Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(7):1207-1217. doi:10.7150/jca.20964 This issue Cite
Research Paper
Synergistic antitumor effects of cMet inhibitor in combination with anti-VEGF in colorectal cancer patient-derived xenograft models
Xiangheng Chen1,2,3, Zhonghai Guan2, Jun Lu2, Haohao Wang2, Zhongkun Zuo1, Fei Ye1, Jiangsheng Huang1 ![]() , Lisong Teng2
, Lisong Teng2 ![]()
1. Department of Minimally Invasive Surgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan, P. R. China;
2. Department of Surgical Oncology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, P.R. China;
3. Department of Gastrointestinal Surgery, The First Affiliated Hospital of University of South China, Hengyang, Hunan, P. R. China;
Received 2017-5-10; Accepted 2018-2-27; Published 2018-3-14
Abstract
cMet signaling pathway is involved in the resistance to anti-VEGF therapy and cMet overexpression is associated with tumor progression and poor prognosis. In this study, the expression of cMet in 146 Chinese colorectal cancer (CRC) patients was examined by immunohistochemistry staining. Our data demonstrated that cMet overexpression rate was 42.5% (62/146) and cMet overexpression was closely correlated with distant metastasis of CRC. Using CRC patient-derived xenograft (PDX) mouse models we investigated antitumor activity of a novel selective cMet inhibitor volitinib alone or in combination with anti-VEGF inhibitor apatinib in vivo. Our results showed that combination treatment significantly inhibited tumor growth in two PDX models. While volitinib treatment alone induced moderate improvement in tumor growth inhibition, combination treatment synergistically reduced microvessel density, suppressed proliferation, and increased apoptosis in PDX models. Further analysis showed synergistic inhibition of MAPK and PI3K/Akt pathways by volitinib and apatinib. Taken together, our data provide a rationale to targeting both cMet and VEGF in the treatment of cMet overexpressing CRC in clinical trials.
Keywords: Colorectal cancer, cMet, PDX, Volitinib, Apatinib
Introduction
Colorectal cancer (CRC) is a leading cause of cancer-related mortality worldwide [1]. Despite many improvements in screening, surgery and adjuvant therapy for CRC in recent years [2, 3], the overall outcome of these malignant tumors is not satisfactory, owing to tumor recurrence and distant metastasis [4]. To improve the prognosis of CRC patients, there is a great need to identify additional new targets to establish more effective treatments for CRC.
Vascular endothelial growth factor (VEGF), a key pro-angiogenic growth factor, plays a pivotal role in tumor angiogenesis and its overexpression is often associated with tumor growth and metastases [5]. VEGF inhibitors, including anti-VEGF monoclonal antibodies (such as bevacizumab), VEGF-binding proteins (such as aflibercept), and small molecule VEGFR tyrosine kinase inhibitors (such as regorafenib), have produced a significant improvement in progression-free survival for patients with several types of cancers including metastatic CRC [6-8]. Apatinib is a novel oral small-molecule tyrosine kinase inhibitor that blocks VEGFR2. Previous studies demonstrated that apatinib had antitumor activity in a wide variety of solid tumors in vitro and in vivo [9, 10]. Apatinib was approved in China for patients with metastatic gastric cancer in 2014 [11]. In addition, it is currently being evaluated in Phase II/III clinical trials for multiple solid cancers.
Anti-VEGF targeted therapies bring significant advances in the treatment of CRC. However, the improvements in overall survival (OS) have been modest because patients exhibit acquired resistance to VEGF inhibition [12]. Interestingly, emerging evidence has demonstrated that tumor vascular pruning caused by VEGF inhibition led to the induction of hypoxia and the upregulation of hypoxia-inducible factor-1α (HIF-1α) and cMet expression, which increases the invasive and metastatic behavior of tumor cells [13, 14]. Therefore, a strategy that simultaneously targets VEGF and cMet appears to be promising in preclinical and clinical studies for the treatment of CRC.
cMet, known as the receptor for hepatocyte growth factor, is a member of the receptor tyrosine kinase family and plays a key role in embryogenesis, tumor survival, angiogenesis and metastasis [15, 16]. cMet is overexpressed, amplified, or mutated in a wide variety of solid tumors [17, 18], and is correlated with poor prognosis [19, 20].cMet contributes to tumor aggressiveness and resistance to therapy targeted against VEGF and epidermal growth factor receptor (EGFR) [21, 22]. Volitinib (savolitinib, AZD6094) is an orally bioavailable inhibitor of cMet. Previous studies indicated that volitinib exhibited antitumor effects in patient-derived xenograft (PDX) models of gastric carcinoma and papillary renal cell carcinoma [23, 24]. However, the effects of volitinib in preclinical models of CRC have yet to be investigated.
In present study, we first investigated the expression and prognostic value of cMet in a cohort of Chinese CRC patients. Next we evaluated the antitumor effects of volitinib in combination with VEGF inhibitor apatinib in PDX models of CRC.
Materials and Methods
Drugs
Volitinib was kindly provided as a gift from Hutchison MediPharma, Inc. (Shanghai, China). Apatinib was obtained from Jiangsu Hengrui Medicine, Co. (Lianyungang, China). Volitinib and apatinib were diluted in 0.5% (w⁄v) carboxymethyl cellulose.
Patients and tissue samples
This study included 146 patients with colorectal carcinoma who were surgically treated between 2008 and 2009 at the Department of Surgical Oncology, The First Affiliated Hospital, College of Medicine, Zhejiang University. Written informed consent was provided from all patients and the study protocol was approved by Ethics Board at the First Affiliated Hospital, College of Medicine, Zhejiang University. Formalin-fixed, paraffin-embedded CRC specimens of all patients were obtained from the pathology department. Histological subtype was determined after a review of tumor sections by two trained pathologists. Tumor stage was assessed by reference to the criteria of the TNM-system of the UICC, 7th edition.
Immunohistochemistry (IHC)
Paraffin embedded CRC tissue sections of 4 µM in thickness were subjected to IHC using rabbit anti-human cMet monoclonal antibody (clone SP44) (Spring Biosciences, Pleasanton, CA, USA). Staining procedures were conducted according to the manufacturer's protocol.
CRC xenograft specimens were fixed in 4% para-formaldehyde (PFA) overnight at 4°C, dehydrated and embedded in paraffin, and stained with hematoxylin and eosin (H&E) according to routine protocols. Immunostaining was performed using the following primary antibodies: HER2 (Abcam, Cambridge, United Kingdom), VEGF (Abcam, Cambridge, United Kingdom) and Ki-67 (Abcam, Cambridge, United Kingdom). Staining procedures were conducted according to the manufacturers' protocols.
The intensity of cMet, HER-2, and VEGF staining was evaluated as previously reported [25-27]. cMet positive expression (or high expression) was defined as IHC scores of 2+ or 3+, and negative expression (or low expression) was defined as IHC scores of 0 or 1+. For Ki67, only nuclear immunoreactivity was considered positive. The percentage of Ki67-stained nuclei was determined by counting at least five randomly chosen microscopic fields (magnification 40 ×) and 1,000 total cells. All samples were analyzed and evaluated independently by two board-certified clinical pathologists blinded to the clinical and molecular characteristics of the patients.
Fluorescence in situ hybridization (FISH)
cMet gene copy numbers were assessed by FISH using the ZytoLight SPEC MET/CEN7 Dual Color Probe (Zytovision, Bremerhafen, Germany), consisting of an orange fluorochrome direct labeled CEN 7 probe specific for the alpha satellite centromeric region of chromosome 7 and a green fluorochrome labeled SPEC MET probe specific for MET gene. FISH assays were performed as previously described [28]. All samples were visualized and scored using the CytoVision platform (Leica Biosystems). For each specimen, the MET to CEP 7 ratio was determined by counting the green (MET) and orange (CEP 7) signals in a minimum of 100 nuclei. Samples were defined as MET amplified if MET: CEP 7 ratio was >2.0 [29].
Establishment of PDX models and treatment protocol
Five-to-six-week-old female BALB/c nude mice were purchased from Shanghai Slac Laboratory Animal Corporation (Shanghai, China), and housed in a temperature-controlled animal facility on a 12/12 h light/dark schedule with food and water ad libitum. All procedures were performed according to the NIH Guide for Care and Use of Laboratory Animals and were approved by the Institute Animal Care and Use Committee of Zhejiang University [approval ID:SYXK(ZHE)2005-0072]. PDX mouse models were established using fresh CRC tissues surgically removed from patients as previously described [30]. Tumor size was measured using a digital caliper. Tumor volume (mm3) was calculated by the following formula: V= a x b2/2, where V represents the tumor volume, and a and b are the longest and the shortest tumor diameter, respectively. Xenografts from the third generation were used for the experiments, once the tumor volume reached 100-200 mm3. Mice with third generation xenografts were randomized into four groups (5 mice per group): (A) Vehicles, 0.5% (w⁄v) carboxymethyl cellulose, orally gavage, daily; (B) Volitinib, 12.5 mg/kg, oral gavage, daily; (C)Apatinib, 100 mg/kg, oral gavage, daily; and (D) Volitinib, 12.5 mg/kg + Apatinib, 100 mg/kg, oral gavage, daily. Mice were treated for 28 days and monitored twice daily for signs of toxicity, moribund mice were euthanized by CO2 asphyxiation and deaths were recorded. Body weight and subcutaneous tumor size were measured once weekly. Relative tumor growth inhibition (TGI) was calculated using the following formula: (1-T/C) %, where T is relative tumor growth of treated group, and C is relative tumor growth of control group. The mice were sacrificed after 28 days of treatment. Tumors were excised, weighed, and either processed for paraffin embedding or snap-frozen and stored in liquid nitrogen for further experiments.
Mutation analysis by next-generation sequencing (NGS)
Tumor samples were harvested from euthanized mice, and pathologically reviewed to ensure that no significant tumor necrosis had occurred before extraction of DNA. Genomic DNA was extracted using the QIAamp Mini kit (Qiagen, Germany) according to the manufacturer's instructions. The Ion AmpliSeq V2 Cancer Hotspot NGS Panel (Life Technologies, Carlsbad, CA) was used to identify multiple gene mutations (Genechem Inc, Shanghai, China). Ampliseq V2, a commercially available predesigned gene panel, was used to construct sample libraries that cover not only the most commonly tested genes EGFR, KRAS, NRAS and BRAF but also other mutations in genes such as TP53, APC, MET, NOTCH1 and PIK3CA[31].
Fluorescent immunohistochemistry
Tumor-bearing mice were anesthetized and the tumors harvested. Tumor specimens were then fixed in 4% PFA overnight at 4 °C, rinsed several times with PBS, infiltrated with 30% sucrose, frozen in Optimal Cutting Temperature (OCT) compound (Sakura Finetek, Inc., Torrance, CA, USA) and frozen for cryostat sectioning. To evaluate vessel density, 5-µM thick sections of tumor samples were stained with CD31 antibody (Biolegend, San Diego, CA, USA). Sections were mounted using Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were acquired under a fluorescence microscope (BX51; Olympus). For CD31 immunostaining quantification, approximately 6 randomly selected fields of 3 to 4 samples per treatment at x200 magnification were examined in a blind manner using Image J 1.43 freeware (NIH). Results are expressed as the average per treatment of blood vessels number and CD31-positive area ± SD.
TUNEL assay
Apoptosis was assessed in paraffin-embedded xenograft sections of 5 µM in thickness. Apoptotic cells were visualized using the In situ Cell Death Detection Kit (Roche Applied Sciences, Indianapolis, IN, USA) according to the manufacturer's procedures. Slides were imaged and analyzed using Image J in a blind manner. TUNEL-positive apoptotic cells versus total cells were counted in at least five randomly chosen microscopic fields (magnification 20×).
Western blotting
Western blotting was performed with a SDS-PAGE electrophoresis system. Briefly, tissue protein samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk, membranes were incubated overnight at 4℃ with appropriate primary antibodies (Cell Signaling Technology, Danvers, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were detected with an enhanced chemiluminescence detection system (Millipore, Billerica, MA, USA).
Statistical analysis
OS was defined as time from operation to death, irrespective of cause. Survival analysis was done using the Kaplan-Meier method, and the log-rank test was used to compare the difference between survivals. Correlation between cMet expression and clinicopathological factors was estimated by the chi-square test and Fisher exact test. The difference in multiple groups was analyzed by one-way analysis of variance (ANOVA). Stata software version 17.0 (StataCorp, LP) and GraphPad Prism software version 5.0 (GraphPad Software, Inc.) were used, and P< 0.05 was considered statistically significant.
Results
The relationship between cMet expression and clinicopathological features of CRC
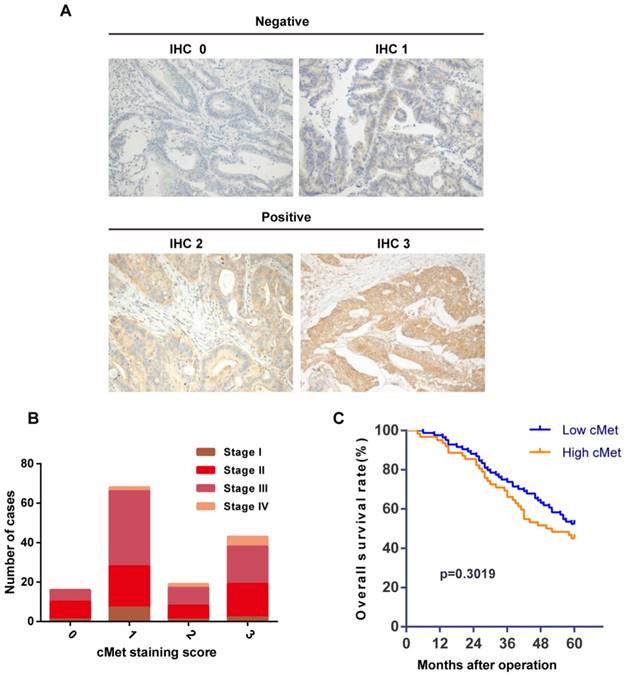
We investigated the relationship between cMet expression and clinicopathological features in a cohort of 146 Chinese CRC patient tumors (Table 1). Immunostaining showed cMet expression in the cytoplasm/membrane of tumor cells (Figure 1A). cMet positive expression (IHC score 2+ or 3+) was found in 62 cases (42.5%) of CRC tumors and negative expression (IHC score 0 or 1+) was detected in 84 cases (57.5%). Significant positive correlations were detected between cMet expression and distant metastasis (Table 1, P=0.037). However, there was no significant correlation between cMet expression with other clinicopathological parameters such as age, gender, tumor location, tumor size, tumor differentiation, or carcinoembryonic antigen (CEA), although there was an increasing trend in cMet expression with advancing TNM stage (Figure 1B).To assess the predictive role of cMet for prognosis, Kaplan-Meier curves with a log-rank test for OS were performed. There was no significant difference in OS between cMet high and cMet low groups after the operation (Figure 1C, log-rank test, P=0.3019).
Correlation between cMet expression and clinicopathological features of 146 CRC patients
| Clinicopathological features | Patients | cMet expression | p-value | |
|---|---|---|---|---|
| n=146 | Low(n=84) | High(n=62) | ||
| Age (years) | 0.251a | |||
| <65 | 74 | 46(62.2%) | 28(37.8%) | |
| ≥65 | 72 | 38(52.8) | 34(47.2%) | |
| Gender | 0.240a | |||
| Male | 95 | 58(61.1%) | 37(38.9%) | |
| Female | 51 | 26(51.0%) | 25(49.0%) | |
| Tumor location | 0.210a | |||
| Right colon | 45 | 22(48.9%) | 23(51.1%) | |
| Left colon | 30 | 16(53.3%) | 14(46.7%) | |
| Rectum | 71 | 46(57.5%) | 25(42.5%) | |
| Tumor size | 0.476a | |||
| <5 cm | 78 | 47(60.3%) | 31(39.7%) | |
| ≥5 cm | 68 | 37(54.4%) | 31(45.6%) | |
| Tumor differentiation | 0.692a | |||
| Well-Moderate | 66 | 39(59.1%) | 27(40.9%) | |
| Moderate-Poor | 54 | 32(59.3%) | 22(40.7%) | |
| Poor-Mucious | 26 | 13(50.0%) | 13(50.0%) | |
| Depth of invasion | 0.239b | |||
| T1-T2 | 13 | 10(76.9%) | 3(23.1%) | |
| T3-T4 | 133 | 74(55.6%) | 59(44.4%) | |
| Lymph node metastasis | 0.340a | |||
| N0 | 71 | 38(53.5%) | 33(46.5%) | |
| N1-2 | 75 | 46(61.3%) | 29(38.7%) | |
| Distant metastasis | 0.037*b | |||
| M0 | 137 | 82(59.9%) | 55(40.1%) | |
| M1 | 9 | 2(22.2%) | 7(77.8%) | |
| TNM stage | 0.839a | |||
| I-II | 65 | 38(58.5%) | 27(41.5%) | |
| III-IV | 81 | 46(56.8%) | 35(43.2%) | |
| CEA dosage | 0.835a | |||
| Unknown | 14 | 7(50.0%) | 7(50.0%) | |
| <5 ng/mL | 67 | 39(58.2%) | 28(41.8%) | |
| ≥5 ng/mL | 65 | 38(58.5%) | 27(41.5%) | |
| Vascular invasion | 0.480a | |||
| Absent | 117 | 69(59.0%) | 48(41.0%) | |
| Present | 29 | 15(51.7%) | 14(48.3%) | |
cMet expression and Kaplan-Meier analysis of overall survival in CRC patients. (A) Representative immunohistochemical staining of cMet in CRC samples. cMet positive expression was defined as IHC 2+ or IHC 3+, and negative expression was defined as IHC 0 or IHC 1+. Magnification: ×200. (B) There was a increasing trend in cMet expression with advancing TNM stage of CRC, although this was not statistically significant. (C) Kaplan-Meier Curve showed no significant difference in the overall survival between patients with low and high cMet expression (log-rank = 1.066, P=0.3019).
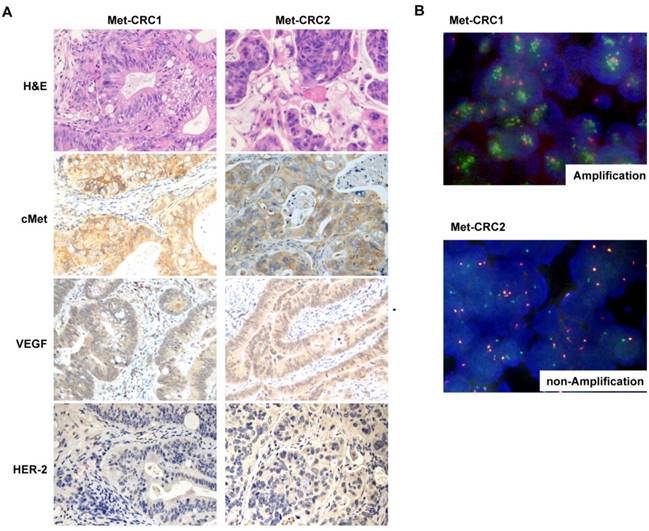
Histological and molecular characterization of Met-CRC1 and Met-CRC2
Tissue sections from two CRC PDX models were examined by cMet IHC and FISH analysis. The Met-CRC1 model was found to harbor cMet overexpression (IHC 3+) and cMet amplification (FISH+), and the Met-CRC2 model showed cMet overexpression (IHC 2+) but no cMet amplification (FISH-) (Figure 2A, 2B). IHC analysis showed that both Met-CRC1 and Met-CRC2 models were positive for VEGF expression but negative for HER-2 expression (Figure 2A).
Histological and molecular characterization of Met-CRC1 and Met-CRC2 xenografts. (A) H&E staining and immunohistochemical staining for cMet, VEGF, and HER2. Positive expression was indicated by brown staining, and the nuclei was counterstained as blue. Magnification, x200. (B) Representative images of FISH. cMet gene copy numbers were evaluated by FISH analysis (green signal, Met; orange signal, CEP7).
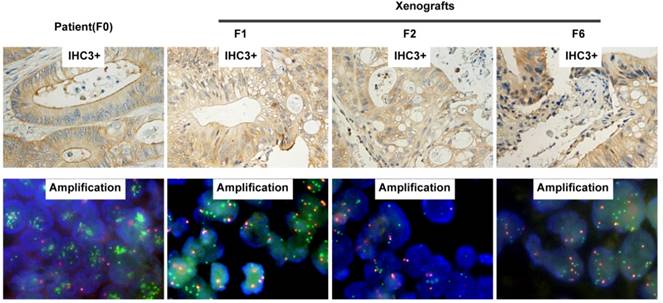
H&E staining of the third generation xenografts of Met-CRC1 and Met-CRC2 showed that overall morphology and tumor differentiation of xenografts were similar to the original patient tumor tissues (Figure 2A). Further analysis showed that cMet overexpression and gene amplification were continuously maintained in all generations of Met-CRC1 model up to the 6th passage (Figure 3), consistent with our previous data[30, 32].
Representative images of cMet expression and amplification by IHC and FISH on patient (F0) and corresponding serial passages of xenograft models of Met-CRC1. For IHC images, magnification is x200.
Genetic characterization of Met-CRC1 and Met-CRC2
To investigate the genetic characteristics of CRC, we used the Ion AmpliSeq Cancer Panel V2 to identify the most frequent mutations in 50 commonly mutated oncogenes and tumor suppressor genes. We analyzed somatic DNA mutations of nine key genes selected for their importance in CRC (Table 2). KRAS mutation (G12D), NOTCH1, TP53 and APC mutation were detected in two CRC PDX models, while NRAS, EGFR, BRAF and MET were wild-type. In addition, mutation in PIK3CA at position Q546K was observed in Met-CRC1 model.
Mutation profiling of the two CRC PDX models
| Samples | Genes involved in EGFR signal | Genes involved in colorectal cancer | |||||||
|---|---|---|---|---|---|---|---|---|---|
| KRAS | NRAS | EGFR | BRAF | PIK3CA | NOTCH1 | MET | TP53 | APC | |
| Met-CRC1 | p.G12D | WT | WT | WT | p.Q546K | p.R1598H | WT | p.P33R | p.R1450X |
| p.S1588N | p.P72R | ||||||||
| p.E1583D | |||||||||
| p.M1580T | |||||||||
| p.M1580L | |||||||||
| Met-CRC2 | p.G12D | WT | WT | WT | WT | p.R1598H | WT | p.R150W | p.C1560X |
| p.S1588N | p.R123W | p.C1578X | |||||||
| p.R243W | |||||||||
| p.P33R | |||||||||
| p.P72R | |||||||||
Anti-tumor activity of volitinib alone or in combination with apatinib in CRC PDX models
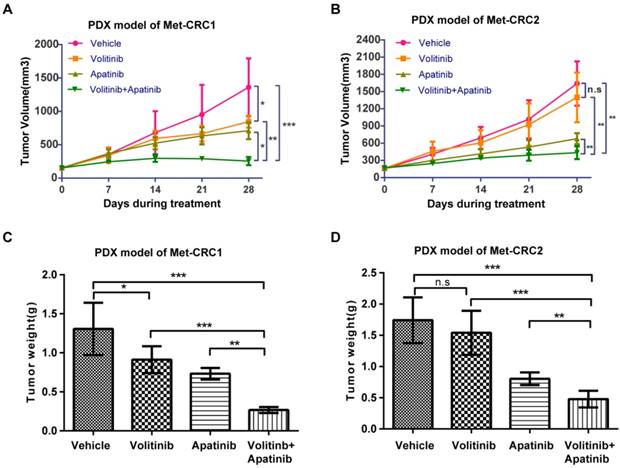
We next evaluated the anti-tumor activity of volitinib alone or in combination with apatinib in PDX models Met-CRC1 and Met-CRC2. Volitinib was orally administered at 12.5 mg/kg daily and apatinib was administered at 100 mg/kg daily for 28 days. 28 days later, in Met-CRC1 which harbored cMet gene amplification, volitinib single-agent treatment only produced mild to moderate tumor growth inhibition compared with vehicle (TGI=37.9%, P< 0.05), but significantly stronger anti-tumor effect was observed in the group receiving combination therapy (TGI=81.3%, Figure 4A). However, in Met-CRC2 which lacked cMet gene amplification, volitinib alone treatment did not induce significant improvement in tumor growth inhibition compared with vehicle (TGI=14.8%, Figure 4B), but combination therapy significantly inhibited tumor growth (TGI=73.4%, P< 0.01). At the end of treatment, tumor weights in the combination group of both models were significantly decreased compared with vehicle (Figure 4C, 4D). Overall, all treatments were well tolerated. No obvious side effects or weight loss was observed during the experimental period in either xenograft model (data not shown).
Anti-tumor efficacy of volitinib alone or in combination with apatinib in Met-CRC1 and Met-CRC2 xenografts. Mice (5 mice/group) were treated with vehicle, volitinib, or apatinib alone or in combination. (A, B) Tumor growth curve of Met-CRC1 and Met-CRC2 xenografts. Each line represents the average tumor volume (mm3) of each treatment group ± SD. (C, D) Endpoint tumor weights of Met-CRC1 and Met-CRC2 xenografts. Each line represents the average tumor weight (g) of each treatment group ± SD. *P< 0.05; **P< 0.01; ***P< 0.001; n.s., no statistical difference (one-way analysis of variance).
Effects of volitinib in combination with apatinib on tumor angiogenesis
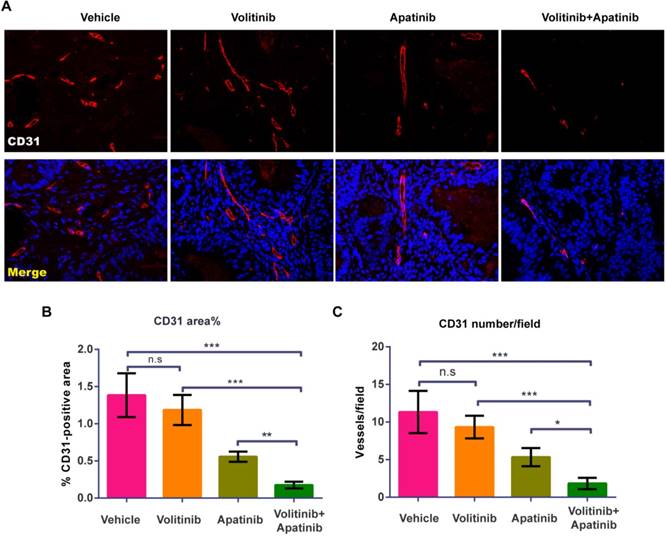
To evaluate the effects of volitinib in combination with apatinib on tumor angiogenesis, we performed CD31 staining on tumors of Met-CRC1. Vasculature was nearly absent in volitinib in combination with apatinib treated group (Figure 5A). Compared with the vehicle treated group, the percentage of vessel areas in tumors was decreased in the combination treated group (1.38 ± 0.30% vs 0.18 ± 0.05%, P< 0.0001) (Figure 5B). In addition, the mean blood vessel number in tumors was reduced by volitinib in combination with apatinib, compared with the vehicle-treated control (11.30 ±2.8 vs 1.83 ± 0.75, P< 0.0001) (Figure 5C).
Volitinib in combination with apatinib significantly inhibited tumor angiogenesis in Met-CRC1 xenografts. (A) Representative images of blood vessels stained with CD31 antibody. Immunofluorescence staining of CD31 (green), the nuclei was stained as blue. Magnification: ×200. (B, C) The percentage of CD31-positive areas per field and averaged blood vessel number per field were determined. Bars, mean±SD. *P< 0.05; **P< 0.01; ***P< 0.001; n.s., no statistical difference (one-way analysis of variance).
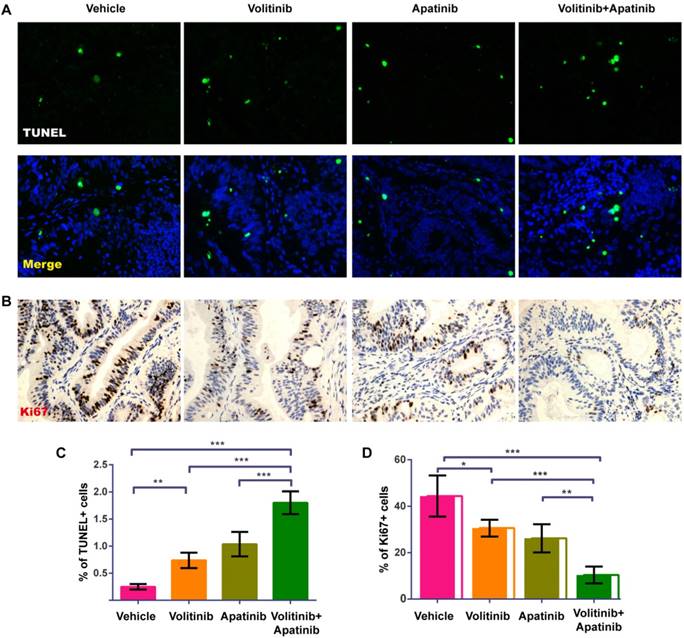
Effects of combined treatment with volitinib and apatinib on CRC apoptosis and proliferation
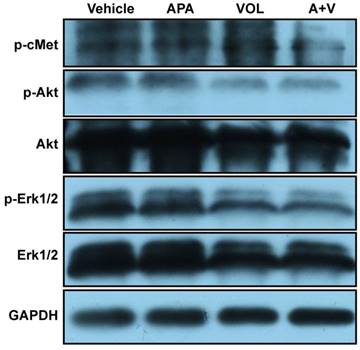
To analyze the effects of combined treatment with volitinib and apatinib on apoptosis and proliferation, we performed TUNEL analysis (Figure 6A) and Ki67 immunostaining (Figure. 6B) on tumors of Met-CRC1. Significantly increased TUNEL staining was found in Met-CRC1 xenografts treated with volitinib alone or in combination with apatinib, compared with vehicle controls (Figure 6C). Significantly decreased expression of Ki67 was also observed in tumors treated with volitinib, apatinib alone or the combination of volitinib and apatinib, compared with vehicle-treated tumors (Figure 6D). These results suggested that the combination of volitinib and apatinib inhibited tumor growth by increasing apoptosis and decreasing cell proliferation. Furthermore, we measured the protein levels of Akt, p-Akt, Erk, p-Erk and p-cMet in Met-CRC1 xenografts harvested at the end of treatment. Combination treatment led to reduced levels of p-cMet, p-Akt and p-Erk, while no significant differences were found in apatinib treatment group (Figure 7).
Volitinib in combination with apatinib significantly increased apoptosis and decreased proliferation in Met-CRC1 xenografts.(A)Representative images of TUNEL-stained (green) sections with DAPI counterstain (blue). Magnification: ×200. (B)Representative images of proliferative cells stained with Ki-67 antibody. Magnification: ×200. (C) Quantitative analysis of apoptotic cells. (D) Quantitative analysis of Ki67 positive cells. Bars, mean±SD. *P< 0.05; **P< 0.01; ***P< 0.001 (one-way analysis of variance).
Volitinib in combination with apatinib reduced the levels of p-cMet, p-Akt and p-Erk in Met-CRC1 xenografts. Mice were treated with vehicle, apatinib (APA), volitinib (VOL) or apatinib and volitinib (A+V). Representative blots showing the levels of indicated proteins in the tumors harvested at the end of treatment. GAPDH was used as a loading control.
Discussion
Anti-VEGF has become an established therapeutic approach to prevent solid tumor growth in patients, and several clinical trials have demonstrated the clinical benefits of anti-VEGF agents in cancers [6, 8]. However, the benefit of anti-VEGF agents in clinical therapy is marginal and transient, and inevitably tumors develop drug resistance [14, 33]. Therefore, the development of new treatment strategies is urgently required.
The cMet pathway is involved in the resistance to VEGF therapy and cMet is overexpressed in some tumors including CRC [20, 29]. To date, several studies have evaluated cMet expression in CRC [17, 34], but the results were inconsistent. In our study, cMet overexpression rate was 42.5% (62/146), similar to the rate reported by Al-Maghrabi et al [35]. Previous studies have demonstrated that cMet was related to metastatic potential and poor prognosis in multiple tumors [20]. However, the role of cMet in CRC has not yet been elucidated. In the present study, cMet overexpression was closely correlated with distant metastases, suggesting that cMet plays a key role in CRC progression. However, no significant association was found between cMet expression and overall survival, a result that was inconsistent with previous reports [36, 37]. This discrepancy may be attributed to the small sample size and the different immunostaining scoring system. In addition, we found that cMet expression was not significantly associated with the depth of invasion, node metastasis, TNM stages, or vascular invasion. In particular, cMet expression tended to be higher with advancing TNM stage of CRC, although this was not statistically significant. These seemingly inconsistent findings may be due to the small sample size of this study.
The aggressive nature of cMet overexpressing CRC and the lack of an appropriate in vivo model for preclinical studies make CRC clinically challenging. PDX, so-called Avatar models [38], have been increasingly and widely used in various types of cancers for translational research in recent years [39]. Our group previously established PDX models of colon and gastric carcinoma and successfully used them to evaluate novel anti-cancer drugs [30, 32]. Using these established CRC PDX models, we performed IHC and FISH analysis and found thatMet-CRC1 model had both cMet overexpression and cMet gene amplification, while Met-CRC2 model had cMet overexpression but no cMet gene amplification. Detailed NGS data analysis confirmed that both CRC PDX models had KRAS mutation. Interestingly, cMet overexpression was detected in clinical samples of EGFR-resistant lung cancer and VEGF-resistant glioblastoma multiforme patients [33, 40]. cMet has been shown to facilitate resistance to anti-VEGF by promoting the growth, survival and metastasis of tumor cells [14, 21]. Combination therapy strategy of cMet inhibitors and other signal transduction inhibitors may be a feasible approach to overcome drug resistance.
Volitinib (AZD6094, Savolitinib) is a highly selective inhibitor against cMet and is currently being evaluated in clinical trials in a number of tumor types [23, 24]. In this study, we evaluated the anti-tumor effects of volitinib alone or in combination with apatinib in CRC PDX models. Our results showed that the combination treatment significantly decreased the growth of tumors of both PDX models regardless of the status of cMet amplification. However, only in cMet amplification Met-CRC1 xenografts, volitinib alone treatment induced significant improvement in tumor growth inhibition compared with vehicle. These differences in efficacy implied that the amplification of cMet may serve as a biomarker to identify sensitivity to volitinib treatment in CRC. Immunohistochemical analysis demonstrated that combination treatment significantly reduced microvessel density, suppressed proliferation, and increased apoptosis compared to either volitinib or apatinib alone, indicating that the observed synergistic effect might be attributed to the dual inhibition on tumor signaling and tumor microenvironment. To our knowledge, this is the first report on synergistic antitumor effects of the combination of cMet inhibitor and anti-VEGF in preclinical colorectal cancer models.
The molecular mechanisms of the synergistic effect of combination treatment remain to be elucidated. One explanation is the potential vascular normalization effect of anti-angiogenic targeted therapy in solid tumors as previously described [41, 42], which may improve both the delivery and effectiveness of concurrent therapies. However, this concept of vascular normalization needs to be validated in clinical trials. Another explanation could involve vessel pruning, hypoxia, crosstalk pathway, and/or increased expression of cMet upon anti-angiogenic treatment [14, 21]. In present study, we detected reduced levels of p-Erk and p-Akt following combination treatment with volitinib and apatinib. Thus we speculated that co-targeting cMet and VEGFR2 may exhibit a synergetic tumor inhibition through inhibiting both MAPK-Erk and PI3K-Akt pathways.
However, it is worth noting that cMet positive expression rate was only 42.5% in our cohorts of CRC patients. Therefore, therapeutically targeting cMet may only benefit a small portion of CRC patients. Further studies are needed to develop additional new targets for effective treatments of CRC patients.
In conclusion, we observed that cMet was widely expressed in Chinese CRC patients, and cMet overexpression was associated with distant metastasis of CRC. Furthermore, we reported the potential anti-tumor activity of a novel cMet inhibitor volitinib for the blockade of cMet alone and the benefit for combined treatment with anti-VEGF in CRC PDX models. The status of cMet amplification may be a promising biomarker for predicting the response to volitinib treatment. Our data provide support for a rationale to targeting both cMet and VEGF to achieve better therapeutic effect in cMet overexpressing CRC.
Abbreviations
CRC: Colorectal cancer; IHC: Immunohistochemistry; PDX: Patient-derived xenograft; VEGF: Vascular endothelial growth factor; OS: Overall survival; HIF-1α: Hypoxia-inducible factor-1α; EGFR: Epidermal growth factor receptor; HE: Hematoxylin and eosin; PFA: Para-formaldehyde; FISH: Fluorescence in situ hybridization; TGI: Tumor growth inhibition; NGS: Next-generation sequencing; OCT: Optimal cutting temperature; PVDF: Polyvinylidene difluoride; ANOVA: One-way analysis of variance; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; CEA: Carcinoembryonic antigen.
Acknowledgements
This work was supported by National Science and Technology Major Project of the Ministry of Science and Technology of China (Grant No. 2013ZX09506015), National Natural Science Foundation of China (Grant No. 81272676), National Natural Science Foundation of China (Grant No. 81374014).
Author Contributions
Lisong Teng and Jiangsheng Huang conceived and designed the study; Xiangheng Chen, Zhonghai Guan, Jun Lu and Haohao Wang performed the experiment; Zhongkun Zuo and Fei Ye prepared the patient samples; Xiangheng Chen analyzed the data and wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F. et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663-71
3. Bertelsen CA, Neuenschwander AU, Jansen JE, Wilhelmsen M, Kirkegaard-Klitbo A, Tenma JR. et al. Disease-free survival after complete mesocolic excision compared with conventional colon cancer surgery: a retrospective, population-based study. Lancet Oncology. 2015;16:161-8
4. Pawlik TM, Choti MA. Surgical therapy for colorectal metastases to the liver. J Gastrointest Surg. 2007;11:1057-77
5. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579-91
6. Grothey A, Cutsem EV, Sobrero A, Siena S, Falcone A, Ychou M. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303-12
7. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T. et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499-506
8. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42
9. Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y. et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102:1374-80
10. Scott AJ, Messersmith WA, Jimeno A. Apatinib: a promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs Today (Barc). 2015;51:223-30
11. Li J, Qin S, Xu J, Guo W, Xiong J, Bai Y. et al. Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial. J Clin Oncol. 2013;31:3219-25
12. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8): 592-603. Nat Rev Cancer. 2008;8:592-603
13. Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15:167-70
14. Keunen O, Klein G. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011;108:3749-54
15. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF. et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802-4
16. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89-103
17. Gayyed MF, Abd El-Maqsoud NM, El-Hameed El-Heeny AA, Mohammed MF. c-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J Gastrointest Oncol. 2015;6:618-27
18. Lee HE, Kim MA, Lee HS, Jung EJ, Yang HK, Lee BL. et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer. 2012;107:325-33
19. Luo HY, Xu RH. Predictive and prognostic biomarkers with therapeutic targets in advanced colorectal cancer. World J Gastroenterol. 2014;20:3858-74
20. Miyamoto M, Ojima H, Iwasaki M, Shimizu H, Kokubu A, Hiraoka N. et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer. 2011;105:131-8
21. Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V. et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2:270-87
22. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G. et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658-73
23. Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W. et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9:323-33
24. Schuller AG, Barry ER, Jones RD, Henry RE, Frigault MM, Beran G. et al. The MET Inhibitor AZD6094 (Savolitinib, HMPL-504) Induces Regression in Papillary Renal Cell Carcinoma Patient-Derived Xenograft Models. Clin Cancer Res. 2015;21:2811-9
25. Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr. et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:4105-14
26. Li C, Wang L, Zheng L, Zhan X, Xu B, Jiang J. et al. SIRT1 expression is associated with poor prognosis of lung adenocarcinoma. Onco Targets Ther. 2015;8:977-84
27. Liu YJ, Shen D, Yin X, Gavine P, Zhang T, Su X. et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer. 2014;110:1169-78
28. Lacroix L, Post SF, Valent A, Melkane AE, Vielh P, Egile C. et al. MET genetic abnormalities unreliable for patient selection for therapeutic intervention in oropharyngeal squamous cell carcinoma. PloS one. 2014;9:e84319
29. Peng Z, Li Z, Gao J, Lu M, Gong J, Tang ET. et al. Tumor MET Expression and Gene Amplification in Chinese Patients with Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer. Mol Cancer Ther. 2015;14:2634-41
30. Guan Z, Chen X, Jiang X, Li Z, Yu X, Jin K. et al. Establishing a patient-derived colorectal cancer xenograft model for translational research. Int J Clin Exp Med. 2016;9(11):21346-21357
31. Rothe F, Laes JF, Lambrechts D, Smeets D, Vincent D, Maetens M. et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25:1959-65
32. Lu J, Li G, He K, Jiang W, Xu C, Li Z. et al. Luteolin exerts a marked antitumor effect in cMet-overexpressing patient-derived tumor xenograft models of gastric cancer. J Transl Med. 2015;13:42
33. Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K. et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773-83
34. Takeuchi H, Bilchik A, Saha S, Turner R, Wiese D, Tanaka M. et al. c-MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res. 2003;9:1480-8
35. Al-Maghrabi J, Emam E, Gomaa W, Saggaf M, Buhmeida A, Al-Qahtani M. et al. c-MET immunostaining in colorectal carcinoma is associated with local disease recurrence. BMC Cancer. 2015;15:676
36. Gao H, Guan M, Sun Z, Bai C. High c-Met expression is a negative prognostic marker for colorectal cancer: a meta-analysis. Tumour Biol. 2015;36:515-20
37. Voutsina A, Tzardi M, Kalikaki A, Zafeiriou Z, Papadimitraki E, Papadakis M. et al. Combined analysis of KRAS and PIK3CA mutations, MET and PTEN expression in primary tumors and corresponding metastases in colorectal cancer. Mod Pathol. 2013;26:302-13
38. Malaney P, Nicosia SV, Dave V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014;344:1-12
39. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer discovery. 2014;4:998-1013
40. Gou LY, Li AN, Yang JJ, Zhang XC, Su J, Yan HH. et al. The coexistence of MET over-expression and an EGFR T790M mutation is related to acquired resistance to EGFR tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget. 2016;7:51311-51319
41. Vangestel C, Van de Wiele C, Van Damme N, Staelens S, Pauwels P, Reutelingsperger CP. et al. (99)mTc-(CO)(3) His-annexin A5 micro-SPECT demonstrates increased cell death by irinotecan during the vascular normalization window caused by bevacizumab. J Nucl Med. 2011;52:1786-94
42. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205-18
Author contact
![]() Corresponding author: Lisong Teng, email: cw9771edu.cn; Jiangsheng Huang, email: HJS13907313501com.
Corresponding author: Lisong Teng, email: cw9771edu.cn; Jiangsheng Huang, email: HJS13907313501com.