Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(6):998-1005. doi:10.7150/jca.22915 This issue Cite
Research Paper
IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma
Iwona Lugowska1,2#, Pawel Teterycz1#, Michal Mikula3, Maria Kulecka4, Anna Kluska3, Aneta Balabas3, Magdalena Piatkowska3, Michal Wagrodzki5, Andrzej Pienkowski1, Piotr Rutkowski1, Jerzy Ostrowski3,4 ![]()
1. Department of Soft Tissue/Bone Sarcoma and Melanoma, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology; Roentgena 5, 02-781 Warsaw, Poland
2. Early Phase Clinical Trials Unit, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology; Roentgena 5, 02-781 Warsaw, Poland
3. Department of Genetics, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology; Roentgena 5, 02-781 Warsaw, Poland
4. Department of Gastroenterology, Hepatology and Clinical Oncology, Medical Center for Postgraduate Education, Roentgena 5, 02-781 Warsaw, Poland
5. Department of Pathology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology; Roentgena 502-781 Warsaw, Poland
# Equal contribution
Received 2017-9-20; Accepted 2018-1-28; Published 2018-2-28
Abstract
Background. Recent studies have shown that isocitrate dehydrogenase 1/2 (IDH1/2)- activating mutations occur in a variety of cancers, including acute myeloid leukaemia, gliomas, and chondrosarcomas (CHS)s. The effect of IDH1/2 mutation on overall survival (OS) has not been reported in CHS. The aim of our study was to assess the prevalence of known cancer-related gene mutations in CHS, as well as their prognostic role in patient survival.
Methods. DNA from FFPE samples of 80 patients (F:M- 1:1.3; mean age: 58 years; range 27-86) with histologically confirmed CHS (G1:29; G2:34; G3:17) was subjected to library preparation with the Ion AmpliSeq Cancer Hotspot Panel v2 and sequenced on the PGM Ion Torrent.
Results. Among the clinical features only histological grade influenced OS. Deep sequencing identified 1784 single nucleotide variants. Of them, 426 were considered to be pathogenic or probably pathogenic. Activating IDH1/2 mutations were found in 27 patients (34%) including 17 R132 IDH1 (21%), 10 R172 IDH2 (13%) and 3 R140 IDH2 variants (4%). Three patients had concurrent IDH1 and IDH2 mutations. The R140 IDH2 mutant has not been reported to date in CHS patients. OS for CHS patients with IDH1/2 mutations was significantly lower than in patients without mutations (93% vs 64%; p<0.001). No other genetic feature of the Cancer Hotspot Panel had an impact on OS.
Conclusions. In CHS, IDH1/2-mutation status and the histological aggressiveness of the CHS are important predictors for OS. The R140 IDH2 may also be a novel target for the treatment of CHS patients.
Keywords: chondrosarcoma, IDH1/2 mutation, AmpliSeq Cancer Hotspot Panel, next-generation sequencing
Background
Chondrosarcoma (CHS) is a rare bone or joint cancer typically affecting the population aged between 15 and 60 years, with male predominance. CHS can arise de novo, or grow out of a benign bone tumour such as an enchondroma or osteochondroma. Although the molecular background of CHS development is not clear, the increased risk for its development is linked to Ollier's Disease, Maffucci Syndrome, Multiple Hereditary Exostoses, and Paget's disease. Tumours can also arise in sites previously treated with radiation therapy or chemotherapy [1,2]. There are several subtypes of CHS: conventional, clear cell, myxoid, mesenchymal or dedifferentiated. Conventional and clear cell CHS are often low to intermediate grade tumours, but dedifferentiated and mesenchymal CHS are high grade tumours with spreading potential, and need multimodal treatment (10-15% of all CHS cases) [1,3]. The most common clinical manifestations are pain, swelling, or limited movement due to the growing tumour. The diagnosis is based on imaging studies (X-ray, bone, CT, MRI and PET scans), which must be follow by tumour biopsy to confirm the diagnosis. Complete surgical removal of the tumour is the most important treatment strategy, because CHS do not generally respond to chemo- or radiation therapy. Chemotherapy should be considered in mesenchymal or dedifferentiated chondrosarcomas, or in recurrent/metastatic disease. In selected cases, proton radiation therapy may be an option [4-6].
The prognosis of CHS is related to grade and histological subtype; the outcomes of high grade tumours, particularly dedifferentiated or mesenchymal subtypes, are poor due to the risk of recurrence and metastases. Only low grade tumours have a favourable prognosis. The reported 5-year survival ranges for chondrosarcoma grades 1, 2 and 3 are 82% - 99%; 63% - 92% and 5% - 77%, respectively. Five-year survival for dedifferentiated and mesenchymal CHS was reported to be about 24% [5]. Peripheral CHS tumors have a favourable prognosis compared to central CHS and usually affect younger patients [7]. Adverse prognostic factors are also considered, including axial localization of the primary tumour, size above 8 cm, distant metastases at presentation, positive surgical margins, presence of pathological fracture, or older age [8-11]. Recently published data highlights that prognosis may also be influenced by the molecular and genetic profile of CHS. For example, TP53 overexpression and mutations, as well as chromosome 17 alterations where TP53 is localized, are found in high-grade CHS, suggesting a TP53 pathway role in CHS progression [12,13]. The pRB pathway is another cell-cycle-regulating pathway frequently perturbed in CHS, with deletions and methylations of the CDKN2 gene, encoding p16 protein, being the most frequent events [14,15]. The loss of p16 protein expression is correlated with increasing tumour grade and poor prognosis [16]. On the other hand, the markers responsible for the proliferative, “dedifferentiated” phenotype in bone, such as collagen (subtype I, II and X), proteoglycan (aggrecan) [17] or cyclooxygenase-2 overexpression [18], have never been shown to carry prognostic value.
Isocitrate dehydrogenase (IDH) is a NADP+-dependent enzyme that metabolizes the conversion of isocitrate to α-ketoglutarate (αKG). The somatic mutations of IDH1 and IDH2 were initially identified in ~80% of gliomas [19], ~20% of acute myeloid leukaemias (AMLs) [20,21], and 50% of patients with CHS and cartilage tumours [22]. Almost all mutations observed in IDH1 and IDH2 are monoallelic point mutations affecting only a few residues. The mutant IDH enzymes produce 2-hydroxyglutarate (2HG) from αKG. 2HG exhibits a >100-fold increase in IDH1/2-mutated tumour samples and may thus promote tumourigenesis by accumulating the putative oncometabolite [21,23]. IDH2 mutant expression in a murine model of mesenchymal progenitor cells triggered widespread DNA hypermethylation, impaired cell differentiation and induced loss of cell contact, ultimately leading to the formation of undifferentiated sarcomas when cells were grown as xenografts [24].
The identification of prognostic and predictive factors provides insight into tumour biology, and treatment decision-making. We therefore decided to assess the relationship between the mutational status of IDH 1/2 and survival of patients with CHS.
Methods
Ethical approval and Patients
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Local Bioethical Committee of Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology and with the 1964 Helsinki declaration. Informed consent was obtained from all individual participants included in the study.
We retrospectively included 80 patients with histologically confirmed CHS, who were treated at Maria Sklodowska-Curie Memorial Cancer Centre and Institute of Oncology, Warsaw, Poland, between 1996 and 2013. Median age of the patients was 59 years (range: 27-86). Male/female ratio was 1.22:1. The most common location of the primary tumour was the lower extremity, with 30 cases (38%). CHS was located in the upper extremity and axial skeleton in 22 (28%) and 28 patients (35%), respectively. In 43/80 patients (54%), the primary tumour size was greater than 8 cm in diameter; in 20 cases (25%) no information was available on tumour size at diagnosis, because patients were primarily treated outside our Centre. Regarding histological subtype and grade, all tissues were re-evaluated by a referral pathologist (second opinion). We observed 65 conventional (81%), 6 myxoid subtypes (7.5%), 6 dedifferentiated (7.5%) and 3 mesenchymal (4%) CHS. Low grade tumours (G1) accounted for 29 (36%) and high grade tumours were identified in 51 cases (64%): grade 2 - 33 cases (41%); grade 3 - 18 cases (23%). A surgical procedure was performed in 73 cases (91%). In this group there were 49 limb salvage operations (67%). The median follow-up period was 52 months (range: 1-185 months).
DNA isolation and sequencing with the Ion AmpliSeq Cancer Hotspot Panel
DNA from tumour samples was extracted using a QIAamp DNA FFPE Tissue Kit (Qiagen), following the manufacturer's protocol. DNA sample concentrations were measured fluorescently using Qubit (Invitrogen), following the manufacturer's instructions, and stored at -20oC. DNA was subjected to library preparation with the Ion AmpliSeq Cancer Hotspot Panel v2, designed to probe 2,800 COSMIC mutations from 50 oncogenes and tumour suppressor genes, and sequenced on PGM Ion Torrent (Thermo), as described elsewhere [25]. Briefly, molarity was determined for each DNA library and up to 16 libraries were pooled in equimolar concentrations and used for automatic template preparation on the Ion Chef, using reagents from the Ion PGM IC 200 Kit and Ion 318 Chip Kit v2 BC. Sequencing was performed on the Ion Torrent PGM sequencer using 500-flow runs. Data from the PGM runs were processed on the Ion Torrent server using a platform-specific pipeline incorporated in the Torrent Suite v5.0.3 (Thermo) to obtain sequence reads, trim adapter sequences, filter and remove poor signal reads, and assign the reads to a given barcode. The reads were mapped to the hg19 (Homo sapiens) reference genome and adjusted to the specific amplicon target regions of the Ion AmpliSeq Cancer Hotspot Panel v2 deposited in the “CHP2.20131001.hotspots” BED file, available in the Torrent Suite. Coverage analysis (v5.0.2.0) and variantCaller (v5.0.3.5) plug-ins used a set of default parameters optimized for calling somatic variants.
Post-sequencing data analyses
Annotation of variants and their impact on mature protein was conducted by the Variant Effect Predictor [26] while deleteriousness was assessed with SIFT [27], PolyPhen [28] and Condel [29] algorithms. A variant was considered pathogenic if it was predicted as deleterious by these three algorithms, had a high impact on protein coding (stop-gained, frameshift etc.) or was previously reported as pathogenic in ClinVar.
Statistical analysis of the clinical dataset
Overall survival (OS) was calculated from the date of the pre-treatment biopsy or first operation (whichever occurred earlier) to tumour-progression-related death or the latest follow-up date. Disease-free survival (DFS) was calculated from the date of the end of treatment to the date of recurrence or to the latest follow-up date. Univariate analysis of the association between OS and clinical factors was based on the Kaplan-Meier estimator and the log-rank test. To investigate the relationship between categorical parameters and IDH1/2 mutation status, the Fisher's exact test was used. The effect of deleterious variants was assessed with the log-rank test and type I error was controlled with the Benjamini-Hochberg adjustment for multiple comparisons [30]. The analyses were performed with STATA (version 13) and R language (version 3.3.0) [31].
Availability of data
The anonymised sequencing datasets generated during the current study are available in the European Nucleotide Archive repository under the accession number PRJEB20222.
Results
Clinical outcome
In the analysed group of patients, we observed 43 relapses, and 12 deaths due to disease progression. Five-year OS and DFS rates were 85% and 46%, respectively. The clinical prognostic factor significantly influencing OS was tumour grade (p=0.009) and, for DFS, pathological fracture. Other prognostic factors like age, tumour size, histological subtype (conventional vs other), localization of primary tumour (axial vs non-axial) or the presence of soft tissue infiltration were not statistically significant. (Table 1)
Clinical characteristics and survival data according to clinical-pathological features
| N patients (%) | OS | p | DFS | p | ||
|---|---|---|---|---|---|---|
| Age at diagnosis | ||||||
| <40 y | 9 | 11% | 85% | 0.573 | 47% | 0.763 |
| >40 y | 71 | 89% | 83% | 42% | ||
| Sex | ||||||
| male | 44 | 55% | 82% | 0.349 | 40% | 0.387 |
| female | 36 | 45% | 84% | 45% | ||
| Tumour size | ||||||
| <8cm | 17 | 21% | 83% | 0.854 | 57% | 0.399 |
| >8cm | 43 | 54% | 90% | 45% | ||
| missing data | 20 | 25% | ||||
| Site of origin | ||||||
| extremity | 52 | 65% | 82% | 0.867 | 43% | 0.615 |
| axial | 28 | 35% | 85% | 42% | ||
| Histology subtype | ||||||
| conventional | 65 | 81% | 86% | 0.341 | 46% | 0.215 |
| other | 15 | 18% | 72% | 28% | ||
| Grade | ||||||
| 1 (low) | 29 | 36% | 95% | 0.009 | 71% | 0.002 |
| 2 (high) | 33 | 41% | 82% | 32% | ||
| 3 (high) | 18 | 23% | 62% | 23% | ||
| Pathological fracture | ||||||
| present | 64 | 80% | 50% | 0.844 | 47% | 0.010 |
| absent | 9 | 11% | 84% | 25% | ||
| missing data | 7 | 9% | ||||
| Surgery | ||||||
| operable | 72 | 90% | 85% | 0.123 | n/a | |
| non-operable | 0.8 | 10% | 63% | |||
| IDH mutation | ||||||
| Absent | 53 | 66% | 93% | <0.001 | 49% | 0.059 |
| Present | 27 | 34% | 64% | 28% | ||
| IDH 1 mutation | ||||||
| Absent | 63 | 79% | 87% | 0.029 | 47% | 0.159 |
| Present | 17 | 21% | 68% | 25% | ||
| IDH 2 mutation | ||||||
| Absent | 67 | 84% | 86% | 0.009 | 45% | 0.180 |
| Present | 13 | 16% | 67% | 30% | ||
OS: overall survival; DFS: disease free survival
Mutational status
We identified 1,784 unique variants across all 50 genes included in the Cancer Hotspot Panel v2 (Table S1). Among them 614 were considered pathogenic or likely pathogenic according to SIFT, PolyPhen and Condel algorithms, or had a high impact on protein coding (stop-gained, frameshift etc.). Variants likely to be deleterious were submitted to further analysis. The most frequently mutated genes included TP53 - 39 samples (49%), IDH1 - 17 samples (21%), EGFR - 15 samples (19%) and IDH2 - 13 samples (17%). The mean number of variants discovered in a sample was 8.93. In 23 samples (29%) no point mutations were found in regions included in the Cancer Hotspot panel v2. The structure of genetic variance among the investigated samples is shown in Figure 1.
The distribution of mutations according to histological grade. Only samples containing alternative variants are included. MES, DeDiff and MYX abbreviations refer to mesenchymal, dedifferentiated and myxoid chondrosarcoma subtypes, respectively.
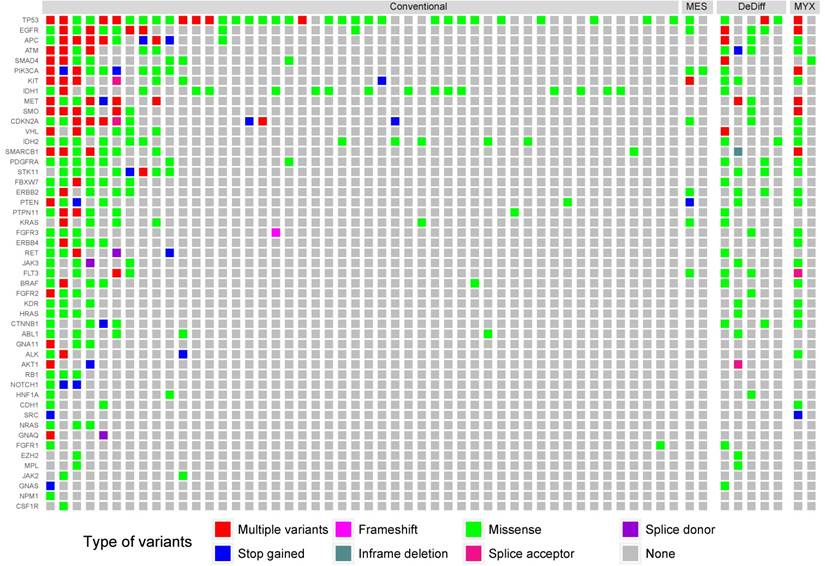
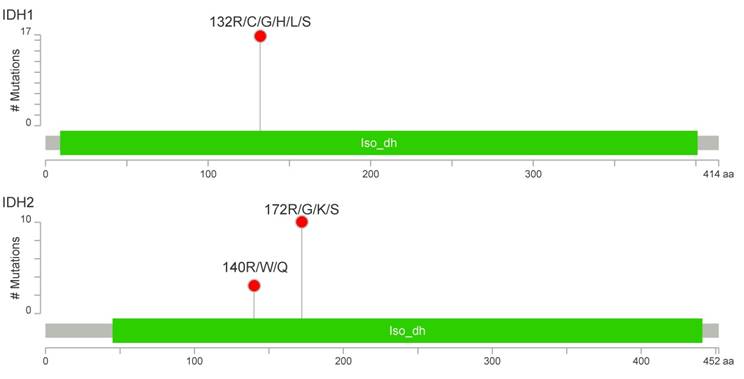
IDH1/2-activating mutations were found in 27 patients (34%), including 17 cases with R132 IDH1 (21%), 10 cases with R172 IDH2 (13%) and 3 cases with R140 IDH2 variants (4%) (Figure 2). The R140 IDH2 mutant has not been reported to date in CHS patients. Interestingly, three patients had a concurrent mutation in IDH1 and IDH2 (Figure S1). The coexistence of IDH1 and IDH2 mutations, and R140 IDH2 mutations was confirmed by the resequencing of appropriate specimens. There was no significant effect on patient survival for any other variants evaluated in the Cancer Hotspot Panel v2.
Number and localization of activating mutations found in IDH1 (above) and IDH2 (below) genes. Visualization was performed with MutationMapper [54].
Correlation between mutational status and clinical outcome in chondrosarcoma
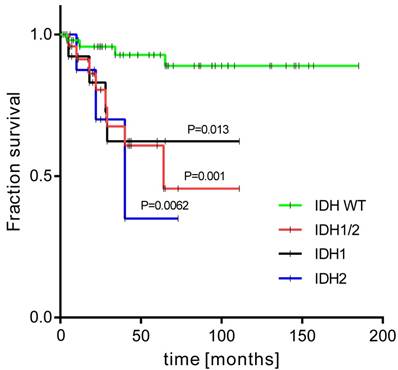
The patients with activating IDH1/2 mutations had a 5-year OS rate of 64% compared to 93% in those without (p < 0.001) (Figure 3). When each IDH gene was analysed alone, the IDH1 mutation carriers had worse OS compared to the IDH1 mutation negative group: 68% vs 87% (p=0.029); and to the IDH2 mutation patients: 67% vs 86% (p=0.009). The relationship between IDH1/2 status and histological grade was not statistically significant, however we observed that the IDH1/2 mutation was present in 21% of G1 tumours, 39% of G2, and was highest in G3 CHS (44%). Notably, the IDH2 mutation was more frequent in high grade tumours (low grade: 7%; high grade: 22%). Interestingly, all three cases of R140 IDH2 variant were high grade. The statistical trends were noted for IDH1/2 mutation status and patient age. Eleven patients were aged under 40 years and only one of them had an IDH1/2 mutation, in contrast to older patients, in whom IDH1/2 mutations were present in 37% of cases (P=0.063).
Kaplan-Meier survival curves of overall survival for groups with and without activating mutations in IDH1/2 genes.
Discussion
Activating mutations in IDH1/2 genes have been shown to exist and play a vital role in tumour development in a variety of neoplasms. They were initially described in brain tumours - gliomas, glioblastomas and astrocytomas [32,33]. Data suggest that IDH1/2 mutations are present in the vast majority of secondary glioblastoma cases [34]. In gliomas and glioblastomas, it has also been shown that the IDH1/2 mutation is associated with better patient prognosis [35,36]. Another group of neoplasms in which IDH1/2 alterations have been described are blood malignancies, namely myelodysplastic syndromes, AML and chronic myelomonocytic leukaemia [37]. IDH1/2 activation mutations are present in about 20% of AMLs and carry a negative prognosis value for patients [20,38].
In our study, activating IDH1/2 mutations were present in 34% of CHS tumours in the Polish population and had adverse prognostic impact on survival. This number is lower than reported in previous studies in CHS. Amary et al. [22] reported 56%, while Tarpey et al. [39] found a prevalence of 59%. Conversely the prevalence of IDH2 mutants was higher in our study than in Amary's work: 7.5% vs ~4.8%. In contrast to our findings, Amary et al. reported no differences in the OS of patients with and without IDH1/2 mutations. However, while previous studies [22,39] reported IDH1/2 mutations mostly in central but not in peripheral chondrosarcomas, all our patients suffered for peripheral chondrosarcomas. Furthermore, recently published study [40] confirmed the presence of IDH1/2 mutations in 87% (20/23) of dedifferentiated chondrosarcomas and in 30% (6/20) of conventional chondrosarcomas; 81% of our patients suffered for conventional chondrosarcoma. We found TP53 mutations in about half, and EGFR in 19% of CHS patients, but there was no statistical relationship between mutations in these genes and OS.
Tamoxifen-inducible expression of mutant IDH1 in chondrocytes resulted in enchondromas: benign cartilage tumours which are precursors to malignant CHS [41]. Arai et al. [42] analysed 13 CHS and found IDH1/2 mutations in six cases. The more aggressive behaviour of CHS was linked to IDH1/2 mutations. It has been proposed that the IDH1/2 mutation status may be used for distinguishing intracranial chondrosarcomas from chordomas and chondroblastic osteosarcoma [43]. We additionally observed that high grade tumours expressed IDH1/2 mutations more often than did low grade CHS; specifically IDH2 was present in 22% of high grade CHS, and only in 7% of low grade tumours. We also showed a statistical trend between IDH1/2 mutations and patient age. It is important to highlight the presence of the R140 IDH2 mutation in 3 CHS samples (all cases were high grade tumours). This genetic feature has not been described in CHS before.
Monotherapy based on specific inhibition of mutant IDH1 using AGI-5198, although decreased levels of 2-HG in a dose dependent manner, may not be sufficient for treatment of inoperable or metastasized chondrosarcoma patients [44]. However, a specific IDH2-R140 inhibitor, AG-221 (Enasidenib) was recently approved by the FDA to treat adult patients with relapsed or refractory AML, after in a phase I/II trial it induced an overall response rate of 40.3% in IDH2-mutated AML patients [45]. Hence this new targeted drug could in the future be tested on IDH2 R140-positive CHS patients.
Regarding clinical factors and their prognostic role, histological grade was the most important factor, which is in line with previous findings by Evans et al. [46] and Giuffrida et al. [47]. Other prognostic factors negatively influencing the prognosis are as follows: metastases, non-operable disease, positive surgical margins, age over 55 years, clinical diameter of the tumour above 8 cm [48]. However, our database had limited information about surgical margins and tumour size and, probably due to the small sample size, we were unable to confirm the statistical differences between OS and the above-mentioned parameters. In our patients only histological grade was a statistically important factor, with a linear trend. Five-year OS was 95% in G1, 82% in G2, and 62% in G3 tumours.
To date performed studies on IDH1/2 mutations prevalence in CHS have used mixed mutation detection technologies and assays. For example, Amary et al. employed mass spectrometry on MassARRAY platform followed by IDH1/2 mutations validation with capillary sequencing [22]. Same group in another study by Tarpey et al. utilized Exome-Seq assay with sequence coverage at a depth of 30× [39]. Moreover, Kerr et al. adapted SNaPshot platform combined with capillary sequencing validation [43] and Arai et al. [42] used capillary sequencing and immunohistochemistry to identify IDH1/2 and IDH1 mutations, respectively. Finally, Chen and colleagues [40] used IDH1/2 RGQ PCR Kit from Qiagen that utilizes primers and probes to perform allele-specific amplification with a readout in a real-time PCR machine. In this study, we used Thermo's AmpliSeq technology coupled with next-generation sequencing (NGS) on Ion Torrent PGM platform and on average we generated sequence coverage of ×1667 and ×685 for IDH1 and IDH2 amplicon, respectively. We believe that such a high coverage offers the sensitivity that outperforms aforementioned assays and ultimately allowed us, for the first time, to identify the IDH2 R140 mutant and concurrent IDH1/2 mutation in CHS cases. While the presence of IDH1 and IDH2 mutations is frequently thought to be mutually exclusive, 3 out of 27 CHS patients (11%) in our study had concurrent IDH1/2 mutations. Interestingly, such cases have previously only rarely been identified in mutational studies focused on IDH1/2: in gliomas (4 out of 739 IDH1/2 mutation-positive cases - 0.005%) [49] and AMLs (2 out of 125 IDH1/2 mutation-positive cases - 0.016%) [50]. Such a high frequency of concurrent IDH1/2 mutations observed among CHS patients could be due to the higher sensitivity of the NGS compared to the Sanger sequencing method, used in the aforementioned studies on gliomas and AML samples. It is difficult to detect somatic variants at frequency levels below 10% with Sanger sequencing [51], while NGS permits identification of variants at frequencies of 0.1% or lower [52,53]. Hence the co-occurrence of IDH1/2 mutations in gliomas and AMLs could also be higher than previously thought.
Conclusions
Findings on the adverse prognostic value of IDH1/2 mutations opens new clinical possibilities in the management of CHS patients and could help modify potential treatment and surveillance strategies. It also provides a theoretical basis for pursuing IDH1/2 mutation as a druggable target in CHS treatment. However, preclinical and clinical trials are warranted to prove this hypothesis and test the efficacy of IDH1/2 mutant inhibitors in CHS treatment.
Abbreviations
CHS: chondrosarcoma; IDH: isocitrate dehydrogenase; OS: overall survival; DFS: disease free survival; NGS: next-generation sequencing.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This study was funded by The National Centre for Research and Development (grant STRATEGMED1/233574/15/NCBR/2015 - EPTHERON).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Heymann D. Bone Cancer: Primary Bone Cancers and Bone Metastases. Elsevier. 2014:730-736
2. Dorfman HD, Czerniak B. Bone cancers. Cancer. 1995;75:203-10 doi: 10.1002/1097-0142(19950101)75:1+<203:AID-CNCR2820751308>3.0.CO;2-V
3. Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone: a clinicopathologic analysis with emphasis on histologic grading. Cancer. 1977;40:818-31
4. Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987-93 doi: 10.2106/JBJS.F.00288
5. Grimer RJ, Gosheger G, Taminiau A, Biau D, Matejovsky Z, Kollender Y, San-Julian M, Gherlinzoni F, Ferrari C. Dedifferentiated chondrosarcoma: prognostic factors and outcome from a European group. Eur J Cancer. 2007;43:2060-5 doi: 10.1016/j.ejca.2007.06.016
6. Mitchell AD, Ayoub K, Mangham DC, Grimer RJ, Carter SR, Tillman RM. Experience in the treatment of dedifferentiated chondrosarcoma. J Bone Joint Surg Br. 2000;82:55-61
7. Ahmed AR, Tan T-S, Unni KK, Collins MS, Wenger DE, Sim FH. Secondary chondrosarcoma in osteochondroma: report of 107 patients. Clin Orthop Relat Res. 2003:193-206 doi: 10.1097/01.blo.0000069888.31220.2b
8. Fiorenza F, Abudu A, Grimer RJ, Carter SR, Tillman RM, Ayoub K, Mangham DC, Davies AM. Risk factors for survival and local control in chondrosarcoma of bone. J Bone Joint Surg Br. 2002;84:93-9
9. Eefting D, Schrage YM, Geirnaerdt MJA, Le Cessie S, Taminiau AHM, Bovée JVMG, Hogendoorn PCW, EuroBoNeT consortium. Assessment of interobserver variability and histologic parameters to improve reliability in classification and grading of central cartilaginous tumors. Am J Surg Pathol. 2009;33:50-7 doi: 10.1097/PAS.0b013e31817eec2b
10. van Maldegem AM, Gelderblom H, Palmerini E, Dijkstra SD, Gambarotti M, Ruggieri P, Nout RA, van de Sande MAJ, Ferrari C, Ferrari S, Bovée JVMG, Picci P. Outcome of advanced, unresectable conventional central chondrosarcoma. Cancer. 2014;120:3159-64 doi: 10.1002/cncr.28845
11. Mavrogenis AF, Angelini A, Drago G, Merlino B, Ruggieri P. Survival analysis of patients with chondrosarcomas of the pelvis. J Surg Oncol. 2013;108:19-27 doi: 10.1002/jso.23351
12. Terek RM, Healey JH, Garin-Chesa P, Mak S, Huvos A, Albino AP. p53 mutations in chondrosarcoma. Diagn Mol Pathol. 1998;7:51-6
13. Bovée JVMG, Hogendoorn PCW, Wunder JS, Alman BA. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer. 2010;10:481-8 doi: 10.1038/nrc2869
14. Schrage YM, Lam S, Jochemsen AG, Cleton-Jansen A-M, Taminiau AHM, Hogendoorn PCW, Bovée JVMG. Central chondrosarcoma progression is associated with pRb pathway alterations: CDK4 down-regulation and p16 overexpression inhibit cell growth in vitro. J Cell Mol Med. 2009;13:2843-52 doi: 10.1111/j.1582-4934.2008.00406.x
15. Asp J, Inerot S, Block JA, Lindahl A. Alterations in the regulatory pathway involving p16, pRb and cdk4 in human chondrosarcoma. J Orthop Res. 2001;19:149-54 doi: 10.1016/S0736-0266(00)00022-X
16. van Beerendonk HM, Rozeman LB, Taminiau AHM, Sciot R, Bovée JVMG, Cleton-Jansen A-M, Hogendoorn PCW. Molecular analysis of the INK4A/INK4A-ARF gene locus in conventional (central) chondrosarcomas and enchondromas: indication of an important gene for tumour progression. J Pathol. 2004;202:359-66 doi: 10.1002/path.1517
17. Aigner T, Müller S, Neureiter D, Illstrup DM, Kirchner T, Björnsson J. Prognostic relevance of cell biologic and biochemical features in conventional chondrosarcomas. Cancer. 2002;94:2273-81 doi: 10.1002/cncr.10461
18. Schrage YM, Machado I, Meijer D, Briaire-de Bruijn I, van den Akker BE, Taminiau AHM, Kalinski T, Llombart-Bosch A, Bovée JVMG. COX-2 expression in chondrosarcoma: a role for celecoxib treatment? Eur J Cancer. 2010;46:616-24 doi: 10.1016/j.ejca.2009.11.002
19. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-73 doi: 10.1056/NEJMoa0808710
20. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058-66 doi: 10.1056/NEJMoa0903840
21. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225-34 doi: 10.1016/j.ccr.2010.01.020
22. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC. et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334-43 doi: 10.1002/path.2913
23. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44 doi: 10.1038/nature08617
24. Lu C, Venneti S, Akalin A, Fang F, Ward PS, DeMatteo RG, Intlekofer AM, Chen C, Ye J, Hameed M, Nafa K, Agaram NP, Cross JR. et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27:1986-98 doi: 10.1101/gad.226753.113
25. Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, Ostrowski J. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med Genomics. 2015;8:19. doi: 10.1186/s12920-015-0092-2
26. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069-70 doi: 10.1093/bioinformatics/btq330
27. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812-4
28. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet. 20130 7: Unit7.20. doi: 10.1002/0471142905.hg0720s76
29. González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440-9 doi: 10.1016/j.ajhg.2011.03.004
30. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological). 1995;57:289-300
31. Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. ISBN 3-900051-07-0. 2014
32. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu I-M, Gallia GL, Olivi A, McLendon R, Rasheed BA. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-12 doi: 10.1126/science.1164382
33. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8 doi: 10.1038/nature07385
34. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012;108:11-27 doi: 10.1007/s11060-011-0793-0
35. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre J-Y. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150-4 doi: 10.1200/JCO.2009.21.9832
36. Chen J-R, Yao Y, Xu H-Z, Qin Z-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients with Glioblastomas. Medicine (Baltimore). 2016;95:e2583. doi: 10.1097/MD.0000000000002583
37. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): An update. Mutat Res Rev Mutat Res. 2016;769:47-62 doi: 10.1016/j.mrrev.2016.04.009
38. Marcucci G, Maharry K, Wu Y-Z, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH. et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348-55 doi: 10.1200/JCO.2009.27.3730
39. Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O'Meara S, Davies H, Nik-Zainal S, Beare D, Butler A, Gamble J. et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923-6 doi: 10.1038/ng.2668
40. Chen S, Fritchie K, Wei S, Ali N, Curless K, Shen T, Brini AT, Latif F, Sumathi V, Siegal GP, Cheng L. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum Pathol. 2017;65:239-46 doi: 10.1016/j.humpath.2017.05.015
41. Hirata M, Sasaki M, Cairns RA, Inoue S, Puviindran V, Li WY, Snow BE, Jones LD, Wei Q, Sato S, Tang YJ, Nadesan P, Rockel J. et al. Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc Natl Acad Sci USA. 2015;112:2829-34 doi: 10.1073/pnas.1424400112
42. Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012;29:201-6 doi: 10.1007/s10014-012-0085-1
43. Kerr DA, Lopez HU, Deshpande V, Hornicek FJ, Duan Z, Zhang Y, Rosenberg AE, Borger DR, Nielsen GP. Molecular distinction of chondrosarcoma from chondroblastic osteosarcoma through IDH1/2 mutations. Am J Surg Pathol. 2013;37:787-95 doi: 10.1097/PAS.0b013e31827ab703
44. Suijker J, Oosting J, Koornneef A, Struys EA, Salomons GS, Schaap FG, Waaijer CJF, Wijers-Koster PM, Briaire-de Bruijn IH, Haazen L, Riester SM, Dudakovic A, Danen E. et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015;6:12505-19 doi: 10.18632/oncotarget.3723
45. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, Kantarjian HM, Collins R, Patel MR. et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722-31 doi: 10.1182/blood-2017-04-779405
46. Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone. A clinicopathologic analysis with emphasis on histologic grading. Cancer. 1977;40:818-31 doi: 10.1002/1097-0142(197708)40:2<818::AID-CNCR2820400234>3.0.CO;2-B
47. Giuffrida AY, Burgueno JE, Koniaris LG, Gutierrez JC, Duncan R, Scully SP. Chondrosarcoma in the United States (1973 to 2003): an analysis of 2890 cases from the SEER database. J Bone Joint Surg Am. 2009;91:1063-72 doi: 10.2106/JBJS.H.00416
48. Marulli G, Duranti L, Cardillo G, Luzzi L, Carbone L, Gotti G, Perissinotto E, Rea F, Pastorino U. Primary chest wall chondrosarcomas: results of surgical resection and analysis of prognostic factors. Eur J Cardiothorac Surg. 2014;45:e194-201 doi: 10.1093/ejcts/ezu095
49. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469-74 doi: 10.1007/s00401-009-0561-9
50. Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K, Horst H-A, Germing U, Döhner H. et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636-43 doi: 10.1200/JCO.2010.28.3762
51. Rohlin A, Wernersson J, Engwall Y, Wiklund L, Björk J, Nordling M. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat. 2009;30:1012-20 doi: 10.1002/humu.20980
52. Harismendy O, Schwab RB, Bao L, Olson J, Rozenzhak S, Kotsopoulos SK, Pond S, Crain B, Chee MS, Messer K, Link DR, Frazer KA. Detection of low prevalence somatic mutations in solid tumors with ultra-deep targeted sequencing. Genome Biol. 2011;12:R124. doi: 10.1186/gb-2011-12-12-r124
53. Li M, Stoneking M. A new approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012;13:R34. doi: 10.1186/gb-2012-13-5-r34
54. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088
Author contact
![]() Corresponding author: Jerzy Ostrowski, MD, PhD; Cancer Center-Institute, Roentgena 5, 02-781 Warsaw, Poland, Tel: +48 225462575, E-mail: jostrowcom.pl
Corresponding author: Jerzy Ostrowski, MD, PhD; Cancer Center-Institute, Roentgena 5, 02-781 Warsaw, Poland, Tel: +48 225462575, E-mail: jostrowcom.pl