Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(7):1145-1151. doi:10.7150/jca.24035 This issue Cite
Research Paper
Phase I/II clinical trial of everolimus combined with gemcitabine/cisplatin for metastatic triple-negative breast cancer
In Hae Park1, Sun-Young Kong2, Youngmee Kwon1, Min Kyeong Kim4, Sung Hoon Sim1, Jungnam Joo3, Keun Seok Lee1 ![]()
1. Center for Breast Cancer, National Cancer Center, Korea
2. Department of Laboratory Medicine, National Cancer Center, Korea
3. Biometric Research Branch, National Cancer Center, Korea
4. Department of Cancer Biomedical Science, Graduate School of Cancer Science, National Cancer Center, Korea
Received 2017-11-27; Accepted 2018-1-8; Published 2018-3-8
Abstract
Background: The PI3K/AKT/mTOR pathway is an important oncogenic driver in triple-negative breast cancer (TNBC). This study investigated the clinical efficacy and safety of the combination of gemcitabine and cisplatin with everolimus (GPE) in patients with metastatic TNBC.
Methods: In phase I, we assessed the maximum tolerated dose (MTD) of GPE in metastatic TNBC patients. Then, using a seamless design, we conducted a randomized phase II trial to compare GPE to GP in terms of progression-free survival (PFS) and toxicity. In addition, we investigated the mutational status of PIK3CA (E542K, E545K, H1047R) in tumor tissues (n=14) and cell-free DNA (cfDNA) from blood samples (n=23) using droplet digital PCR.
Results: In phase I (n=9), we found that the MTD of GPE was gemcitabine 800 mg/m2 and cisplatin 30 mg/m2 on days 1 and 8 every 3 weeks along with everolimus 5 mg daily. Phase II was terminated early after 14 patients had been enrolled because of slow recruitment and concerns about efficacy. Results of the combined analysis of phases I and II showed the objective response rate (ORR) of GPE (n=16) was 31.3% and the median PFS was 5.5 months (95% CI, 3.5-7.5). Stomatitis and hematologic toxicities were observed most frequently in the GPE arm. PIK3CA mutations were identified in 8 (57.1%) tumor samples and 17 (73.9%) cfDNA samples; there was no significant association between PIK3CA mutation status and response to GPE treatment.
Conclusions: Although the majority of patients with metastatic TNBC demonstrated PIK3CA mutations in cfDNA, the addition of everolimus to gemcitabine/cisplatin did not have a synergistic effect in these patients. Further studies are needed to determine the most effective way to target the PI3K/AKT/mTOR pathway in TNBC patients.
Keywords: Triple negative breast cancer, mTOR inhibitor, cell free DNA, PIK3CA mutation
Introduction
Triple-negative breast cancer (TNBC) has aggressive subtypes with poor prognoses [1]. TNBC tumors are defined as estrogen receptor (ER) and/or progesterone receptor (PgR)-negative and human epidermal growth factor receptor 2 (HER2)-negative, and account for 11-23% of all breast cancers [1,2]. Until now, chemotherapy has remained the standard treatment, with the most common chemotherapeutic regimens for advanced TNBC based on anthracycline- or taxane-containing combination therapies [3]. Platinum agents also are commonly used in combination with other chemotherapeutic agents such as taxane, ifosfamide, or gemcitabine. In a phase II study of patients with metastatic TNBC, the combination of carboplatin and gemcitabine showed an overall response rate of 32% [4]. According to our previous data, even a low dose of gemcitabine and cisplatin had clinical efficacy with overall survival of 7.6 months in heavily pretreated metastatic TNBC patients [5].
Dysregulation of the PI3K pathway by PIK3CA mutations has been found frequently in ER-positive breast cancer [6, 7]. Meanwhile, the loss of phosphatase and tensin homolog (PTEN) expression, known to activate the PI3K pathway, more frequently noted in TNBC subtypes [8]. In addition, PIK3CA and PTEN have been identified in recent deep sequencing studies as the most prevalent clonal mutation sites next to TP53 in TNBC [9]. Therefore, the PI3K/AKT/mTOR pathway is considered to be a promising target in TNBC. Everolimus is a selective inhibitor of mammalian target of rapamycin (mTOR), which is an important down-stream effector in the PI3K/AKT/mTOR pathway. Previously, it showed the promising results combined with endocrine therapy in ER positive metastatic breast cancer and approved as the second line therapy in patients who failed prior endocrine therapy [7]. Everolimus has also shown efficacy against the basal-like subtype of TNBC cell lines and enhanced the sensitivity of breast cancer cell to cytotoxic drugs, which resulted in synergistic inhibition of cell proliferation and apoptosis [10-12].
On the basis of these prior data, we investigated the feasibility of gemcitabine, cisplatin, and everolimus (GPE) combination therapy in metastatic TNBC and determined the maximum tolerated dose (MTD) in a phase I study. Then, we addressed the safety and efficacy of this combination therapy in a subsequent phase II study.
Patients and methods
Patients
Eligible patients had metastatic TNBC confirmed by histologic diagnosis and immunohistochemistry (IHC). Patients were included if they had adequate performance status and organ function and were excluded if they had an impairment of gastrointestinal function or symptomatic deterioration of lung function. All patients had been previously treated with anthracycline- and taxane-based chemotherapeutic regimens. In addition, patients who previously had received gemcitabine or inhibitors of mTOR or PI3K were excluded from the study. All patients provided written informed consent and the study was approved by the institutional review board at National Cancer Center, Korea. This study has been registered in ClinicalTrials.gov (NCT01939418).
Study design and treatment
This study was a single institution phase Ib/II trial (Figure 1). In phase Ib, the primary objective was to identify the recommended dose (RD) of GPE in patients with metastatic TNBC. Subsequently, phase II was conducted to investigate the efficacy of GPE compared to GP (gemcitabine plus cisplatin) in terms of progression-free survival (PFS). A traditional 3+3 design was used in the phase Ib study; the starting doses of cisplatin 30 mg/m2 and gemcitabine 800 mg/m2 iv on days 1 and 8, and everolimus 5 mg po qod (dose level 1) were administered every 21 days. If one of three patients at a given dose level developed dose-limiting toxicity (DLT), then at least three more patients would enter at the same dose level. If none of three patients or less than two of six patients developed DLT at a certain dose, the next patient would be treated with the higher dose (Table 1). After we determined the RD in the phase Ib study, the phase II study was conducted. Patients were recruited according to the same eligibility criteria and randomly assigned to the GPE or GP arms. Patients received gemcitabine and cisplatin intravenously on days 1 and 8 every 3 weeks and everolimus at a fixed dose once a day or every other day without resting until they experienced unacceptable toxicity, disease progression, or upon investigator decision.
Study design
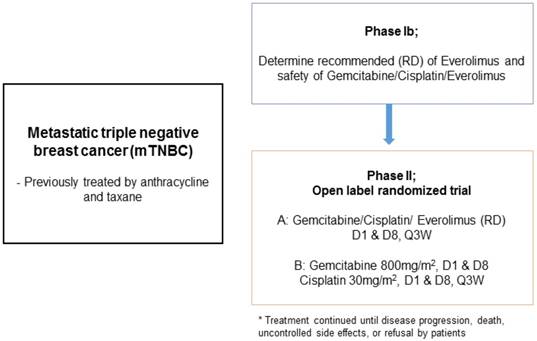
Patient characteristics, PI3KCA mutation status, and treatment response.
| Number | Age | No. of previous chemotherapy | Visceral metastasis | Time from diagnosis to enrollment (mo) | Treatment | PI3KCA mutation (Tissue) | PI3KCA mutation (cfDNA) | Response |
|---|---|---|---|---|---|---|---|---|
| Ph1-1 | 52 | 1 | None | 1.6 | GPE (1) | E545K, H1047R | H1047R | PR |
| Ph1-2 | 37 | 1 | None | 4.0 | GPE (1) | NA | H1047R | PR |
| Ph1-3 | 60 | 2 | Liver | 6.9 | GPE (1) | NA | None | SD |
| Ph1-4 | 50 | 1 | Lung | 13.5 | GPE (2) | E542K, E545K, H1047R | E545K H1047R | PR |
| Ph1-5 | 56 | 1 | Lung | 17.9 | GPE (2) | E542K, E545K | H1047R | PR |
| Ph1-6 | 72 | 2 | Liver | 53.9 | GPE (2) | NA | H1047R | SD |
| Ph1-7 | 51 | 1 | Lung | 3.1 | GPE (2) | NA | H1047R | SD |
| Ph1-8 | 47 | 1 | Lung | 9.4 | GPE (2) | E545K, H1047R | H1047R | SD |
| Ph1-9 | 50 | 1 | None | 19.8 | GPE (2) | E542K | H1047R | CR |
| Ph2-1 | 50 | 1 | Lung | 9.0 | GP | None | E545K | PD |
| Ph2-2 | 49 | 2 | None | 16.4 | GP | E545K | H1047R | SD |
| Ph2-3 | 45 | 2 | Liver | 8.6 | GP | NA | H1047R | PR |
| Ph2-4 | 50 | 1 | None | 8.8 | GPE | None | H1047R | SD |
| Ph2-5 | 37 | 2 | Liver, Lung | 9.0 | GPE | None | H1047R | PD |
| Ph2-6 | 51 | 2 | None | 10.0 | GPE | E545K | E542K H1047R | SD |
| Ph2-7 | 54 | 1 | Lung | 5.5 | GPE | NA | None | SD |
| Ph2-8 | 65 | 1 | Lung | 12.1 | GP | E545K H1047R | E542K H1047R | PD |
| Ph2-9 | 45 | 1 | None | 18.3 | GPE | None | None | SD |
| Ph2-10 | 54 | 1 | None | 6.3 | GPE | None | None | SD |
| Ph2-11 | 62 | 2 | None | 13.0 | GP | NA | None | CR |
| Ph2-12 | 50 | 1 | Lung | 7.6 | GP | NA | E542K | PR |
| Ph2-13 | 59 | 1 | Lung | 5.6 | GPE | NA | None | SD |
| Ph2-14 | 47 | 1 | None | 3.8 | GP | None | H1047R | PR |
GP, gemcitabine, cisplatin; GPE, gemcitabine, cisplatin, everolimus; (#), dose level in phase I; NA, no available tissue; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
Assessments and biomarker analysis
The RD for the subsequent phase II study was defined as the MTD that was the highest dose of GPE attained without unacceptable side effects. DLT was defined as side effects during treatment that were severe enough to prevent a further increase in dosage of GPE, or to prevent continuation of treatment at any dosage level. PFS was defined as the time from the date of randomization to the date of first documented tumor progression or death due to any cause, whichever occurred first. The secondary objectives consisted of overall survival (OS), objective response rate (ORR), and toxicity profiles. OS was calculated from the date of randomization to the date of death or date of last follow-up. For the evaluation of treatment response, tumor assessment was conducted every two cycles and recorded according to RECIST criteria version 1.1. Toxicity profiles were graded according to NCI-CTCAE version 4.0 and were presented as the ratio of the number of occurrences to the safety population.
Sample collection and PI3KCA mutation detection by ddPCR
Pre-treatment blood samples (5 mL) were obtained in serum separator tubes (SST), and serum was obtained by centrifugation of the SST at 2000 ×g for 10 min. Serum was aliquoted and stored at -80℃ until analysis. CfDNA from 800 µl of serum was extracted with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. cfDNA was quantified using the Qubit 2.0 Fluorometer with the Qubit dsDNA HS (High Sensitivity) Assay Kit (Life Technologies, Carlsbad, CA, USA). For formalin-fixed paraffin-embedded (FFPE) tumor samples, genomic DNA was extracted using the QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany). DNA quantification was performed with a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA).
PIK3CA mutations were detected by droplet digital PCR (ddPCR) in singleplex assays using inventoried assays for PIK3CA H1047R (dHsaCP2000077), E545K (dHsaCP2000075), and E542K (dHsaCP2000073) (BioRad, Hercules, CA, USA) on a BioRad QX200 Droplet Digital PCR System. Each assay run included a mutation-positive template and no-template controls. The fractional abundance (%) of the PIK3CA mutation was calculated from the number of FAM-positive events (mutation-positive) over total-positive events (both FAM and HEX positive for mutation and wild type, respectively) using QuantaSoft Version 1.7.4.0917 (BioRad) and 0.1% and more was evaluated as mutation positive.
Statistical analysis
In the phase Ib study, the required sample size was approximately 9-12 patients. Baseline patient characteristics, the type and grade of toxicities, and any adverse events were summarized using descriptive statistics. The primary objective of phase II was PFS estimated by the Kaplan-Meier method followed by comparison using log-rank test. We set the median PFS of the GP arm to 4.0 months, which was inferred from the data of O'Shaughnessy (13). Considering 4.0 months vs. 5.6 months median PFS between the two arms (HR=1.4), the phase II study required 103 treatment failures (events) when both the type I and II errors were set to 0.2. The trial was planned for 3 years of accrual and 1 year of follow-up. Based on these figures, to observe 103 events during the follow-up, approximately 52 patients per arm were needed. Considering 5% drop out, 55 patients per arm needed to be recruited.
Results
Phase Ib and phase II clinical trials
At first, three patients entered at dose level 1 and none developed DLT. Two patients experienced grade 3 neutropenia and recovered with conservative care. Then, six patients were enrolled at dose level 2. Among the six patients, one developed grade 4 neutropenia, therefore, the RD for the phase II trial was determined at dose level 2 (gemcitabine 800 mg/m2, cisplatin 30 mg/m2, and everolimus 5 mg daily). Of nine patients, five (55.6%) patients achieved objective response (complete response [CR] =1, partial response [PR] =4) and four patients showed stable disease (SD) for more than 4 months.
The phase II clinical trial was then conducted using a seamless design after completion of phase Ib; however, phase II was closed early due to slow enrollment and concerns about efficacy. Fourteen patients participated in the phase II trial (n=7 each for GP and GPE), and combined analysis was conducted on all 23 patients from the phase Ib and phase II trials. Sixteen of the patients had received gemcitabine, cisplatin, and everolimus (GPE) and three of those patients had received everolimus (5 mg) every other day. The median age of the patients was 50 years (range, 37-72 years) and median time from diagnosis of metastasis to enrollment was 9.0 months (range, 1.6-53.9 months).
Table 2 summarizes the patient characteristics and clinical response to treatment. All patients had received anthracycline and taxane-based prior chemotherapy either in adjuvant or in metastatic settings and 13 (56.5%) patients had visceral metastasis before treatment. Among patients (n=16) who received GPE treatment, the ORR (CR + PR) was 31.3% (n=5), while 62.5% (n=10) showed stable disease. Conversely, in patients who received GP treatment, ORR was 57.1% (CR=1, PR=3), SD 14.3% (n=1), and PD 28.6% (n=2). The median PFS was 5.7 months (95% confidential interval [CI] 2.4-9.0) and median OS was 19.1 months (95% CI, 7.5-30.7) in all patients (Figure 2A, 2B). These results were not significantly different when compared to the patients who received GPE treatment (median PFS=5.5 months, 95% CI, 3.5-7.5; median OS=19.1 months, 95% CI, 5.3-32.9). There was one extreme responder to everolimus (Ph1-9) who achieved CR after four cycles of GPE treatment. She has remained on everolimus monotherapy and has been progression-free for more than 3 years.
Toxicity profiles of phase Ib & II study
| Phase Ib | Phase II | |||||||
|---|---|---|---|---|---|---|---|---|
| GPE (dose level 1) 5 mg QOD (n=3) | GPE (dose level 2) 5 mg QD (n=6) | GPE (n=7) | GP (n=7) | |||||
| G1-2 | G3-4 | G1-2 | G3-4 | G1-2 | G3-4 | G1-2 | G3-4 | |
| Hematologic Toxicity | ||||||||
| Neutropenia | 0 | 2 | 0 | 3 | 2 | 14 | 1 | 15 |
| Anemia | 1 | 0 | 3 | 0 | 2 | 4 | 2 | 7 |
| Thrombocytopenia | 0 | 0 | 2 | 0 | 3 | 1 | 6 | 1 |
| Non-Hematologic Toxicity | ||||||||
| Lethargy | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 |
| Anorexia | 3 | 0 | 2 | 0 | 2 | 0 | 1 | 0 |
| Stomatitis | 1 | 0 | 4 | 1 | 9 | 1 | 0 | 0 |
| Hyperglycemia | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
Toxicity: G1-2: Grade 1-2; G3-4: Grade 3-4; GPE: gemcitabine, cisplatin, everolimus; GP: gemcitabine, cisplatin.
Survival analysis for all participants (phase I/II)
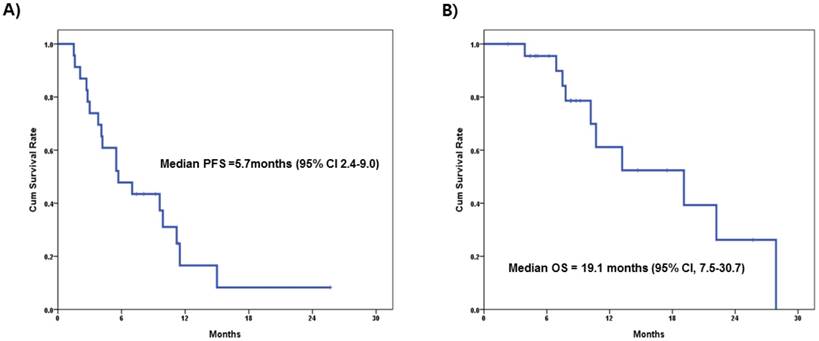
In terms of adverse events, hematologic toxicities including anemia and neutropenia were most notable in the GPE arm. One patient with grade 3 hyperglycemia was noted in the GPE arm, which was controlled with anti-diabetic drugs. The other remarkable toxicity was stomatitis, which was attributed to everolimus.
PI3KCA mutation status in cfDNA and archived tumor samples
Of the 23 patients, 14 (60.9%) had available archived tumor samples for PIK3CA mutation assay. PIK3CA cfDNA analysis was performed successfully in all patients. PIK3CA hot spot (HS) mutations were identified in 8 (57.1%) tumor samples and 17 (73.9%) cfDNA samples (Supplementary Table 2). The most prevalent HS mutation was E545K (50%) in tumor tissue and H1047R (65.2%) in cfDNA. The overall concordance in PI3KCA mutation status between archival tumor and cfDNA sample pairs was 42.9% (6/14). Among five patients without PIK3CA HS mutations in archival tumor samples, the H1047R mutation was newly detected in cfDNA samples of three patients (Supplementary Table 1). Conversely, all eight patients with mutations in archival tumor samples also had PIK3CA HS mutations in cfDNAs even though they were not all the same genotypes.
The primary tumor sample of the extreme responder, Ph1-9, was evaluated both by ddPCR and whole exome sequencing (WES, Supplementary Methods). The E542K hot spot mutation was detected by ddPCR (1.40%, 0.2/12.0), but was not found by WES, while a PTEN frame shift deletion (677delC, S227fs, allele frequency 76/318) was detected by WES. In addition, the H1047R mutation, not E542K, was detected in the cfDNA of this patient.
Correlation between PIK3CA HS mutation status and treatment efficacy
The clinical response rate was assessed according to everolimus treatment and PIK3CA mutation status in cfDNA in all patients participating in the phase I/II trials (Table 3). More patients achieved SD with the treatment including everolimus regardless of PIK3CA mutation status (11/16, 68.8%). Five patients harboring wild type PIK3CA showed SD with GPE treatment. Among patients with PIK3CA mutations (n=17), eight (47.1%) patients achieved objective response to treatment: five (5 of 11 patients, 45.5%) patients with GPE treatment and three (3 of 6 patients, 50%) with GP treatment (Table 3).
Clinical response in patient subgroups according to PIK3CA mutation status and everolimus treatment
| PIK3CA mutation | Response | Treatment arm | |
|---|---|---|---|
| GPE, n | GP, n | ||
| Wild type (n=6) | CR+PR | 0 | 1 |
| SD | 5 | 0 | |
| PD | 0 | 0 | |
| Mutant type (n=17) | CR+PR | 5 | 3 |
| SD | 5 | 1 | |
| PD | 1 | 2 | |
CR: complete response; PR: partial response; SD: stable disease; PD: progressive disease; GPE: gemcitabine, cisplatin, everolimus; GP: gemcitabine, cisplatin.
Discussion
The combination of targeted agents with cytotoxic chemotherapy for treating cancers refractory to standard treatment is an active area of investigation. Advanced TNBC typically responds poorly and shows rapid resistance to currently used chemotherapeutic agents. Previously, the mTOR inhibitor, everolimus, was demonstrated to potentiate the effect of commonly-used cytotoxic drugs such as paclitaxel, doxorubicin, gemcitabine, and cisplatin [12, 14]. In hormone receptor-positive metastatic breast cancer patients, everolimus showed prolonged PFS when combining with exemestane [7]. In addition, its combination with trastuzumab and several cytotoxic agents like paclitaxel and vinorelbine showed promising antitumor activity in HER2-overexpressing metastatic breast cancer [15, 16].
In this study, we assessed the feasibility of the combination of gemcitabine, cisplatin, plus everolimus in a phase I trial in metastatic TNBC patients. The RD was determined to be gemcitabine 800 mg/m2 iv and cisplatin 30 mg/m2 iv on days 1 and 8 every 3 weeks and everolimus 5 mg daily po. We assumed that the doses of the two cytotoxic drugs were acceptable as a combination therapy based on our previous report [5]. Although the dose of everolimus was lower than that used in monotherapy, it did not necessarily mean that the lower dose of everolimus lacked efficacy. Actually, a similar phase I trial with the same drugs reported that an even lower dose of the drugs in combination (gemcitabine 600 mg/m2, cisplatin 12.5 mg/m2 on days 1 and 8 every 3 weeks, and everolimus 5 mg three times per week) showed clinical efficacy in certain types of cancer [17].
According to our results, the majority of patients did not achieve an objective response to everolimus combination treatment even though they had PIK3CA HS mutations in cfDNA. In addition, more patients experienced hematologic toxicities and stomatitis compared to patients who received GP treatment. Recently, a similar, modest efficacy of everolimus in combination with cytotoxic drugs in TNBC patients was reported [16]. The combination of everolimus with carboplatin showed a clinical benefit rate of 36% and a median PFS of 3 months in metastatic TNBC patients [16]. Another study examined the response to the addition of everolimus to the standard neoadjuvant chemotherapy, paclitaxel [12]. In this study, the everolimus combination did not increase the pathologic CR compared to standard chemotherapy (30.4% vs. 25.9%, P=0.76).
As a conjunctive study, we investigated the impact of three of the most prevalent PIK3CA-activating HS mutations in cfDNA on the efficacy of everolimus in patients with metastatic TNBC. Many studies showed that the feasibility of detecting various tumor specific mutations in cfDNA such as EGFR mutations in patients with lung cancer, KRAS mutations with colorectal and pancreatic cancer in patients, and BRAF mutation in patients with melanoma [18-21]. Previously, the proportion of TNBC patients harboring PIK3CA mutations was reported to be 10 to 22% in primary tissue by sequencing analysis [9, 22]. We evaluated PIK3CA mutation status in primary tumor tissue using ddPCR because all of the tumor samples were archived FFPE and DNA quantity and quality were very low. Even though the number of samples were small, 57.1% of tumor tissue and more than 70% of cfDNA samples were found to have PIK3CA HS mutations, which was much higher than previous data. We assumed that such discordant results could be caused by different analyzing methods, digital PCR and study population (samples). The digital PCR method is known to be highly sensitive and capable of accurately detecting amplified loci present at ratios less than 1:100, which may be more suitable for highly fragmented small quantities of DNA and RNA to detect specific alterations than other sequencing methods, as in our setting. Next, all patients in this study had metastatic disease and were retrospectively analyzed. Specifically, blood samples for cfDNA were collected just before study treatment. Recent study showed that PIK3CA mutation rate was several fold higher in relapsed ER negative cancer and the additional mutations were substantially increasing as breast cancer evolution [23]. Similarly, our study also showed a higher mutation rate of PIK3CA in plasma samples than in primary tissues. Although our study had many limitations with small number of patients, it may be suggested that cfDNA analysis is helpful to timely reflect the current mutation status when considering the nature of tumors that evolve continuously.
According to our results, the PIK3CA HS mutations alone did not seem to be adequate biomarkers for the mTOR inhibitor everolimus in metastatic TNBC. This study had a limitation not to include other several biomarkers for PI3K pathway activation such as PTEN loss, other mutations and/or amplifications/deletions in PI3K-associated genes [22, 24]. There needs to be more comprehensive studies to elucidate biomarkers of PI3K pathway alteration using advanced methods.
In conclusions, the addition of everolimus to gemcitabine/cisplatin did not have a synergistic effect in patients with metastatic TNBC, though the majority of patients demonstrated PIK3CA HS mutations in cfDNA. Further studies are needed to determine the most effective way to target the PI3K/AKT/mTOR pathway in TNBC patients and new strategies for effective combinations of PI3K-targeted drugs with conventional therapy in the future.
Supplementary Material
Supplementary method and tables.
Acknowledgements
We thank the patients who participated in this study and clinical research nurses for their contribution to the study.
Funding
This work was supported by Novartis. The funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Ethics Committee approval and Patient Consent
This study was approved by the Institutional Review Board, National cancer center, Korea and the reference number was NCCCTS-13-670. Signed informed consent for this trial was obtained from all patients.
Availability of data and material
Data are available on request from the PI of this study, In Hae Park, National cancer center. Korea.
Author contributions
All authors reviewed and provided critical comments on drafts of the manuscript and gave final approval to submit for publication. IH Park and J Joo contributed to the study conception and design. IH Park, S Kong, Y Kwon, MK Kim, SH Sim, and KS Lee contributed to acquisition of data. IH Park, S Kong, and KS Lee contributed to interpretation of results. IH Park and MK Kim provide administrative and technical support. IH Park and KS Lee were involved in study supervision. IH Park and S Kong performed or supervised the analysis. J Joo provided statistical expertise.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429-34
2. Thike AA, Cheok PY, Jara-Lazaro AR, Tan B, Tan P, Tan PH. Triple-negative breast cancer: clinicopathological characteristics and relationship with basal-like breast cancer. Mod Pathol. 2010;23(1):123-33
3. Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast. 2010;19(5):312-21
4. Maisano R, Zavettieri M, Azzarello D, Raffaele M, Maisano M, Bottari M, Nardi M. Carboplatin and gemcitabine combination in metastatic triple-negative anthracycline- and taxane-pretreated breast cancer patients: a phase II study. J Chemother. 2011;23(1):40-3
5. Kim JS, Park IH, Lee KS, Ro J. Outcomes of palliative weekly low-dose gemcitabine-cisplatin chemotherapy in anthracycline-and taxane-pretreated metastatic breast cancer patients. J Breast Cancer. 2014;17(4):339-43
6. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25(48):6416-22
7. Baselga J, Campone M, Piccart M, Burris H, Rugo H, Sahmoud T. et al. Everolimus in postmenopausal hormone receptor positive advanced breast cancer. N Eng J Med. 2012;366(6):520-9
8. Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X. et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2004;65(7):2554-59
9. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y. et al. The clonal and mutational evolution spectrum of primary triple negative breast cancers. Nature. 2012;486(7403):395-9
10. Liu H, Zang C, Schefe JH, Schwarzlose-Schwarck S, Regierer AC, Elstner E. et al. The mTOR inhibitor RAD001 sensitizes tumor cells to the cytotoxic effect of carboplatin in breast cancer in vitro. Anticancer Res. 2011;31(9):2713-22
11. Yunokawa M, Koizumi F, Kitamura Y, Katanasaka Y, Okamoto N, Kodaira M. et al. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012;103(9):1665-71
12. O'Reilly T, McSheehy PM, Wartmann M, Lassota P, Brandt R, Lane HA. Evaluation of the mTOR inhibitor, everolimus, in combination with cytotoxic antitumor agents using human tumor models in vitro and in vito. Anticancer Drugs. 2011;22(1):58-78
13. O'Shaughnessy J, Schwarzberg LS, Danso MA, Miller KD, Rugo HS, Neubauer M. et al. Phase III study of iniparib plus gemcitabine and carbiplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32(34):3840-7
14. Chiang CT, Yeh PY, Gao M, Chen CW, Yeh LC, Feng WC. et al. Combinations of mTORC1 inhibitor RAD001 with gemcitabine and paclitaxel for treating non-Hodgkin lymphoma. Cancer Lett. 2010;298(2):195-203
15. Andre F, O'Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G. et al. Everolimus for woment with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15(6):580-91
16. Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP. et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomized, double-blind, multicenter trial. Lancet Oncol. 2015;16(7):816-29
17. Costello BA, Borad MJ, Qi Y, Kim GP, Northfelt DW, Erlichman C, Alerts SR. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Invest New Drugs. 2014;32(4):710-6
18. Kimura H, Kasahara K, Kawaishi M, Kunitoh H, Tamura T, Holloway B, Nishio K. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res. 2006;12(13):3915-21
19. Sorenson GD. Detection of mutated KRAS2 sequences as tumor markers in plasma/serum of patients with gastrointestinal cancer. Clin Cancer Res. 2000;6(6):2129-37
20. Ryan BM, Lefort F, McManus R, Daly J, Keeling PW, Weir DG, Kelleher D. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: strong prognostic indicator in postoperative follow up. Gut. 2003;52(1):101-8
21. Daniotti M, Vallacchi V, Rivoltini L, Patuzzo R, Santinami M, Arienti F. et al. Detection of mutated BRAFV600E variant in circulating DNA of stage III-IV melanoma patients. Int J Cancer. 2007;120(11):2439-44
22. Kriegsmann M, Endris V, Wolf T, Pfarr N, Stenzinger A, Loibl S. et al. Mutational profiles in triple negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget. 2014;5(20):9952-65
23. Yates L, Knappskog S, Wedge D, Farmery J, Gonzalez S, Martincorena I. et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32:169-84
24. Markman B, Atzori F, Perez-Garcia J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21(4):683-91
Author contact
![]() Corresponding author: Keun Seok Lee, Center for Breast Cancer, National Cancer Center, Korea. Email: ksleere.kr; Tel : +82-31-920-1623
Corresponding author: Keun Seok Lee, Center for Breast Cancer, National Cancer Center, Korea. Email: ksleere.kr; Tel : +82-31-920-1623