Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(7):1267-1276. doi:10.7150/jca.24013 This issue Cite
Research Paper
Increased AURKA Gene Copy Number Correlates with Poor Prognosis and Predicts the Efficacy of High-dose Interferon Therapy in Acral Melanoma
Junya Yan*, Jiayi Yu*, Xiaowen Wu, Tianxiao Xu, Huan Yu, Jie Dai, Meng Ma, Huan Tang, Longwen Xu, Zhihong Chi, Lu Si, Xinan Sheng, Chuanliang Cui, Yan Kong ![]() , Jun Guo
, Jun Guo ![]()
Key laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Renal cancer and Melanoma, Peking University Cancer Hospital & Institute, Beijing, China.
* contributed equally to this work.
Received 2017-11-25; Accepted 2018-2-28; Published 2018-3-15
Abstract
Background: AURKA kinase is an essential serine/threonine kinase for mitosis and chromosome stability. The aberrant amplification and overexpression of AURKA are commonly observed in various types of cancer, including cutaneous melanoma. However, the status and the clinical significance of AURKA copy number (CN) in acral melanoma (AM) have not been fully elucidated.
Methods: Four hundred and seventy-two AM samples were included in the study. AURKA CN was examined using the QuantiGenePlex DNA Assay. We analysed the relationship of AURKA CN to clinicopathological characteristics and survival of patients with AM.
Results: In this study, AURKA copy gain (set as more than 2.0 copies) was detected in 24.6% (116/472) of the samples. We did not observe any obvious correlation between clinicopathological characteristics and AURKA copy gain of the patients. However, patients with AURKA copy gain had a significantly shorter overall survival time (OS) and progression-free survival time (PFS) than those with normal AURKA CN (OS: P = 0.022; PFS: P < 0.001). Furthermore, multivariate Cox regression analysis showed that AURKA copy gain was an independent poor prognostic factor for patients with AM undergoing adjuvant interferon therapy.
Conclusions: This study suggested that AURKA copy gain is an adverse prognostic factor for AM. Furthermore, AURKA copy gain may be a useful biomarker to predict the outcome of interferon therapy in patients with AM.
Keywords: acral melanoma, AURKA copy number, interferon, prognosis
Introduction
Acral melanoma (AM) is a rare subtype of melanoma in Caucasians, which accounts for only 5% [1]. However, the proportion of AM is more than 50% in the Asian populations [2]. For patients in stage II/III of AM, surgical resection and high-dose interferon (HD-IFN) adjuvant treatment, are the main therapeutic regimens [3]. However, validated molecular biomarkers for predicting outcomes in patients with AM undergoing HD-IFN adjuvant therapy are lacking. For patients in stage IV of AM, standard chemotherapy (dacarbazine) cannot significantly improve overall survival (OS) [4]. Recently, individualized targeted therapy targeting BRAF and KIT mutations and anti-checkpoint immunotherapy have improved the clinical outcome of patients with metastatic AM [5-8]. However, the frequencies of BRAF and KIT mutations in Chinese patients with AM are 15.5% and 11.9% [9, 10], respectively, which leaves more than 70% patients without validated targeted therapy. Furthermore, anti-immune checkpoint drugs against PD-1/PD-L1 and CTLA-4 did not showed high overall response rate (ORR) because of lack of proper predictive biomarkers [11, 12]. Therefore, identification and characterization of molecular prognostic biomarkers and their application in combination with traditional treatment are urgently required.
AURKA (Aurora kinase A) belongs to the Aurora kinase family, and encodes an evolutionarily conserved serine/threonine kinase. During mitosis, AURKA is involved in centrosome maturation and separation, as well as bipolar spindle formation, ensuring that chromosomes are properly segregated and cytokinesis is smoothly executed. Therefore AURKA plays an essential role in cell cycle. Overexpression and aberrant amplification of AURKA have been observed in several types of cancers, such as cancers of the breast, colon, ovary, and cutaneous melanoma [13-16]. Recent studies have shown that AURKA amplification and overexpression are associated with poor prognosis in diverse cancers [17-19]. Considering this, a series AURKA kinase inhibitors (AKIs) have been applied both in vivo and in vitro. More importantly, certain AKIs have yielded positive results in clinical trials [20-22]. A phase II clinical trial using alisertib, an investigational AURKA inhibitor, in patients with multiple advanced solid tumors showed that nine of 49 (18%) women with breast cancer and ten of 48 (21%) participants with small-cell lung cancer demonstrated a partial response [20]. However, it remains to be determined whether patients with AURKA copy gain will respond better to AURKA inhibitors.
A previous study showed that AURKA copy gain was detected in 37.5% (6/16) patients with AM [23]. However, the data was mainly derived from a small-cohort of Caucasian patients and was not verified in Asian patients with AM. More importantly, large-scale screening of AURKA copy gain in AM samples has not been reported.
In this study, we collected 472 melanoma tissue from patients with AM and examined the CN of AURKA. Thus, this study represents the first systematic analysis of AURKA CN in patients with AM. Survival analysis revealed that AURKA copy gain was significantly correlated with clinical outcome, indicating that AURKA copy gain could be an independent predictive factor for prognosis in patients with AM treated with HD-IFN.
Materials and Methods
Study populations
From January 2005 to December 2015, 472 patients with pathological diagnosis of AM were recruited for this retrospective study from the Peking University Cancer Hospital. Two hundred and fifty-six out of 472 patients received HD-IFN adjuvant treatment as follows: 15 × 106 U/m2 d1-5/w × 4w + 9 × 106 U tiw × 48w. All clinical and pathological data were obtained after reviewing medical records, including age, sex, TNM (tumour-node-metastases) stage, thickness (Breslow), ulceration and survival (follow-up persisted until March 2017, or until the missing of follow-up or death of patients). Written informed consent was obtained from all participants. This study was approved by our Institutional Review Board and was conducted according to the Declaration of Helsinki Principles.
QuantiGenePlex DNA assay
Tissue homogenates were prepared using the QuantiGene sample processing kit for 472 (1 sample per patient) formalin-fixed, paraffin-embedded tissues (FFPE; Panomics of Affymetrics, Santa Clara, CA, USA) according to the manufacturer's instructions. Briefly, 5 to 8 pieces of deparaffinized sections (4-10 μm) were incubated with 150 μl homogenizing solution supplemented with 1.5 μl proteinase K (50 μg/μl) at 65°C for 6 h. The tissue homogenate was separated from debris by brief centrifugation and transferred to a new tube.
The branched DNA (bDNA) assay was performed using the QuantiGenePlex DNA kit (Panomics) according to the manufacturer's instructions. Briefly, the homogenate DNA was sheared using Covaris S2 (Covaris, Woburn, MA, USA) with the following settings: duty cycle 5%, intensity 3, cycles/burst 200, 80 s. For each assay well, 40 μl homogenate was denatured with 2.5 M NaOH (final concentration 0.18 M) in the presence of a DNA probe. The neutralized tissue homogenate was transferred to each well of hybridization plates containing the working bead mix. All samples were in processed in duplicates. The hybridization plate was sealed and incubated at 54°C ± 1°C in shaking incubator (600 rpm) for 18-22 h. The unbound samples were washed away using the Bio-plex Pro II wash station (Bio-Rad, Hercules, CA, USA). Then, the beads were sequentially hybridized with DNA pre-amplifier, DNA amplifier, labelled probe and SAPE (streptavidin-conjugated R-phycoerythrin). Fluorescence intensities were measured using the Bio-plex 100 system (Bio-Rad).
The mean fluorescence intensities of the duplicates were calculated for all genes. Background values were subtracted from each probe set signal. Values of tested genes were normalized to the geometric means of RPPH1, RPP30 and RPLP0. For each test gene, the normalized signal was divided by the signal of the reference DNA sample (G1521, Promega, Madison, WI, USA), and the values were multiplied by the known copy number (usually 2 copies) of each gene in the reference genome.
Immunohistochemistry
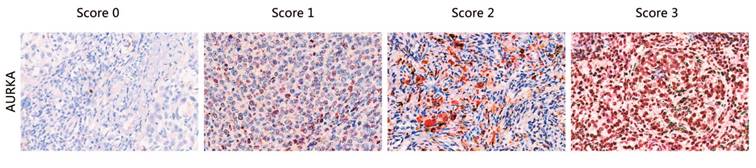
FFPE tissue sections were examined by immunohistochemistry (IHC) using a polyclonal antibody against AURKA (Cell Signaling). A standard Strept-avidin horseradish immunoperoxidase method was used for AURKA staining. Primary antibodies were diluted in buffer containing 10% normal goat serum. The tissue sections were deparaffinized with Xylene for 30 min and rehydrated in decreasing concentrations of ethanol. Endogenous peroxidases were blocked with 30% H2O2 diluted in phosphate-buffered saline (PBS) for 15 min. For antigen retrieval, slides were heated in a pressure cooker in EDTA (pH 8.5) for 2 min and 30 s, followed by cooling to room temperature (RT) in the same buffer. For antigen blocking, the slides were blocked with normal goat serum with 1 h. After washing, slides were incubated with the primary antibody overnight at 4℃ (dilution 1:500). Three 5-minute washes in buffer were conducted after each incubation. The slides were then incubated with secondary antibody, anti-rabbit/mouse antibody (DAKO) (30 min at RT), followed by staining with AEC for 5-30 min at RT until coloration was achieved, counterstaining with hematoxylin followed by staining, and sealing with water-soluble encapsulating agent. Staining intensity and percentage were independently scored by three pathologists as 0, 1, 2, and 3 (“0” as negative, and “1”, “2” and “3” as positive; typical staining is shown in Fig. 1), without the knowledge of the copy number variations in the sample.
Typical staining of AURKA proteins. The staining score for each sample, counting the intensity and density of the staining, was graded as 0, 1, 2, and 3 (“0” as negative, and “3” as the strongest; or “0” as negative, and “1”, “2” and “3” as positive).
RNA extraction and RT-RCR analysis
Total RNA was isolated from FFPE tissue sections using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer's instructions. RNA was quantified by measuring the A260/A280 absorbance rations (Nano-Drop Technologies, Wilmington, DE). RNA quality was evaluated using a 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). Total RNA was reverse transcribed to cDNA using a ReverTra Ace qRCR RT kit (TOYOBO), according to the manufacturer's protocol. AURKA mRNA level was evaluated by RT-PCR on a 7500 Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) using SYBR Green (Sangon Biotech, Shanghai, China) according to the manufacturer's instructions. Forward and reverse primer sequences were as follows: AURKA, 5'-ATTGCAGATTTTGGGTGGT-3' and 5'-AAACTTCAGTAGCATGTTCCTGTC-3'; GAPDH (internal control), 5'-GAAGGTGAAGGTCGGAGTC-3' and 5'-GAAGATGGTGATGGGATTTC-3'. The delta-Ct method was used to determine relative gene expression level.
Statistical analysis
The association between AURKA copy gain and clinicopathlogic characteristics were analysed using the Pearson's χ2 test or Fisher exact test. For the survival analyses, OS and progression-free survival (PFS) were analysed using the Kaplan-Meier method with the log-rank test for assessing differences in survival probability between groups. For patients with AM treated with HD-IFN, Kaplan-Meier estimates of time-to-event relapse-free survival (RFS) were calculated. Log-rank tests were used to estimate the statistical significance between the time-dependent outcomes of RFS. The Cox regression model was used to evaluate the prognostic factors. Two-sided P values less than 0.05 were considered to be statistically significant. All statistical analyses were performed using the SPSS 16.0 software.
Results
Patient characteristics
Four hundred and seventy-two patients with AM were included in this study and their clinicopathological characteristics are listed in Table 1. The patients, 245 males (51.9%) and 227 females (48.1%), were aged 15 to 92 years (median age, 55.0 years). According to the American Joint Committee on Cancer classification, tumour thickness was classified as follow: < 1 mm in 13 (2.8%) patients, 1-2 mm in 82 (17.4%) patients, 2-4 mm in 166 (35.2%) patients, and > 4 mm in 193 (41.0%) patients. Three hundred and twenty (67.8%) patients showed ulceration at the time of diagnosis. Histological examination was performed on formalin-fixed tissues in all case, and tumours were diagnosed and classified according to the TNM classification. Briefly, there were 47 (10.0%) case of stage I, 145 (30.6%) cases of stage II, 123 (26.1%) cases of stage III, and 157 (33.3%) cases of stage IV.
Relationship between AURKA CN and patients' clinicopathological characteristics.
| Variable | Calculated AURKA CN in tumor tissue | Patient number with calculated AURKA CN | ||||
|---|---|---|---|---|---|---|
| n (%) | Median | P value | ≤ 2.0 | > 2.0 | P value | |
| n (%) | n (%) | |||||
| Total | 472 | 1.67 | 356 (75.4) | 116 (24.6) | ||
| Age (years) | ||||||
| < 55 | 229 (48.5) | 1.71 | 0.226* | 172 (75.1) | 57 (24.9) | 0.878+ |
| ≥ 55 | 243 (51.5) | 1.61 | 184 (75.7) | 59 (24.3) | ||
| Gender | ||||||
| Female | 227 (48.1) | 1.69 | 0.426* | 167 (73.6) | 60 (26.4) | 0.367+ |
| Male | 245 (51.9) | 1.63 | 189 (77.1) | 56 (22.9) | ||
| Ulceration | ||||||
| Yes | 320 (67.8) | 1.68 | 0.243* | 239 (74.7) | 81 (25.3) | 0.305+ |
| No | 139 (29.4) | 1.63 | 110 (79.1) | 29 (20.9) | ||
| Unknown | 13 (2.8) | |||||
| Thickness | ||||||
| < 1 mm | 13 (2.8) | 1.79 | 0.538# | 10 (76.9) | 3 (23.1) | 0.550+ |
| 1-2 mm | 82 (17.4) | 1.73 | 66 (80.5) | 16 (19.5) | ||
| 2-4 mm | 166 (35.2) | 1.6 | 129 (77.7) | 37 (22.3) | ||
| >4 mm | 193 (41.0) | 1.67 | 141 (73.1) | 52 (26.9) | ||
| Unknown TNM stages | 18 (3.8) | |||||
| I | 47 (10.0) | 1,74 | 0.462# | 40 (85.1) | 7 (14.9) | 0.188+ |
| II | 145 (30.6) | 1.71 | 107 (73.8) | 38 (26.2) | ||
| III | 123 (26.1) | 1.63 | 97 (78.9) | 26 (21.1) | ||
| IV | 157 (33.3) | 1.63 | 112 (71.3) | 45 (28.7) | ||
*Wilcoxon rank-sum test.
# Kruskal-Wallis rank test.
+Pearson chi-squared test.
AURKA CN
The median AURKA CN in patients with AM was 1.67 (ranged from 0.22 to 8.04), which was not significantly affected by analysed clinicopathological characteristics (Table 1). Using 2.0 copies as the cut-off in AURKA CN evaluation, gene copy gain was detected in 116 cases (24.6%), including 98 patients with 2.0 to 2.99 AURKA copies and the remaining 18 patients with 3.0 to 8.04 copies (data not shown).
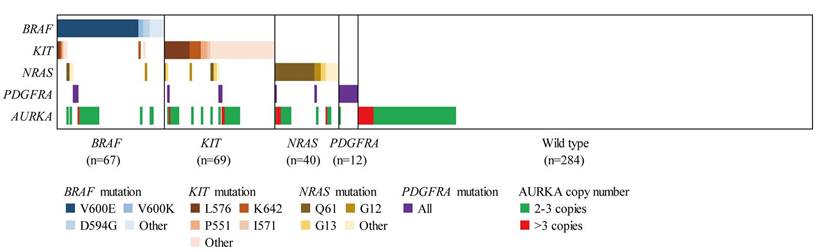
In addition, we analysed the correlation of AURKA copy gain to the mutations of therapeutic targets (including NRAS, BRAF, KIT and PDGFRA) in AM (Table 2 and Fig. 2). Among the 116 AM samples with AURKA copy gain, 17 (15.7%), 18 (16.5%), 22 (20.2%) and 4 (3.7%) showed mutations of NRAS, BRAF, KIT, and PDGFRA, respectively. This indicated that AURKA copy gain may not be mutually exclusive to mutations in the well-established target genes. Further analysis of the incidence of NRAS, BRAF, KIT and PDGFRA mutation in the patients of our cohort showed no significant differences in gene mutation frequency between patients with and without AURKA copy gain.
Associations between AURKA CN and NRAS, BRAF, KIT, and PDGFRA gene mutation status.
| Gene status | Total | AURKA CN | ||
|---|---|---|---|---|
| ≤2.0 | >2.0 | P value | ||
| n (%) | n (%) | n (%) | ||
| NRAS mutations | ||||
| Yes | 53 (11.2) | 36 (67.9) | 17 (32.1) | 0.178* |
| No | 386 (81.8) | 295 (76.4) | 91 (23.6) | |
| Unknown | 33 (7.0) | |||
| BRAF mutations | ||||
| Yes | 67 (14.2) | 49 (73.1) | 18 (26.9) | 0.608* |
| No | 380 (80.5) | 289 (76.1) | 91 (23.9) | |
| Unknown | 25 (5.3) | |||
| KIT mutations | ||||
| Yes | 77 (16.3) | 55 (71.4) | 22 (28.6) | 0.381* |
| No | 365 (77.3) | 278 (76.2) | 87 (23.8) | |
| Unknown | 30 (6.4) | |||
| PDGFRA mutations | ||||
| Yes | 20 (4.2) | 16 (80.0) | 4 (20.0) | 0.629* |
| No | 424 (89.8) | 319 (75.2) | 105 (24.8) | |
| Unknown | 28 (6.0) | |||
*Pearson chi-squared test.
AURKA copy gain and MAPK pathway gene mutation pattern of AM (n=472). Patients harboring at least one gene mutation of BRAF, KIT, NRAS, PDGFRA and AURKA copy gain are presented.
Association between AURKA CN and AURKA expression levels
To determine the association between AURKA CN and AURKA expression levels, FFPE tissue sections from 124 patients with AM were examined using IHC (Table 3). Of the 124 samples analysed, the overall rate of detection of AURKA was 41.1%. For the 28 cases with AURKA copy gain, the detection rate for AURKA was 60.7%, which was significantly higher than that (35.4%) in cases (34/96) with normal AURKA CN.
Correlation of AURKA CN to AURKA expression.
| IHC scorea | AURKA CN | |
|---|---|---|
| > 2.0 | ≤ 2.0 | |
| 0 | 11 | 62 |
| 1 | 10 | 21 |
| 2 | 5 | 8 |
| 3 Positive/total | 2 17/28 | 5 34/96 |
| Positive rate (%) | 60.7 | 35.4 |
| P valueb | 0.017c | - |
Abbreviations: CN, copy number; IHC, immunohistochemistry.
a. Staining intensity and percentage were independently scored by three pathologists as 0, 1, 2, and 3 (“0” as negative, and “1”, “2” and “3” as positive).
b Significance evaluated by chi-square tests.
c Positive rate of AURKA in cases with AURKA copy gain (CN > 2.0) versus that in cases with normal AURKA CN (CN ≤ 2.0).
In addition, we also examined AURKA mRNA expression levels of 124 AM patients. Relative AURKA mRNA expression ranged from 0.0298 to 8.574 (median, 0.219). When we dichotomized AURKA CN into gain and normal, AURKA mRNA expression level was higher in patients with AURKA copy gain than with normal AURKA CN (median mRNA: 0.155 vs. 0.645, P = 0.022; Fig. S1a). Furthermore, dividing into two subgroups according to the protein expression levels (negative and positive), AURKA mRNA median levels were 0.130 and 0.412, respectively. There was significant correlation between AURKA mRNA and protein expression (P = 0.014; Fig. S1b). These findings indicated that the gain in AURKA CN correlates with high levels of AURKA expression.
Prognostic significance of AURKA copy gain for overall survival and progression-free survival of patients with AM
We analysed the prognostic significance of AURKA copy gain for OS and PFS. Survival data were collected for patients (n = 472) with AM. The median follow-up period was 29.3 (range: 0.9-136.0) months. We found that the median OS for patients with AURKA copy gain was significantly shorter than that for patients with normal AURKA CN (48.5 vs. 59.8 months; P = 0.022; Fig. 3a). In addition, patients with AURKA copy gain had a significantly shorter median PFS compared to those with normal AURKA CN (13.3 vs. 30.0 months; P < 0.001; Fig. 3b).
Kaplan-Meier analysis of overall survival and progression-free survival of AM patients according to clinical stage and AURKA CN: (a and b) for the overall AM patients group, (c and d) for the patients with stage I/II group, and (e and f) for the patients with stage III/IV group.
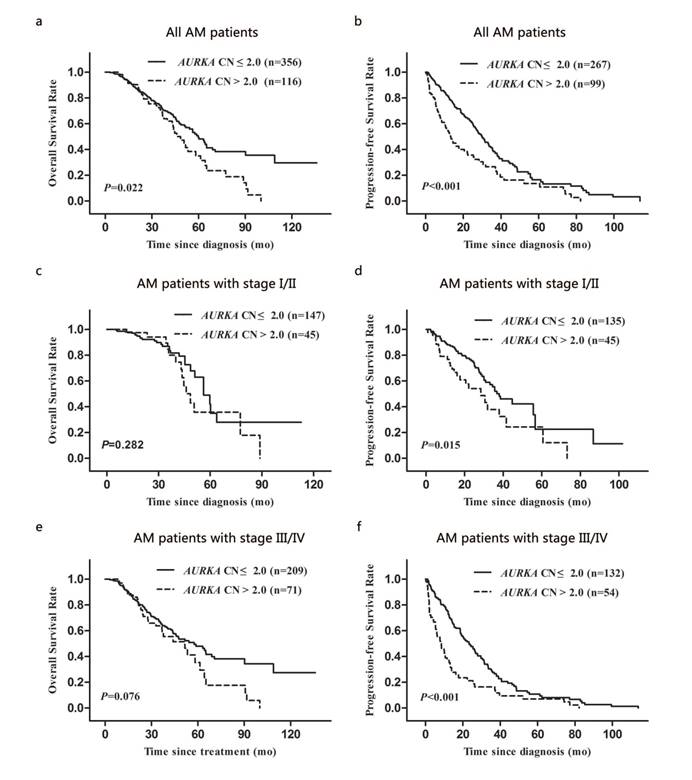
To assess the prognostic value of AURKA copy gain during different clinical stages, we performed a stratified analysis by TNM stage (I/II and III/IV). Patients with AURKA copy gain had significantly shorter periods of PFS than those with normal AURKA CN at stage I/II and stage III/IV (P = 0.015 and P < 0.001, respectively; Fig. 3d and 3f). However, the status of AURKA copy gain was not associated with OS in stage I/II and stage III/IV (P = 0.282 and P = 0.076, respectively; Fig. 3c and 3e).
Multivariate Cox regression analysis revealed that AURKA copy gain was an independent prognostic factor for PFS (HR: 1.98; 95% CI: 1.50-2.61; P < 0.001; Table 4). Among other clinicopathological factors, clinical stage was an independent prognostic factor for OS and PFS (Table 4).
Univariate and multivariate analysis of prognostic factors for OS and PFS of patients with AM.
| Variable | OS | PFS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||||||
| HR | 95% CI* | P value | HR | 95% CI | P value | HR | 95% CI | P value | HR | 95% CI | P value | |
| Age (<55 years/≥55 years) | 1.10 | 0.80-1.48 | 0.591 | 0.99 | 0.77-1.28 | 0.929 | ||||||
| Gender (female/male) | 1.04 | 0.76-1.43 | 0.788 | 1.15 | 0.89-1.49 | 0.274 | ||||||
| Ulceration (yes/no) | 1.11 | 0.79-1.56 | 0.557 | 1.22 | 0.93-1.62 | 0.154 | ||||||
| Thickness (≤ 2 mm/>2 mm) | 0.80 | 0.57-1.12 | 0.196 | 0.83 | 0.63-1.09 | 0.181 | ||||||
| TNM stage (I + II/III +IV) | 0.62 | 0.45-0.86 | 0.004 | 1.58 | 1.10-2.27 | 0.012 | 0.47 | 0.37-0.61 | <0.001 | 2.21 | 1.68-2.89 | <0.001 |
| AURKA CN (gain/normal) | 1.54 | 1.06-2.22 | 0.022 | 0.094 | 2.18 | 1.58-3.01 | <0.001 | 1.98 | 1.50-2.61 | <0.001 | ||
| NRAS mutations (yes/no) | 1.81 | 1.12-2.92 | 0.015 | 1.65 | 1.10-2.47 | 0.015 | 1.25 | 0.81-1.94 | 0.313 | |||
| BRAF mutations (yes/no) | 0.78 | 0.52-1.18 | 0.241 | 0.99 | 0.69-1.43 | 0.954 | ||||||
| KIT mutations (yes/no) | 1.36 | 0.88-2.09 | 0.166 | 1.37 | 0.93-2.01 | 0.11 | ||||||
| PDGFRA mutations (yes/no) | 1.24 | 0.54-2.84 | 0.616 | 1.12 | 0.59-2.11 | 0.737 | ||||||
HR, hazard ratio; CI, confidence interval; OS, overall survival; PFS, progression-free survival; TNM, tumor-node-metastases.
The bold values refer to P < 0.05.
Predictive value of AURKA copy gain for outcomes in patients with AM undergoing high-dose interferon therapy
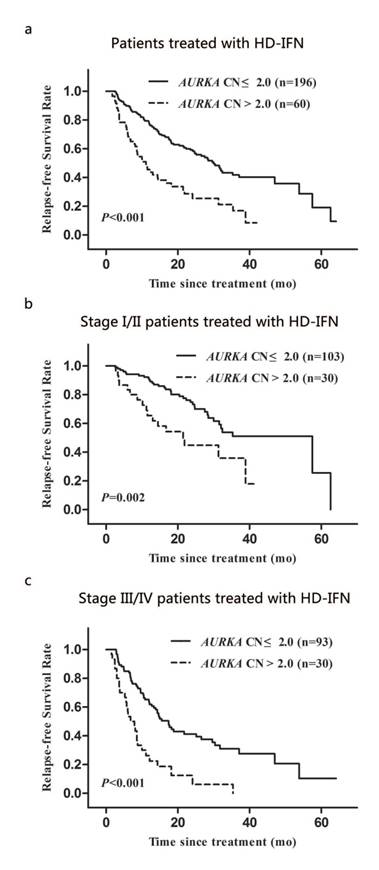
Two hundred and fifty-six out of 472 patients with AM received HD-IFN adjuvant treatment. We determined the relapse-free survival time (RFS) of these patients, which is defined as the time from the date of treatment to first recurrence (local or distant). The median RFS for patients with AURKA copy gain was significantly shorter than that for patients with normal AURKA CN (11.1 months vs. 30.0 months; P < 0.001; Fig. 4a). We also analysed the association between AURKA copy gain and RFS during different clinical stages. Significant differences in RFS was observed between patients with AURKA copy gain and with normal AURKA CN in stage I/II or III/IV of the disease (P = 0.002 and P < 0.001, respectively; Fig. 4b and 4c). Multivariate Cox regression analysis showed that AURKA copy gain was an independent prognostic factor for RFS (HR: 2.93; 95% CI: 2.01-4.27; P < 0.001; Table 5). Of other parameters, age and clinical stages were independent prognostic factors for RFS (Table 5).
Kaplan-Meier analysis of relapse-free survival of AM patients treated with HD-IFN according to clinical stage and AURKA CN: (a) for the overall AM patients treated with HD-IFN group, (b and c) for the patients with stage I/II group and stage III/IV group, respectively.
Univariate and multivariate analysis of prognostic factors for RFS of AM patients treated with HD-IFN.
| Variable | RFS | |||||
|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||
| HR | 95% CI | P value | HR | 95% CI | P value | |
| Age (<55 years/≥55 years) | 1.25 | 0.89-1.76 | 0.192 | |||
| Gender (female/male) | 1.16 | 0.83-1.62 | 0.391 | |||
| Ulceration (yes/no) | 1.16 | 0.80-1.67 | 0.439 | |||
| Thickness (≤ 2 mm/>2 mm) | 0.64 | 0.45-0.91 | 0.014 | 0.417 | ||
| TNM stage (I + II/III +IV) | 0.37 | 0.26-0.52 | <0.001 | 2.72 | 1.91-3.88 | <0.001 |
| AURKA CN (gain/normal) | 3.33 | 2.10-5.26 | <0.001 | 2.82 | 1.94-4.10 | <0.001 |
| NRAS mutations (yes/no) | 1.24 | 0.69-2.22 | 0.469 | |||
| BRAF mutations (yes/no) | 1.72 | 1.00-2.94 | 0.051 | 0.344 | ||
| KIT mutations (yes/no) | 1.14 | 0.70-1.85 | 0.61 | |||
| PDGFRA mutations (yes/no) | 2.31 | 0.70-7.67 | 0.17 | |||
HR, hazard ratio; CI, confidence interval; RFS, relapse-free survival; TNM, tumor-node-metastases.
Discussion
Owing to delayed detection and aggressive biologic behavior, patients with AM appear to have an overall worse prognosis than those with other cutaneous melanomas [24]. Therefore, novel molecular biomarkers that can improve the definition of prognosis and direct therapeutic regimen selection for AM are urgently required. To our knowledge, the current study is the first to investigate the association between AURKA CN and its prognostic significance in patients with AM on a large scale.
Data from two previous studies showed that the frequencies of AURKA copy gain in cutaneous melanoma and acral melanoma are 13.1% and 37.5% [16, 23], respectively. In our cohort, the frequency of AURKA copy gain in AM is 24.6% (116/472), which is significantly different from that observed in the previous study, possibly due to differences in methodology and scoring criteria. One group used fluorescence in situ hybridization (FISH) and defined 12.1% melanomas as AURKA FISH-positive, although the result strongly depended on the cut-off criteria applied [16]. In addition, the subtype bias between Caucasian and Asian patients with melanoma may be another reason for this variation.
In our study, Cox univariate and multivariate analysis indicated that AURKA copy gain is an independent prognostic factor for PFS, but not for OS. Further analysis was performed to determine the relationship between AURKA copy gain and subsets of patients in different clinical stages of the disease. AURKA copy gain was significantly associated with PFS but not OS in the stage I/II and stage III/IV subgroup. These data suggested that AURKA copy gain correlated with the recurrence or metastasis of AM, indicating a more aggressive phenotype. Some recent investigations indicated that AURKA could promote progression of epithelial-mesenchymal transition (EMT), leading to tumour metastasis [25-27]. Further investigations are required to determine the role of AURKA copy gain during AM recurrence or metastasis.
Overexpression and amplification of AURKA kinase are generally detected in various cancers, which are associated with poor prognosis [13-19]. However, the association between AURKA copy gain and AURKA mRNA/protein levels is not fully elucidated. In our cohort, the IHC and RT-PCR results suggested that AURKA copy gain correlates with high levels of AURKA expression. A recent study indicated that AURKA mRNA level was higher in breast cancer patients exhibiting AURKA amplification but no significant correlation was indicated between AURKA amplification and protein levels [28]. This suggested that AURKA copy gain correlates with AURKA expression to a certain extent. However, a comprehensive analysis of AURKA at the levels of gene expression, gene copy number and protein in melanoma is still required. Additionally, biochemical studies of AURKA copy gain may assist in ascertaining its functional effects.
Considering the prognostic significance of AURKA copy gain in patients with AM, AURKA copy gain may be a potent target for targeted therapy of AM. In preclinical trials, AKIs could suppress cell proliferation, migration, and invasion of cancer cells, inhibiting the progress and growth of many cancers including malignant melanoma [29-32]. Interestingly, certain AKIs have been used in clinical trials [20-22]. Alisertib, an AKI, was used successfully in phase III clinical trials in patients with different types of advanced cancer [20]. However, the status of AURKA CN in melanoma was not examined. Further investigations are required to evaluate the effect of AKIs on AM models with or without AURKA copy gain. Furthermore, a recent study revealed that AKIs could enhance the effects of BRAF and MEK inhibitors in melanoma treatment [33]. Therefore, AKI-based single or combinations therapy may provide a new strategy for the treatment of advanced AM.
Interferon was the only approved agent for the adjuvant treatment of melanoma before 2014. According to a meta-analysis of 17 randomized controlled trials (RCTs) involving 8122 patients with high-risk cutaneous melanoma, interferon adjuvant treatment showed significant DFS (HR = 0.82) benefit and marginal OS (HR = 0.89) benefit [34]. More importantly, clinical trials have demonstrated the success of HD-IFN adjuvant treatment for AM in Asian patients [35]. However, drug toxicity might influence the quality of life to certain extent. Therefore, new predictive factors are required to identify patients who can benefit maximally from the HD-IFN adjuvant treatment. In our study, Cox univariate and multivariate analysis indicated that AURKA copy gain was significantly associated with the outcome of patients treated with HD-IFN. Therefore, AURKA copy gain might be a predictive biomarker to expedite decisions on whether to receive HD-IFN adjuvant treatment or switch to alternative options. The AURKA kinase downregulates p53, via phosphorylation of both Ser215 and Ser315 residues [36, 37]. Previous studies revealed that IFN-mediated induction of p53 contributes to tumour suppression [38]. These studies explained why patients with normal AURKA CN benefitted partially from HD-IFN therapy. Further investigations are required to extensively explore the specific mechanisms.
Our study has one limitation. The current study is retrospective, and focused only on AM. Thus, prospective well-designed studies focused on various subtypes of melanoma are required.
In summary, we demonstrated that a small population (24.6%) of patients with AM contains AURKA copy gain. Our study also demonstrated that AURKA copy gain correlates with poor prognosis in AM. Furthermore, AURKA copy gain may be a useful biomarker to predict the outcome of interferon therapy in patients with AM.
Abbreviations
AM: acral melanoma; CN: copy number; OS: overall survival; PFS: progression-free survival; HD-IFN: high-dose interferon; ORR: overall response rate; AKI: AURKA kinase inhibitor; IHC: immunohistochemistry; FFPE: formalin-fixed paraffin-embedded; RT-PCR: real-time quantitative reverse transcription-polymerase chain reaction; RFS: relapse-free survival; HR: hazard ratio; CI: confidence interval; EMT: epithelial-mesenchymal transition.
Supplementary Material
Supplementary figures S1.
Acknowledgements
This work was supported by grants from the Major State Basic Research Development Program of China (2013CB911004), National Natural Science Foundation of China (81672696; 81772912), Beijing Municipal Natural Science Foundation (7152033), Beijing Baiqianwan Talents Project, Beijing Municipal Administration of Hospitals Clinical medicine Development of special funding support (ZYLX201603), and Beijing Municipal Science & Technology Commission (Z151100003915074).
Authors' contribution
JG and YK are responsible for the study design. JYY and JYY performed the experiments and draft the manuscript. XWW, TXX, HY, JD, MM, HT, LWX, ZHC, LS, XNS and CLC participated in the data analysis and interpretation. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McLaughlin CC, Wu XC, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103:1000-7
2. Chi Z, Li S, Sheng X, Si L, Cui C, Han M. et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11:85
3. Coit DG, Thompson JA, Algazi A, Andtbacka R, Bichakjian CK, Carson WE. et al. Melanoma, Version 2.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2016;14:450-73
4. Mouawad R, Sebert M, Michels J, Bloch J, Spano JP, Khayat D. Treatment for metastatic malignant melanoma: old drugs and new strategies. Crit Rev Oncol Hematol. 2010;74:27-39
5. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-16
6. Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y. et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-9
7. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
8. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521-32
9. Si L, Kong Y, Xu X, Flaherty KT, Sheng X, Cui C. et al. Prevalence of BRAF V600E mutation in Chinese melanoma patients: large scale analysis of BRAF and NRAS mutations in a 432-case cohort. Eur J Cancer. 2012;48:94-100
10. Kong Y, Si L, Zhu Y, Xu X, Corless CL, Flaherty KT. et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17:1684-91
11. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017
12. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275-87
13. Ferchichi I, Sassi HS, Baccar A, Marrakchi TR, Cremet JY, Ben RK. et al. Assessment of Aurora A kinase expression in breast cancer: a tool for early diagnosis. Dis Markers. 2013;34:63-9
14. Casorzo L, Dell'Aglio C, Sarotto I, Risio M. Aurora kinase A gene copy number is associated with the malignant transformation of colorectal adenomas but not with the serrated neoplasia progression. Hum Pathol. 2015;46:411-8
15. Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X. et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014;33:539-49
16. Koynova DK, Jordanova ES, Milev AD, Dijkman R, Kirov KS, Toncheva DI. et al. Gene-specific fluorescence in-situ hybridization analysis on tissue microarray to refine the region of chromosome 20q amplification in melanoma. Melanoma Res. 2007;17:37-41
17. Xu J, Wu X, Zhou WH, Liu AW, Wu JB, Deng JY. et al. Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One. 2013;8(e):56919
18. Dotan E, Meropol NJ, Zhu F, Zambito F, Bove B, Cai KQ. et al. Relationship of increased aurora kinase A gene copy number, prognosis and response to chemotherapy in patients with metastatic colorectal cancer. Br J Cancer. 2012;106:748-55
19. Chen J, Lin Q, Wen JY, Li X, Ma XK, Fan XJ. et al. Prognosis value of mitotic kinase Aurora-A for primary duodenal adenocarcinoma. Tumour Biol. 2014;35:9361-70
20. Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O. et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16:395-405
21. Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul HM. et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol. 2015;33:2399-404
22. Schöffski P, Besse B, Gauler T, de Jonge MJ, Scambia G, Santoro A. et al. Efficacy and safety of biweekly i.v. administrations of the Aurora kinase inhibitor danusertib hydrochloride in independent cohorts of patients with advanced or metastatic breast, ovarian, colorectal, pancreatic, small-cell and non-small-cell lung cancer: a multi-tumour, multi-institutional phase II study. Ann Oncol. 2015;26:598-607
23. Puig-Butillé JA, Badenas C, Ogbah Z, Carrera C, Aguilera P, Malvehy J. et al. Genetic alterations in RAS-regulated pathway in acral lentiginous melanoma. Exp Dermatol. 2013;22:148-50
24. Rex J, Paradelo C, Mangas C, Hilari JM, Fernández-Figueras MT, Ferrándiz C. Management of primary cutaneous melanoma of the hands and feet: a clinicoprognostic study. Dermatol Surg. 2009;35:1505-13
25. Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK. et al. The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One. 2012;7(e):40378
26. Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu L. et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis. 2008;29:1930-7
27. Liu X, Li Z, Song Y, Wang R, Han L, Wang Q. et al. AURKA induces EMT by regulating histone modification through Wnt/β-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget. 2016;7:33152-64
28. Yamamoto S, Yamamoto-Ibusuki M, Yamamoto Y, Fujiwara S, Iwase H. A comprehensive analysis of Aurora A; transcript levels are the most reliable in association with proliferation and prognosis in breast cancer. BMC Cancer. 2013;13:217
29. Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L. et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080-9
30. Jani JP, Arcari J, Bernardo V, Bhattacharya SK, Briere D, Cohen BD. et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9:883-94
31. Pirker C, Lötsch D, Spiegl-Kreinecker S, Jantscher F, Sutterlüty H, Micksche M. et al. Response of experimental malignant melanoma models to the pan-Aurora kinase inhibitor VE-465. Exp Dermatol. 2010;19:1040-7
32. Xie L, Meyskens FL. The pan-Aurora kinase inhibitor, PHA-739358, induces apoptosis and inhibits migration in melanoma cell lines. Melanoma Res. 2013;23:102-13
33. Caputo E, Miceli R, Motti ML, Taté R, Fratangelo F, Botti G. et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J Transl Med. 2014;12:216
34. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2010;102:493-501
35. Mao L, Si L, Chi Z, Cui C, Sheng X, Li S. et al. A randomised phase II trial of 1 month versus 1 year of adjuvant high-dose interferon α-2b in high-risk acral melanoma patients. Eur J Cancer. 2011;47:1498-503
36. Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J. et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175-82
37. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F. et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55-62
38. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H. et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516-23
Author contact
![]() Corresponding author: Key laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Renal cancer and Melanoma, Peking University Cancer Hospital & Institute, No. 52 Fucheng Road, Haidian District, Beijing, 100142, China. E-mail addresses: guoj307com (J. Guo), k-yan08com (Y. Kong).
Corresponding author: Key laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Renal cancer and Melanoma, Peking University Cancer Hospital & Institute, No. 52 Fucheng Road, Haidian District, Beijing, 100142, China. E-mail addresses: guoj307com (J. Guo), k-yan08com (Y. Kong).