Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(6):1078-1087. doi:10.7150/jca.23596 This issue Cite
Research Paper
SULT1E1 inhibits cell proliferation and invasion by activating PPARγ in breast cancer
Yali Xu1, Xiaoyan Lin1, Jiawen Xu1, Haiyan Jing1, Yejun Qin1 ![]() , Yintao Li2
, Yintao Li2 ![]()
1. Department of Pathology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, P.R. China
2. Department of Medical Oncology, Shandong Cancer Hospital and Institute, Jinan, Shandong, P.R. China
Received 2017-10-31; Accepted 2018-1-29; Published 2018-2-28
Abstract
Sulfotransferase family 1E member 1 (SULT1E1) is known to catalyze sulfoconjugation and play a crucial role in the deactivation of estrogen homeostasis, which is involved in tumorigenesis and the progression of breast and endometrial cancers. Our previous study has shown that the protein levels of SULT1E1 were decreased in breast cancer; however, the underlying mechanism is still poorly understood. In this study, we explored the functional and molecular mechanisms by which SULT1E1 influenced breast cancer. Here, we identified that overexpression of SULT1E1 inhibited breast cancer cell growth through inducing apoptosis and arresting cell cycle progression. Furthermore, enforced expression of SULT1E1 suppressed tumor cell migration and invasion. Moreover, we found that the activation of PPARγ was required for SULT1E1-mediated downregulation of C-myc, Cyclin D1, MMP-2 and MMP-9 as well as for cell apoptosis, migration and invasion. In addition, the overexpression of SULT1E1 significantly inhibited tumor growth in vivo. Taken together, our findings indicated that SULT1E1 performed its tumor suppressor characteristics by activating PPARγ, which provided a novel target for patients with breast cancer.
Keywords: breast cancer, SULT1E1, PPARγ, proliferation, invasion
Introduction
Breast cancer is the most universal of malignant tumors in females and accounts for 23% of all diagnosed cancers[1]. Although current anti-tumor strategies, including surgery, radiotherapy, hormonal therapy, or a combination thereof, have greatly improved in recent years, recurrence and distant metastasis are the leading causes of mortality in breast cancer patients[1-3]. Although significant improvement in the molecular mechanisms of breast cancer development and progression have been made[4-7], the mechanisms underlying breast cancer remain largely unknown. Therefore, gaining a better understanding of the molecular mechanisms of breast cancer and identifying a novel therapeutic target can lead to better treatments.
Sulfotransferase family 1E member 1 (SULT1E1), known as estrogen sulfotransferase, is a key enzyme that catalyzes the sulfation of estrogen and estradiol, leading to their inactivation by inhibiting their ability to bind to the estrogen receptor [8-10]. Previous studies have shown that SULT1E1 negatively regulated adipogenesis by inactivating ERK1/2 MAPK and attenuating insulin signaling in mice [11]. Furthermore, SULT1E1 promoted human adipogenesis by deactivating estrogen in humans [12]. SULT1E1 was found to be correlated with breast and endometrial cancer, and the inhibition of SULT1E1 might lead to increased estrogen levels that promote carcinogenesis [13, 14]. High SULT1E1 levels have been found in the tumor tissues of patients with breast cancer and have been associated with a poor prognosis for breast cancer in women [15-17]. Our previous study also found that SULT1E1 levels were higher in breast cancer tissues than in normal breast tissues[18]. However, the overexpression of SULT1E1 inhibits proliferation in vitro and tumorigenesis in vivo[18]. SULT1E1 also significantly suppresses cell proliferation and induces cell apoptosis. However, the potential roles and molecular mechanism of SULT1E1 remain poorly understood.
In this study, we analyzed the effects of SULT1E1 on proliferation, angiogenesis, invasion and metastasis of breast cancer. In addition, the mechanisms of SULT1E1 regulation in breast cancer cell apoptosis and invasion were investigated. Our results indicate that SULT1E1 inhibits cell invasion by upregulating the expression of PPARγ, supporting the notion that SULT1E1 may offer new possibilities for the treatment of breast cancer.
Materials and Methods
Cell lines and cultures
The human breast cancer cell lines MCF-7 and T47D were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Invitrogen) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA). Cells were cultured in a humidified incubator at 37°C with 5% CO2.
Lentivirus vectors construction and transfection
To upregulate SULT1E1 expression, the PCR-amplified human SULT1E1 coding sequence was inserted into the EcoR I/Xho I site of the lentivirus expression vector pLV-CMV-hSULT1E1-EGFP-Puro. Lentiviruses were produced by HEK293T cells with vector or SULT1E1 using the Lipofectamine® 2000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Lentiviral particles were harvested 48 h after transfection and filtered using a 0.45-mm filter (Sartius, Gottingen, Germany). For infection, cells were transduced with 1 × 109 TU lentivirus and then cultured in medium with a concentration of 2 mg/ml puromycin (Sigma, USA) for 72 h prior to selection.
The small interfering RNA (siRNA) targeting PPARγ was obtained from GenePharma (Shanghai, China). SiRNA sequences targeting human PPARγ are 5ʹ-GATAAGCTTCAATCTGATT-3ʹ. MCF-7 and T47D cells were transfected with 50 nM negative control small interfering RNA (NC) or PPARγ siRNA using Lipofectamine® 2000 Transfection Reagent. Forty-eight hours after transfection, cells were harvested for Western blot analysis.
Cell viability and colony formation assay
Cell proliferation ability was measured using the Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan). Cells were seeded in 96-well plates at a density 3 × 104 cells/mL in a volume of 100 µL/well. Ten microliters of CCK-8 was added per well at 24, 48 and 72 h and incubated for 1 h. The absorbance of each sample was measured at a wavelength of 450 nm by using a SoftMaxPro5 Microplate Reader (Molecular Devices, California, USA).
To complete the colony formation assay, 1x103 cells per well were plated in 6-well plates and cultured for an additional 14 days in complete growth media. The medium was removed, and colonies (a colony was defined as >50 cells) were stained with 0.5% crystal violet for 15 min, photographed and counted.
Flow cytometric analysis
For the cell apoptosis assay, cell apoptosis was tested using FITC Apoptosis Detection Kit I (BD Pharmingen). Cells were harvested and resuspended in 1× binding buffer. Then, cells were incubated with 5 μl FITC Annexin V and 5 μl propidium iodide for 15 min in a dark place at room temperature. Cell apoptosis was measured by flow cytometry (FACScan®; BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software.
For cell cycle analysis, cells were synchronized by serum starvation for 24 h. Then, the cells were harvested and fixed in 70% ice cold ethanol. RNase A was added and incubated at 37°C for 15 min. Finally, cells were stained with propidium iodide (50 μg/ml) for 30 min at 4°C. Cellular DNA content was analyzed using FlowJo software.
Migration and invasion assay
Cell migration and invasion assay was performed using 8-μm pore size membranes (Corning Costar Corp, Cambridge, MA, USA). Cells (8 × 105 in 600 µl serum-free media) were placed into the upper well of the Transwell chamber for migration assays (without Martrigel, BD Biosciences, Franklin Lakes, NJ, USA) and invasion assays (with Martrigel). Next, 800 μl of media containing 10% FBS was added to the lower chamber as a chemoattractant. After a 36-h incubation, the cells that migrated or invaded to the lower compartment were fixed with methanol and stained with 0.5% crystal violet. Cells on the undersides of the filters were imaged with an inverted IX51 microscope (OLYMPUS, Japan) and counted using ImageJ software.
Tube formation assay
Stable SULT1E1 overexpressed breast cancer cells were cultured with serum-free media. Twelve hours later, the supernatant was collected and stored at -80°C until use. For the tube formation assay, 6×104 HUVECs per well were seeded in a 24-well plate coated with Matrigel containing medium with the above supernatant. Tube structures in 6 randomly selected fields were captured and analyzed using ImageJ software 6 h later.
Western blot analysis
Total proteins were extracted from cells and separated using 10% SDS-PAGE gels. Antibodies for GAPDH, PPARγ, C-myc, CyclinD1, MMP-2 and MMP-9 were purchased from Cell Signaling Technology, and SULT1E1 was obtained from Abnova and used as a loading control. The procedure of Western blot analysis was performed as previously described[18].
In vivo tumor growth model
Four-week-old female BALB/c athymic nude mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and maintained under a specific pathogen-free environment. All animal research was conducted in accordance with the National Institutes of Health guidelines approved by the Animal Care and Use Ethic Committee of Shandong University under permit number 2015A048, and all efforts were made to minimize animal suffering. Approximately 1 × 107 logarithmically growing T47D cells transduced with the vector or SULT1E1 in 100 μlRPMI 1640 were subcutaneously injected into the footpad of the left hind limb of each mouse. Tumor size was measured every week with a Vernier caliper and calculated by the formula = Length × Width2/2. After 6 weeks, the mice were sacrificed, and the tumors were dissected and weighed.
Statistical analysis
The statistical analysis was performed using GraphPad Prism 5.0 (SPSS Inc, Chicago, IL, USA). All data are expressed as the mean ± SD from three independent experiments. Student's t-test or a one-way analysis of variance (one-way ANOVA) was employed to determine statistical significance. The level of significance was set at p< 0.05.
Results
Overexpression of SULT1E1 inhibited cell proliferation and induced cell apoptosis
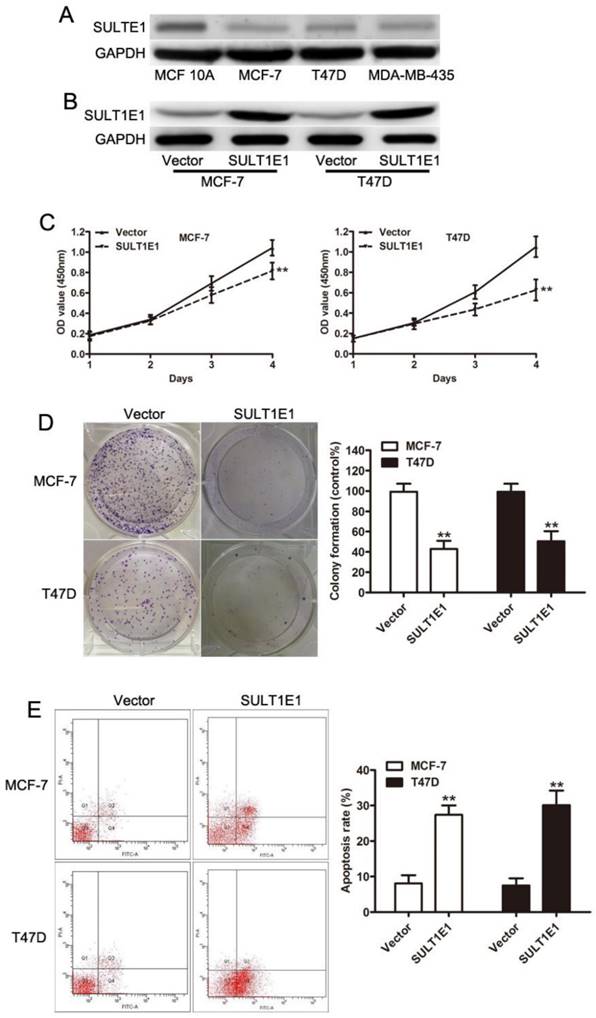
To elucidate the role of SULT1E1 in tumor progression, we enforced the SULT1E1 expression using lentivirus in MCF-7 and T47D cells. Western blot assays revealed that SULT1E1 was efficiently upregulated in both breast cancer cells (Figure 1A). In vitro experiments showed that overexpression of SULT1E1 significantly decreased cell proliferation (Figure 1B) and impaired colony formation ability (Figure 1C) in MCF-7 and T47D cells. To elucidate the role of SULT1E1 in apoptosis, annexin V-FITC/PI double staining was performed to detect the apoptosis of breast cancer cells after SULT1E1 overexpression. Flow cytometry data showed a dramatic increase in annexinV staining in MCF-7 and T47D cells transduced with SULT1E1 compared to the vector. These data together indicated that overexpression of SULT1E1 inhibited the proliferation of breast cancer cells.
The effects of SULT1E1 on breast cancer cells proliferation in vitro. (A) Western blot analysis of SULT1E1 levels in MCF-10A, MCF-7, T47D and MDA-MB-435 cells. (B) Western blotting of SULT1E1 following the transduction of MCF-7 and T47D cells with lentvirus. (C) Overexpression of SULT1E1 dramatically decreased the proliferation ability of breast cancer cells by CCK-8 assay. (D) Colony formation assay showed SULT1E1 remarkably decreased colony formation ability of MCF-7 and T47D cells. (E) Flow cytometry analysis showed that overexpression of SULT1E1 increased apoptosis of breast cancer cells.**P < 0.01.
Overexpression of SULT1E1 suppressed angiogenesis and arrested cell cycle
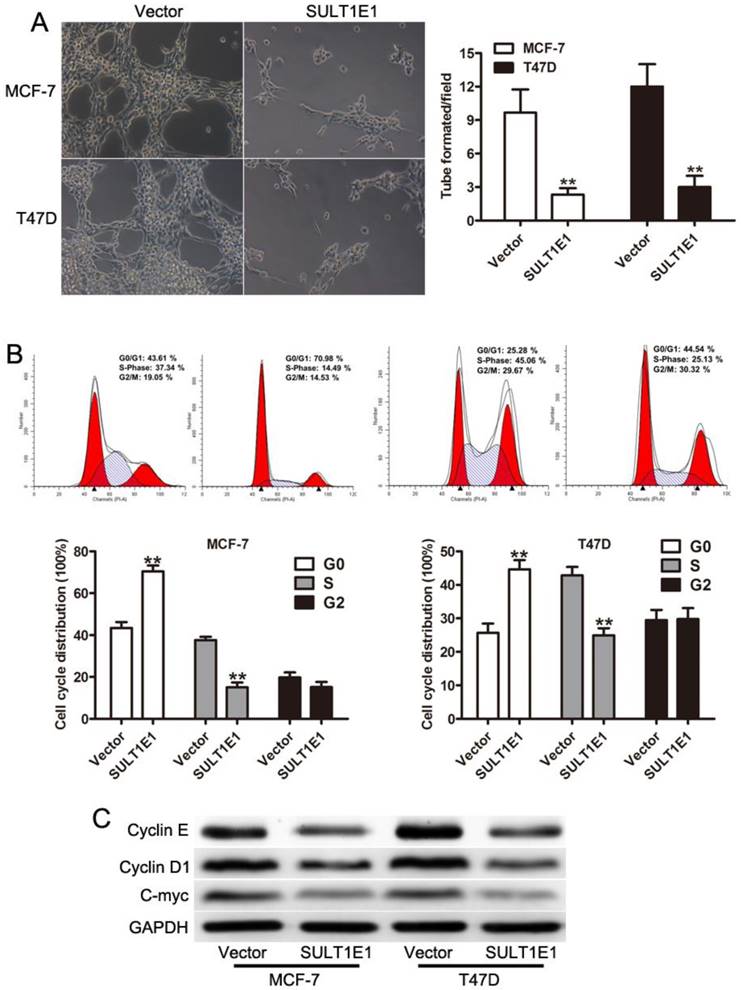
To assess the effect of SULT1E1 on tube formation, HUVEC-1 cells were incubated with conditioned medium (CM) derived from breast cancer cells transduced with vector or SULT1E1. The lengths of tubes were significantly decreased in HUVEC-1 cells incubated with CM from SULT1E1 infected cells compared with control cells (Figure 2A). Cell proliferation was closely associated with the regulation of cell cycle[19, 20]. However, whether the overexpression of SULT1E1 affected the cell cycle remains unknown. We next performed cell cycle analysis and found that the overexpression of SULT1E1 resulted in a higher fraction of cells in the G1 phase and a lower proportion of cells in the S phase (Figure 2B). To evaluate the functions of SULT1E1 on breast cell proliferation, the expression of proliferation-related genes (cyclinD1 and C-myc) was detected by Western blot analysis. The results illustrated that the overexpression of SULT1E1 downregulated cyclinD1 and C-myc expression both in MCF-7 and T47D cells (Figure 2C). These results indicated that SULT1E1 mediated cell proliferation of breast cancer cells by regulating the expression of cyclinD1 and C-myc.
Role of SULT1E1 in cell cycle and endothelial cell tube formation. (A) Overexpression of SULT1E1 reduced branch lengths and numbers of tube formation formed by HUVEC. (B) Cell cycle analysis showed that SULT1E1 could decrease S phase when compared with the control vector cells. (C) Western blotting showed the expression levels of C-myc and CyclinD1 were reduced in MCF-7 and T47D cells transduced with SULT1E1. **P < 0.01.
Overexpression of SULT1E1 inhibited cell migration and invasion by increasing expressions of MMP-2 and MMP-9
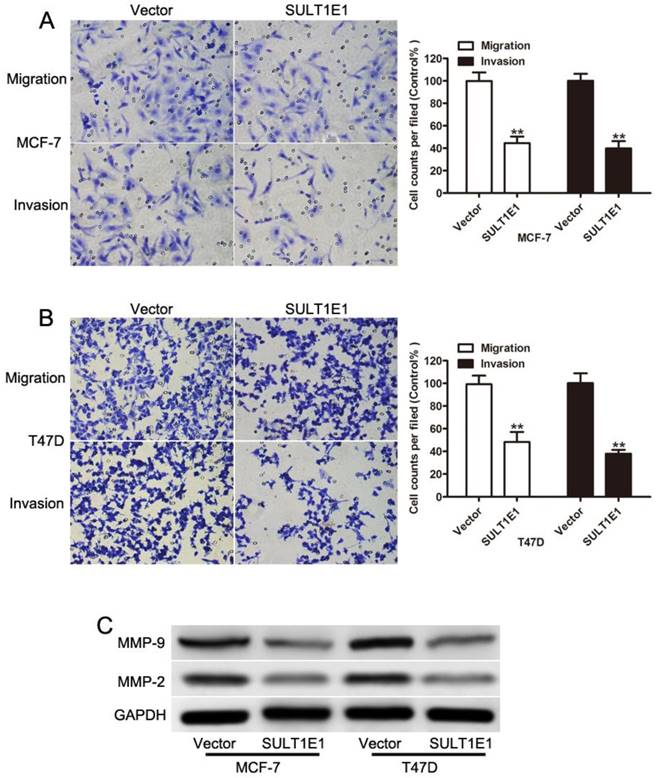
To examine the crucial role of SULT1E1 in breast cancer cell motility, transwell assays were performed to evaluate cell motility in the control and SULT1E1 overexpressed cells. A migration assay showed that ectopic SULT1E1 expression significantly decreased the migration and invasion abilities of MCF-7 (Figure 3A) and T47D (Figure 3B) cells. MMP-2 and MMP-9 have been implicated in tumor invasion and metastasis[21-23]. Our data showed that SULT1E1 could downregulate MMP-2 and MMP-9 expression. Together, these results suggested that SULT1E1 was a negative regulator of breast cancer cell migration and invasion by regulating the MMP-2 and MMP-9 expression.
Effects of SULT1E1 on breast cancer cell migration and invasion. Transwell migration (left) and invasion (right) assays were performed to investigate the effects of SULT1E1 on the migration and invasion abilities of MCF-7 (A) and T47D (B) cell lines. (C) Western blot analysis of MMP-2 and MMP-9 expression in breast cancer cells with or without SULT1E1 overexpression. **P < 0.01.
Inhibition of PPARγ attenuated the effects of SULT1E1 on breast cancer cells
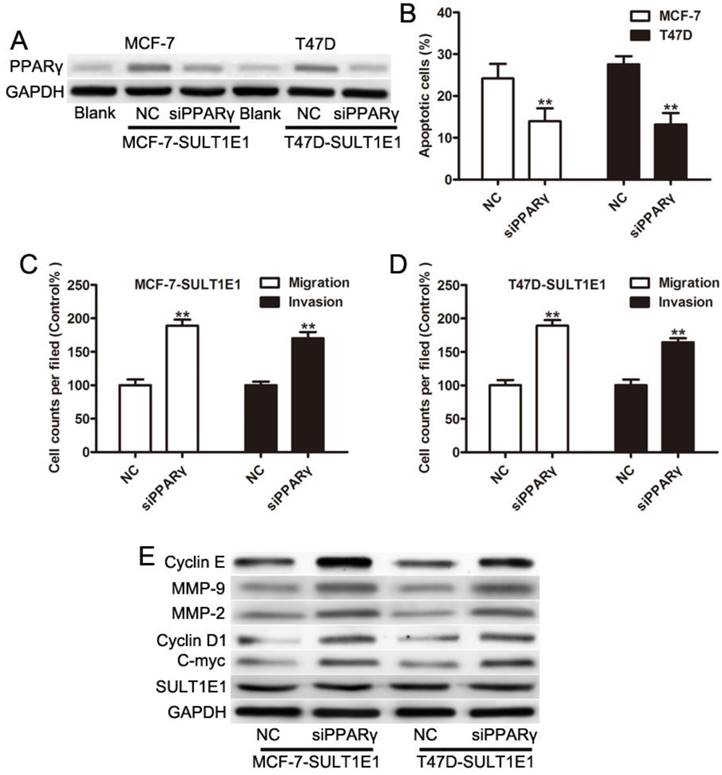
Our previous study showed that SULT1E1 regulated the inflammatory response and lipid metabolism of human endothelial cells via PPARγ[24]. PPARγ has been found to act as a tumor suppressor by inhibiting the development and metastases of primary tumors in many cancers [25-27]. As such, we asked whether PPARγ might be critical for inhibiting motility and invasion in response to SULT1E1. MCF-7 and T47D cells with SULT1E1 overexpression were transfected with negative control (NC) or PPARγ siRNA. Figure 4A shows that the expression levels of PPARγ were significantly reduced in MCF-7 and T47D cells transfected with PPARγ siRNA. A flow cytometry assay showed that cell apoptosis was significantly reversed by PPARγ siRNA (Figure 4B). The inhibition of migration and invasion of SULT1E1 was partially abolished by PPARγ siRNA (Figure 4C and 4D). Western blot analysis also showed that the downregulation of cylinD1, C-myc, MMP-2 and MMP-9 was inverted by PPARγ silencing (Figure 4E). However, there was no change in SULT1E1 protein expressions after PPARγ silencing in MCF-7 and T47D cells (Figure 4E). These findings indicated that SULT1E1 affected the upregulation of PPARγ, which was also essential for the inhibition effects of SULT1E1 on breast cancer cells.
SULT1E1 suppressed breast cancer partly through upregulation of PPARγ. (A) Western blotting showed that PPARγ was knocked down in MCF-7 and T47D cells with SULT1E1 overexpression. (B) Flow cytometry analysis showed that PPARγ silencing in MCF-7 and T47D cells with SULT1E1 overexpression inhibited cell apoptosis. (C) Knockdown of PPARγ inhibited cell migration and invasion as indicated by transwell assays. (D) PPARγ silencing increased the expression of C-myc, CyclinD1, MMP-2, and MMP-9as indicated. **P < 0.01.
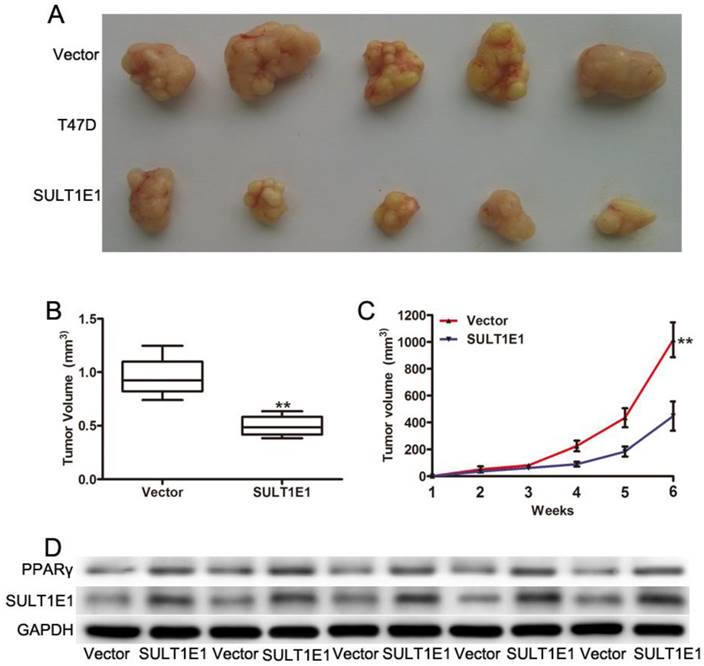
Overexpression of SULT1E1 inhibited tumorigenicity in mouse xenografts
To further investigate the role of SULT1E1 in tumor growth in vivo using a xenograft model in the nude mice, T47D cells transduced with vector or SULT1E1 were injected into the mammary fat pads of 4-week-old female nude mice. After continuous monitoring for 6 weeks, there was a reduction in tumor size in SULT1E1-overexpressed-treated mice compared to vector-treated mice (Figure 5A). The mean tumor weight (Figure 5B) and tumor volume (Figure 5C) of the SULT1E1-overexpressed group were significantly smaller than the control group. Western blot analysis was performed to detect SULT1E1 and PPARγ expression from the xenograft tissue. As shown in Figure 5D, the expression of SULT1E1 and PPARγ was increased in the SULT1E1-overexpressed group compared with the control group. Our data showed that SULT1E1 suppressed tumor growth in xenograft-bearing mouse models.
Overexpression of SULT1E1 inhibits tumor growth in vivo. (A) Representative images of tumors in controls groups and SULT1E1 groups. (B) Bar showed a significant difference in xenografts tumor weight of T47D transduced with vector or SULT1E1. (C) SULT1E1 significantly inhibited tumor growth of T47D cells. (D) Western blotting analysis depicts increased expression of SULT1E1 in xenografts tumor from T47D controls groups or SULT1E1 overexpression T47D group. **P < 0.01.
Discussion
SULT1E1 was first identified in the human liver and located in chromosome 4q13[28]. As reported, SULT1E1 was detected in breast and endometrial tissues and was found to correlate with the risk of breast cancer and endometrial cancer, which are both estrogen-dependent cancers[8, 13]. Accumulating evidence has confirmed that the SULT1E1 single nucleotide polymorphism (SNP) was linked to cancer susceptibility in several malignancies, including breast and endometrial cancers[17, 29]. In this study, we investigated the functions and mechanisms of SULT1E1 in breast cancer. We found that the overexpression of SULT1E1 significantly inhibited cell proliferation by regulating cell apoptosis and cell cycle in vitro. Furthermore, overexpressing SULT1E1 suppressed cell migration and invasion in vitro and tumor growth in vivo.
Previous studies have shown that SULT1E1 was able to mediate cell growth and apoptosis in response to estrogen stimulation[18, 30]. Consistently, we found that the ectopic expression of SULT1E1 could significantly inhibit breast cancer cell proliferation and suppress the growth of xenograft tumors in nude mice, indicating that SULT1E1 functioned as a tumor suppressor. However, previous studies have shown that higher SULT1E1 levels were found in breast cancer tissues compared with that in normal breast tissue, which implied that SULT1E1 might play an oncogenic role in the pathogenesis of breast cancer [18, 29]. In contrast, the expression of SULT1E1 was highly expressed in normal human mammary epithelial cells compared to breast cancer cells [31], which consisted with our data. The discrepancy we speculate that the upregulation of SULT1E1 may duo to body autoimmune inhibition of the tumor progress. But more investigation should be explored. Cell apoptosis and cell cycle were closely associated with cell proliferation[32], and our data indicated that SULT1E1 induced cell cycle at the G0/G1 phase and apoptosis. Cumulative evidence has shown that metastasis was the typical hallmark of cancer, and invasion was the key process in cancer metastasis[33, 34]. In the current study, we found that the overexpression of SULT1E1 could decrease the migration and invasion abilities of MCF-7 and T47D cells. Matrix metalloproteinases (MMPs) played a critical role in tumor cell invasion and metastasis during cancer development. MMP-2 and MMP-9 have been implicated in breast cancer invasion and metastasis[35-37]. Our results demonstrated that the downregulation of SULT1E1 in breast cancer decreased the expression of MMP-2 and MMP-9.
Studies show that PPARγ, a ligand-activated nuclear receptor, has been involved in inflammation, adipocyte differentiation, lipid metabolism, glucose homeostasis, and tumorigenesis[38-40]. Recent studies have shown that PPARγ inhibits cell proliferation and induces apoptosis of breast cancers in vitro and in vivo[41-44]. SULT1E1 has been shown to upregulate PPARγ expression in human umbilical vein endothelial cells (HUVECs)[24]. To further clarify whether PPARγ is involved in SULT1E1-induced cell apoptosis and invasion in breast cancer cells, we knocked down the PPARγ expression in MCF-7 and T47D cells exogenously expressing SULT1E1. Interestingly, the depletion of PPARγ in breast cancer cells resulted in the suppression of motility activity and the activation of apoptosis of the breast cancer cells stably overexpressing SULT1E1. Consistently, the suppressive effect of SULT1E1 on proliferation- and metastasis-related proteins could be rescued by PPARγ siRNA.
Taken together, our results showed that SULT1E1 could inhibit cell proliferation and invasion in vitro and tumor growth in vivo. Further investigation found that SULT1E1 mediated cellular function through the activation of PPARγ. This study may provide a new strategy for future breast cancer therapies.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC. 81402288), a General Financial Grant from the China Postdoctoral Science Foundation (2016M602150), the Natural Science Foundation of Shandong Province (ZR2017QH008), the Shandong Province Key Research and Development Projects (NO.2015GSF118015) and the Research Foundation for Excellent Young and Middle-aged Scientists of Shandong Province (BS2015YY028).
Competing Interests
The authors declare that they have no competing interests.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
3. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-E386
4. Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: Classification, prognostication, and prediction. Lancet. 2011;378:1812-1823
5. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2:101-112
6. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346-352
7. Chiang CH, Chu PY, Hou MF, Hung WC. MiR-182 promotes proliferation and invasion and elevates the HIF-1alpha-VEGF-A axis in breast cancer cells by targeting FBXW7. Am J Cancer Res. 2016;6:1785-1798
8. Song WC. Biochemistry and reproductive endocrinology of estrogen sulfotransferase. Ann N Y Acad Sci. 2001;948:43-50
9. Cole GB, Keum G, Liu J, Small GW, Satyamurthy N, Kepe V, Barrio JR. Specific estrogen sulfotransferase (SULT1E1) substrates and molecular imaging probe candidates. Proc Natl Acad Sci U S A. 2010;107:6222-6227
10. Falany CN. Enzymology of human cytosolic sulfotransferases. FASEB J. 1997;11:206-216
11. Wada T, Ihunnah CA, Gao J, Chai X, Zeng S, Philips BJ, Rubin JP, Marra KG, Xie W. Estrogen sulfotransferase inhibits adipocyte differentiation. Mol Endocrinol. 2011;25:1612-1623
12. Ihunnah CA, Wada T, Philips BJ, Ravuri SK, Gibbs RB, Kirisci L, Rubin JP, Marra KG, Xie W. Estrogen sulfotransferase/SULT1E1 promotes human adipogenesis. Mol Cell Biol. 2014;34:1682-1694
13. Pasqualini JR. Estrogen sulfotransferases in breast and endometrial cancers. Ann N Y Acad Sci. 2009;1155:88-98
14. Mercer KE, Apostolov EO, Gamboa DCG, Yu X, Lang P, Roberts DW, Davis W, Basnakian AG, Kadlubar FF, Kadlubar SA. Expression of sulfotransferase isoform 1A1 (SULT1A1) in breast cancer cells significantly increases 4-hydroxytamoxifen-induced apoptosis. Int J Mol Epidemiol Genet. 2010;1:92-103
15. Utsumi T, Yoshimura N, Takeuchi S, Ando J, Maruta M, Maeda K, Harada N. Steroid sulfatase expression is an independent predictor of recurrence in human breast cancer. Cancer Res. 1999;59:377-381
16. Suzuki M, Ishida H, Shiotsu Y, Nakata T, Akinaga S, Takashima S, Utsumi T, Saeki T, Harada N. Expression level of enzymes related to in situ estrogen synthesis and clinicopathological parameters in breast cancer patients. J Steroid Biochem Mol Biol. 2009;113:195-201
17. Choi JY, Lee KM, Park SK, Noh DY, Ahn SH, Chung HW, Han W, Kim JS, Shin SG, Jang IJ, Yoo KY, Hirvonen A, Kang D. Genetic polymorphisms of SULT1A1 and SULT1E1 and the risk and survival of breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1090-1095
18. Xu Y, Liu X, Guo F, Ning Y, Zhi X, Wang X, Chen S, Yin L, Li X. Effect of estrogen sulfation by SULT1E1 and PAPSS on the development of estrogen-dependent cancers. Cancer Sci. 2012;103:1000-1009
19. Zeng T, Zhu L, Liao M, Zhuo W, Yang S, Wu W, Wang D. Knockdown of PYCR1 inhibits cell proliferation and colony formation via cell cycle arrest and apoptosis in prostate cancer. Med Oncol. 2017;34:27
20. Mazar J, Rosado A, Shelley J, Marchica J, Westmoreland TJ. The long non-coding RNA GAS5 differentially regulates cell cycle arrest and apoptosis through activation of BRCA1 and p53 in human neuroblastoma. Oncotarget. 2017;8:6589-6607
21. Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng WL, Tseng YH, Chen CY, Lin CD, Wu JI, Wang LH, Lin KH. Interleukin-32 increases human gastric cancer cell invasion associated with tumor progression and metastasis. Clin Cancer Res. 2014;20:2276-2288
22. Choi SH, Lee HJ, Jin YB, Jang J, Kang GY, Lee M, Kim CH, Kim J, Yoon SS, Lee YS, Lee YJ. MMP9 processing of HSPB1 regulates tumor progression. PLoS One. 2014;9:e85509
23. Gao H, Zhong F, Xie J, Peng J, Han Z. PTTG promotes invasion in human breast cancer cell line by upregulating EMMPRIN via FAK/Akt/mTOR signaling. Am J Cancer Res. 2016;6:425-439
24. Xu Y, Yang X, Wang Z, Li M, Ning Y, Chen S, Yin L, Li X. Estrogen sulfotransferase (SULT1E1) regulates inflammatory response and lipid metabolism of human endothelial cells via PPARgamma. Mol Cell Endocrinol. 2013;369:140-149
25. Rovito D, Gionfriddo G, Barone I, Giordano C, Grande F, De Amicis F, Lanzino M, Catalano S, Ando S, Bonofiglio D. Ligand-activated PPARgamma downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget. 2016;7:65109-65124
26. Xu B, O'Donnell M, O'Donnell J, Yu J, Zhang Y, Sartor MA, Koenig RJ. Adipogenic differentiation of thyroid cancer cells through the Pax8-PPARgamma fusion protein is regulated by thyroid transcription factor 1 (TTF-1). J Biol Chem. 2016;291:19274-19286
27. Wu K, Yang Y, Liu D, Qi Y, Zhang C, Zhao J, Zhao S. Activation of PPARgamma suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget. 2016;7:44572-44582
28. Aksoy IA, Wood TC, Weinshilboum R. Human liver estrogen sulfotransferase: Identification by cDNA cloning and expression. Biochem Biophys Res Commun. 1994;200:1621-1629
29. Hirata H, Hinoda Y, Okayama N, Suehiro Y, Kawamoto K, Kikuno N, Rabban JT, Chen LM, Dahiya R. CYP1A1, SULT1A1, and SULT1E1 polymorphisms are risk factors for endometrial cancer susceptibility. Cancer-Am Cancer Soc. 2008;112:1964-1973
30. Falany JL, Macrina N, Falany CN. Regulation of MCF-7 breast cancer cell growth by beta-estradiol sulfation. Breast Cancer Res Treat. 2002;74:167-176
31. Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song WC, DeFranco DB, Cheng SY, Xie W. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386
32. Roland CS, Hu J, Ren CE, Chen H, Li J, Varvoutis MS, Leaphart LW, Byck DB, Zhu X, Jiang SW. Morphological changes of placental syncytium and their implications for the pathogenesis of preeclampsia. Cell Mol Life Sci. 2016;73:365-376
33. Nguyen DX, Bos PD, Massague J. Metastasis: From dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274-284
34. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438-1449
35. Marchesin V, Castro-Castro A, Lodillinsky C, Castagnino A, Cyrta J, Bonsang-Kitzis H, Fuhrmann L, Irondelle M, Infante E, Montagnac G, Reyal F, Vincent-Salomon A, Chavrier P. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J Cell Biol. 2015;211:339-358
36. Ren F, Tang R, Zhang X, Madushi WM, Luo D, Dang Y, Li Z, Wei K, Chen G. Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: A systematic review and Meta-Analysis. PLoS One. 2015;10:e135544
37. Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: A tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207-214
38. Reddy AT, Lakshmi SP, Reddy RC. PPARgamma as a novel therapeutic target in lung cancer. PPAR Res. 2016;2016:8972570
39. Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846-855
40. Wang G, Cao R, Wang Y, Qian G, Dan HC, Jiang W, Ju L, Wu M, Xiao Y, Wang X. Simvastatin induces cell cycle arrest and inhibits proliferation of bladder cancer cells via PPARgamma signalling pathway. Sci Rep. 2016;6:35783
41. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419-429
42. Bonofiglio D, Gabriele S, Aquila S, Qi H, Belmonte M, Catalano S, Ando S. Peroxisome proliferator-activated receptor gamma activates fas ligand gene promoter inducing apoptosis in human breast cancer cells. Breast Cancer Res Treat. 2009;113:423-434
43. Catalano S, Mauro L, Bonofiglio D, Pellegrino M, Qi H, Rizza P, Vizza D, Bossi G, Ando S. In vivo and in vitro evidence that PPARgamma ligands are antagonists of leptin signaling in breast cancer. Am J Pathol. 2011;179:1030-1040
44. Rovito D, Gionfriddo G, Barone I, Giordano C, Grande F, De Amicis F, Lanzino M, Catalano S, Ando S, Bonofiglio D. Ligand-activated PPARgamma downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget. 2016;7:65109-65124
Author contact
![]() Corresponding author: Yejun Qin, Department of Pathology, Shandong Provincial Hospital Affiliated to Shandong University, No. 324 Jingwu Road, Jinan, Shandong, 250021, China. Tel: +86-531-68776430. E-mail: qinyejuncom and Yintao Li, Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong University, No. 440 Jiefang Road, Jinan, Shandong, 250117, China. Tel: +86-531-87984777. E-mail: yintaoliedu.cn
Corresponding author: Yejun Qin, Department of Pathology, Shandong Provincial Hospital Affiliated to Shandong University, No. 324 Jingwu Road, Jinan, Shandong, 250021, China. Tel: +86-531-68776430. E-mail: qinyejuncom and Yintao Li, Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong University, No. 440 Jiefang Road, Jinan, Shandong, 250117, China. Tel: +86-531-87984777. E-mail: yintaoliedu.cn