comparing with DGV database, all CNV regions were previously reported overlapped with common CNVs. Among the total 41 CNVs, 27 CNVs were risk variations and the rest 14 were found to be protective variations of PCa. In addition, 25 of the CNVs (19 duplications and 6 deletions) were significantly associated with PCa and were located in gene regions (all P values<0.05). The remaining 16 CNVs (9 duplications and 7 deletions) were located in intergenic regions.
Copy number variations associated with prostate cancer in potential
| Chr. | Start | End | CNV type | Direction | Cases | Control | OR | P value | Genes |
|---|---|---|---|---|---|---|---|---|---|
| 19 | 20079177 | 21077133 | Del | risk | 9 | 0 | 12.60 | 2.39×10-3 | *CR593334,CR614976,LOC284441,ZNF430,ZNF486 |
| 11 | 67258202 | 67488532 | Del | risk | 32 | 2 | 2.08 | 6.77×10-3 | AK091996,BC110365,CR602694 |
| 10 | 59242376 | 59438056 | Del | risk | 9 | 0 | 6.30 | 1.08×10-2 | intergenic |
| 21 | 20315468 | 20449022 | Del | risk | 5 | 0 | INF | 1.27×10-2 | intergenic |
| 11 | 88204734 | 88263638 | Del | risk | 4 | 0 | INF | 3.03×10-2 | GRM5 |
| 16 | 2136084 | 2187363 | Del | risk | 4 | 0 | INF | 3.03×10-2 | CASKIN1,RAB26,SNORD60,TRAF7 |
| 16 | 616990 | 718025 | Del | risk | 4 | 0 | INF | 3.03×10-2 | *AK128777,AL360260,C16orf13,C16orf14,CCDC78 |
| 3 | 4132503 | 4187007 | Del | risk | 7 | 0 | 4.89 | 3.98×10-2 | SUMF1 |
| 6 | 79031809 | 79086086 | Dup | risk | 157 | 33 | 7.02 | 7.86×10-32 | intergenic |
| 11 | 88336310 | 88384073 | Dup | risk | 103 | 11 | 13.72 | 2.39×10-28 | GRM5 |
| 18 | 14737958 | 14828909 | Dup | risk | 45 | 17 | 3.74 | 9.08×10-7 | ANKRD30B |
| 6 | 32601768 | 32664508 | Dup | risk | 19 | 3 | 8.90 | 2.71×10-5 | AK293020,HLA-DRB1,HLA-DRB5 |
| 10 | 22249227 | 22376157 | Dup | risk | 11 | 0 | INF | 6.63×10-5 | DNAJC1 |
| 15 | 19257404 | 19408394 | Dup | risk | 6 | 0 | INF | 5.28×10-3 | A26B1,DQ582260,M84131,POTEB |
| 6 | 80285260 | 80386902 | Dup | risk | 6 | 0 | INF | 5.28×10-3 | LCA5 |
| 4 | 9003188 | 9169160 | Dup | risk | 11 | 3 | 5.13 | 6.25×10-3 | DEFB131,LOC650293 |
| 5 | 109845447 | 109992561 | Dup | risk | 7 | 1 | 9.79 | 1.12×10-2 | FLJ43080 |
| 5 | 32143138 | 32144121 | Dup | risk | 32 | 22 | 2.04 | 1.19×10-2 | KIAA0300,PDZD2 |
| 13 | 63130342 | 63185384 | Dup | risk | 5 | 0 | INF | 1.26×10-2 | intergenic |
| 16 | 32073040 | 32556470 | Dup | risk | 5 | 0 | INF | 1.27×10-2 | BC041879,BC042588,DQ571479,TP53TG3b,X69637 |
| 11 | 58563817 | 58634485 | Dup | risk | 19 | 12 | 2.22 | 2.93×10-2 | AX746988,FAM111B |
| 12 | 85219842 | 85281435 | Dup | risk | 4 | 0 | INF | 3.03×10-2 | MGAT4C |
| 18 | 12062364 | 12122247 | Dup | risk | 4 | 0 | INF | 3.03×10-2 | BX648696,BX649021 |
| 3 | 76005745 | 76061858 | Dup | risk | 4 | 0 | INF | 3.03×10-2 | intergenic |
| 10 | 6717773 | 6783787 | Dup | risk | 7 | 2 | 4.89 | 3.98×10-2 | intergenic |
| 1 | 149608041 | 149614444 | Dup | risk | 7 | 2 | 4.89 | 3.98×10-2 | SELENBP1 |
| 7 | 61631605 | 61631605 | Dup | risk | 45 | 17 | 3.74 | 9.08×10-7 | intergenic |
| 19 | 32620453 | 32675491 | Del | protective | 50 | 421 | 0.15 | 8.96×10-54 | intergenic |
| 11 | 50432844 | 50586426 | Del | protective | 34 | 179 | 0.25 | 2.54×10-16 | intergenic |
| 12 | 36785447 | 36801139 | Del | protective | 7 | 43 | 0.22 | 3.47×10-5 | intergenic |
| 19 | 24228244 | 24354405 | Del | protective | 5 | 26 | 0.27 | 3.13×10-3 | intergenic |
| 9 | 11941222 | 12175185 | Del | protective | 3 | 15 | 0.28 | 3.20×10-2 | intergenic |
| 5 | 99455964 | 99486169 | Dup | protective | 1 | 32 | 0.04 | 8.23×10-7 | intergenic |
| 8 | 145079175 | 145131413 | Dup | protective | 3 | 29 | 0.14 | 9.22×10-5 | PARP10,PLEC1 |
| 8 | 114044747 | 114181033 | Dup | protective | 17 | 57 | 0.41 | 8.19×10-4 | CSMD3 |
| 1 | 187833278 | 187886901 | Dup | protective | 2 | 17 | 0.16 | 4.59×10-3 | intergenic |
| 4 | 162057165 | 162075793 | Dup | protective | 27 | 64 | 0.58 | 1.85×10-2 | intergenic |
| 12 | 22509872 | 22573948 | Dup | protective | 0 | 8 | 0.00 | 2.44×10-2 | KIAA0528 |
| 19 | 21936987 | 21995054 | Dup | protective | 0 | 8 | 0.00 | 2.44×10-2 | ZFS-3,ZNF208 |
| 4 | 145164558 | 145169643 | Dup | protective | 2 | 13 | 0.21 | 3.31×10-2 | GYPA |
| 7 | 53164643 | 53216869 | Dup | protective | 0 | 7 | 0.00 | 4.64×10-2 | intergenic |
P values and ORs are from a regional test at each locus. INF, infinite; Del, deletion; Dup, duplication. *Only five genes in that region were listed.
In order to analyze the global burden of risk CNVs in PCa patients, we compared the amount of patients harboring different types of risk CNVs with respect to 982 controls (Table 3). Risk CNVs were found in 51.5% of the PCa patients compared to 12.5% found in controls (P value=1.1×10-68). When the frequencies of deletions and duplications were analyzed separately, they were still higher in PCa patients than that in controls (9.3% vs. 2.9% in deletions with a P value = 8.2×10-9 and 46.5% vs. 10.3% in duplications with a P value = 1.1×10-68). Similar analysis was also performed in comparing the frequencies of the 14 protective CNVs in PCa patients versus in controls (Table 3). Lower frequencies of the protective CNVs were observed in PCa cases for deletions, duplications and all CNVs, respectively.
Comparisons of the frequencies of risk and protective copy number variations detected in the cases and controls
| Group | Cases(n=753) | Controls(n=982) | P value |
|---|---|---|---|
| Risk CNVs, N | 27 | 14 | |
| Deletions, n (%) | 70(9.3%) | 28(2.9%) | 8.24E-09 |
| Duplications, n (%) | 350(46.5%) | 101(10.3%) | 4.38E-65 |
| All CNVs, n (%) | 385(51.5%) | 123(12.5%) | 1.13E-68 |
| Protective CNVs, N | 11 | 14 | |
| Deletions, n (%) | 70(9.3%) | 512(52.1%) | 2.68E-78 |
| Duplications, n (%) | 51(6.8%) | 208(21.2%) | 7.01E-17 |
| All CNVs, n (%) | 118(15.7%) | 626(63.7%) | 1.84E-89 |
P values were calculated by Chi-test; N, number of CNVs; n, number of patients harboring CNVs.
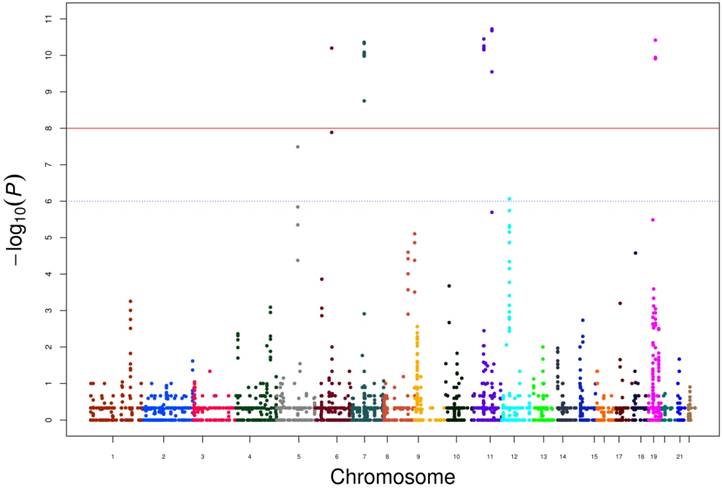
Furthermore, we calculated P values for each CNVs and scores resulting from -log10 (P) are displayed in the genome-wide Manhattan plot in Figure 1. Genes located within or nearby the CNV regions were also used to perform network analysis through cBioportal using 491 TCGA prostate cancer data (Supplementary Figure 1). HLA-DRB1, HLA-DRB5 and GRM5 were clinical actionable genes, among which GRM5 was located at CNV regions and that duplication reached a genome-wide significance in our data. Since the target therapy medications (2-Methyl-6-(phenylethynyl) pyridine, MPEP) toward GRM5 have not been approved by Food and Drug Administration (FDA), our results suggested additional evidence for its potential value as a candidate target therapy for PCa.
Genome-wide Manhatton Plot of all CNVs and scores resulting from -log10 (P).
Discussion
Studies have found several germline CNVs in Caucasian population (deletion at 2p24.3, 12q21.31, 15q21.3 and 20p13) and in African-American population (14q32.33) associated with predisposition of PCa [8, 11, 16, 17]. However, PCa risk-associated CNVs have not been reported in Chinese population. To our knowledge, this is the first genome-wide association study between CNV and PCa in Chinese population by using the published GWAS data from our previous study.
We found 41 CNVs were significantly associated with PCa and these CNVs conferring either risk or protection to PCa. More than half of the PCa risk-associated CNVs were located in gene regions. We used DAVID Bioinformatics Resources 6.8 to perform functional annotation and found that the genes were related with multiple biological processes, cellular components and molecular functions (e.g. protein binding, DNA binding, transferase activity, G-protein coupled receptor activity, virus receptor activity, hydrolase activity, nucleotide binding and so on).
For instance, in the present study, we observed a significant association between 11q14.3 duplication with PCa. GRM5 located in 11q14.3 known as metabotropic glutamate receptor 5 is a Gq protein-coupled receptor that is widely expressed in the brain and activate PLCβ, resulting in intracellular Ca2+ release and protein kinase C (PKC) activation [18]. It was reported that this pathway might relate to the proliferation and migration of tumor cells which included melanoma, oral squamous cell carcinoma, nasopharyngeal carcinoma, breast cancer, colon cancer, sarcoma and various kinds of neurologic tumors. In addition, such biological effect could be inhibited by glutamate antagonists (MPEP) [19-27]. However, it had not been reported in prostate cancer. We also looked into the expression of GRM5 in prostate cancer cell lines in Cancer Cell Line Encyclopedia (CCLE) and found the mRNA expression level was 4.14-4.43 (robust multi-array normalized) among 7 different prostate cancer cell lines, while the expression of GRM5 was nearly 0 in normal prostate tissues in Genotype-Tissue Expression (GTEx) database. In the present study, the frequency of the duplication located in GRM5 region was 13.7% in cases while the frequency was much lower in controls (1.1%). Thus we hypothesized that germline CNV of 11q14.3 may influence the expression of GRM5 and lead to carcinogenesis of PCa. However, the function of GRM5 related CNV increasing PCa risk and the differences between germline and somatic variations of GRM5 should be further investigated.
Higher frequencies of specific prostate cancer risk-associated deletions and duplications had been reported in previous studies [8, 12]. Our results also revealed a relevant contribution of CNVs to PCa-risk with a higher burden of risk CNVs and a lower frequency of protective CNVs in cases compared to controls (all P values <0.05). This provided a genome-wide summary of CNV frequencies in PCa patients and controls which might be helpful to illustrate the importance of CNVs in prostate cancer.
This case-control study was the first genome-wide CNV association study in Chinese population evaluating the relationship between CNVs and PCa. In this study, we analyzed CNV information from GWAS array by using a well-established method. Although, we failed to perform the confirmation evaluation of these CNVs at this stage, we were able to compare our findings with databases by using bioinformatics methods, and we agreed that additional validation studies should be conducted in the future. Moreover, functional studies would be necessary to validate the findings of the association between CNVs and PCa. Nevertheless, our study has provided additional information of carcinogenesis and potential treatment targets of prostate cancer.
Conclusion
41 CNVs were found associated or potentially associated with PCa in the Chinese population. These results provided additional information of genetic risks of PCa. Several CNV regions involved clinical actionable genes that might be potentially targeted by medications. Additional validation and functional studies are still warranted for these results.
Abbreviations
PCa: prostate cancer;
SNP: single nucleotide polymorphisms;
GWAS: genome-wide association study;
CNV: copy-number variations;
ChinaPCa: Chinese Consortium for Prostate Cancer Genetics;
PFB: population frequency of B allele;
MAF: minor allele frequency;
LRR_SD: standard deviation of Log R Ratio;
BAF_drift: measuring departure of the B Allele Frequency from the expected values;
WF: waviness factor;
CNPR: copy number polymorphic regions;
CN: copy number.
Supplementary Material
Supplementary figure S1.
Acknowledgements
We thank all of the subjects included in this study.
This work was in part supported by grants from the Clinical Science and Technology Innovation Project of Shanghai Shen Kang Hospital Development Center to Qiang Ding (SHDC12015105), Precision Medicine Project of National Key Research and Development Plan of China to Jianfeng Xu (2016YFC0902202), the Scientific Research project supported by Huashan Hospital, Fudan University to Yishuo Wu (2016QD079), the “Chen Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation to Rong Na.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Qi D, Wu C, Liu F, Gu K, Shi Z, Lin X. et al. Trends of prostate cancer incidence and mortality in Shanghai, China from 1973 to 2009. Prostate. 2015;75:1662-8
3. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M. et al. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78-85
4. Wang M, Takahashi A, Liu F, Ye D, Ding Q, Qin C. et al. Large-scale association analysis in Asians identifies new susceptibility loci for prostate cancer. Nat Commun. 2015;6:8469
5. Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y. et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46:1103-9
6. Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y. et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704-12
7. Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451-81
8. Liu W, Sun J, Li G, Zhu Y, Zhang S, Kim ST. et al. Association of a germ-line copy number variation at 2p24.3 and risk for aggressive prostate cancer. Cancer Res. 2009;69:2176-9
9. Penney KL, Pyne S, Schumacher FR, Sinnott JA, Mucci LA, Kraft PL. et al. Genome-wide association study of prostate cancer mortality. Cancer Epidemiol Biomarkers Prev. 2010;19:2869-76
10. Yu YP, Song C, Tseng G, Ren BG, LaFramboise W, Michalopoulos G. et al. Genome abnormalities precede prostate cancer and predict clinical relapse. Am J Pathol. 2012;180:2240-8
11. Ledet EM, Hu X, Sartor O, Rayford W, Li M, Mandal D. Characterization of germline copy number variation in high-risk African American families with prostate cancer. Prostate. 2013;73:614-23
12. Emeville E, Broquere C, Brureau L, Ferdinand S, Blanchet P, Multigner L. et al. Copy number variation of GSTT1 and GSTM1 and the risk of prostate cancer in a Caribbean population of African descent. PLoS One. 2014;9:e107275
13. Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G. et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat Genet. 2012;44:1231-5
14. Lin CF, Naj AC, Wang LS. Analyzing copy number variation using SNP array data: protocols for calling CNV and association tests. Curr Protoc Hum Genet. 2013;79(Unit 1):27
15. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57
16. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
17. Demichelis F, Setlur SR, Banerjee S, Chakravarty D, Chen JY, Chen CX. et al. Identification of functionally active, low frequency copy number variants at 15q21.3 and 12q21.31 associated with prostate cancer risk. Proc Natl Acad Sci U S A. 2012;109:6686-91
18. Conn PJ PJ. pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205-37
19. Choi KY, Chang K, Pickel JM, Badger JD 2nd, Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:15219-24
20. Kuribayashi N, Uchida D, Kinouchi M, Takamaru N, Tamatani T, Nagai H. et al. The role of metabotropic glutamate receptor 5 on the stromal cell-derived factor-1/CXCR4 system in oral cancer. PLoS One. 2013;8:e80773
21. Park SY LS, Han IH, Yoo BC, Lee SH, Park JY, Cha IH, Kim J, Choi SW. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol Rep. 2007;17:81-7
22. Low JS, Chin YM, Mushiroda T, Kubo M, Govindasamy GK, Pua KC. et al. A Genome Wide Study of Copy Number Variation Associated with Nasopharyngeal Carcinoma in Malaysian Chinese Identifies CNVs at 11q14.3 and 6p21.3 as Candidate Loci. PLoS One. 2016;11:e0145774
23. Rzeski W, Turski L, Ikonomidou C. Glutamate antagonists limit tumor growth. Proc Natl Acad Sci U S A. 2001;98:6372-7
24. Stepulak A, Sifringer M, Rzeski W, Endesfelder S, Gratopp A, Pohl EE. et al. NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proceedings of the National Academy of Sciences. 2005;102:15605-10
25. Kalariti N LP, Papageorgiou E, Pissimissis N, Koutsilieris M. Regulation of the mGluR5, EAAT1 and GS expression by glucocorticoids in MG-63 osteoblast-like osteosarcoma cells. J Musculoskelet Neuronal Interact. 2007;7:113-8
26. Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M. et al. Expression of glutamate receptor subunits in human cancers. Histochemistry and Cell Biology. 2009;132:435-45
27. Brocke KS SC, Luksch H, Geiger KD, Stepulak A, Marzahn J, Schackert G, Temme A, Ikonomidou C. glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol Ther. 2010;9:455-68
Author contact
![]() Corresponding author: Rong Na, M.D., Department of Urology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, 197 Ruijin Second Road, Shanghai, China. Tel: 86-21-64370045 E-mail: narong.hscom; Qiang Ding, Dr. PH. Department of Urology, Huashan Hospital, Fudan University, 12 Mid-Wulumuqi Road, Shanghai, China. Tel: 86-21-52889999 E-mail: qiangd_urologycom; Jianfeng Xu. 1001 University Place, Evanston, IL 60201, U.S.A. Tel: (224) 264-7501. Email: jxuorg.
Corresponding author: Rong Na, M.D., Department of Urology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, 197 Ruijin Second Road, Shanghai, China. Tel: 86-21-64370045 E-mail: narong.hscom; Qiang Ding, Dr. PH. Department of Urology, Huashan Hospital, Fudan University, 12 Mid-Wulumuqi Road, Shanghai, China. Tel: 86-21-52889999 E-mail: qiangd_urologycom; Jianfeng Xu. 1001 University Place, Evanston, IL 60201, U.S.A. Tel: (224) 264-7501. Email: jxuorg.