Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(8):1329-1336. doi:10.7150/jca.21915 This issue Cite
Research Paper
Verteporfin inhibits papillary thyroid cancer cells proliferation and cell cycle through ERK1/2 signaling pathway
Tian Liao*, Wen-Jun Wei*, Duo Wen*, Jia-Qian Hu, Yu Wang, Ben Ma, Yi-Min Cao, Jun Xiang, Qing Guan, Jia-Ying Chen, Guo-Hua Sun, Yong-Xue Zhu, Duan-Shu Li, Qing-Hai Ji ![]()
Department of Head and Neck Surgery, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China.
* These authors have contributed equally to this work.
Received 2017-7-14; Accepted 2017-10-9; Published 2018-3-29
Abstract
Verteporfin, a FDA approved second-generation photosensitizer, has been demonstrated to have anticancer activity in various tumors, but not including papillary thyroid cancer (PTC). In current pre-clinical pilot study, we investigate the effect of verteporfin on proliferation, apoptosis, cell cycle and tumor growth of PTC. Our results indicate verteporfin attenuates cell proliferation, arrests cell cycle in G2/S phase and induces apoptosis of PTC cells. Moreover, treatment of verteporfin dramatically suppresses tumor growth from PTC cells in xenograft mouse model. We further illustrate that exposure to MEK inhibitor U0126 inactivates phosphorylation of ERK1/2 and MEK in verteporfin-treated PTC cells. These data suggest verteporfin exhibits inhibitory effect on PTC cells proliferation and cell cycle partially via ERK1/2 signalling pathway, which strongly encourages the further application of verteporfin in the treatment against PTC.
Keywords: Papillary thyroid cancer, verteporfin, proliferation, cell cycle, signalling pathway
Introduction
The incidence of thyroid cancer has increased worldwide at a rate higher than any other cancer. Papillary thyroid cancer (PTC) is the most ordinary endocrine malignancy and accounts for 80% cases of differentiated thyroid cancer [1]. In line with the majority of malignancies, thyroid carcinomas are also correlated with specific genetic abnormalities which lead to aberrant cell proliferation. With the rising trends in the incidence of PTC have been well-recognized, the need to develop new treatments is more acute than ever.
Yes associated protein 1 (YAP1) gene is located in the 11q22 amplicon, which is the main nuclear coactivator in the Hippo pathway [2]. Dysregulation of Hippo pathway, as a key oncogenic signal which malignancy cells rely on, could result in permanent activation of pro-tumor signal cascade [3, 4]. Therefore, modulating the Hippo pathway pathway presents an attractive anti-cancer strategy. The small molecule inhibitor of YAP1 [5], verteporfin (VP), a benzoporphyrin derivative approved for photodynamic therapy to eliminate the age-related macular degeneration, may function as a potential candidate for cancer treatment. Indeed, its antitumor effect has been validated in multiple types of cancer, such as pancreatic cancer, breast cancer and colon cancer [5-8], but the role of VP plays in PTC has not yet been known.
Our previous works showed that YAP1, as an oncogene in PTC, could promote cell proliferation of PTC cells via ERK/MAPK signalling pathway [9]. In present study, we demonstrated VP can inactivate ERK1/2 and MEK phosphorylation to inhibit proliferation and arrest G1/S cell cycle of PTC cells. Therefore, we supposed that tumor suppressive function of VP make it be a promising potential strategy for treatment of PTC.
Results
Inhibitory effect of verteporfin on the proliferation of papillary thyroid cancer cells in vitro and in vivo
To access the role of verteporfin on the proliferation of papillary thyroid cancer cells, we firstly treated PTC cell lines K1 and BCPAP with 0.5 μM, 1 μM or 5 μM verteporfin or DMSO. CCK8 proliferation was then used to estimate cell viability of the treated cells. As shown in Fig. 1A and 1B, both K1 and BCPAP cell viability were inhibited by verteporfin treatment, and the inhibitory effect displayed a dose dependent manner.
The effect of verteporfin on the proliferation of PTC cell line K1 and BCPAP. K1 and BCPAP were treated with 0.5 μM, 1 μM or 5 μM verteporfin or DMSO. Cell viability of treated K1 (A) and BCPAP (B) were examined by CCK8 assay. The proliferation related genes (survivin, PCNA, and c-Myc) expression were detected in treated K1 (C) and BCPAP (D) by qPCR. The results are presented as fold-changes in verteporfin treated cells relative to DMSO treated control cells. qPCR results were normalized to β-actin mRNA expression. Western blotting was used to investigate the gene level of proliferation related markers in treated K1 (E) and BCPAP (F). All the experiments were performed in triplicate. (*P<0.05, ***P<0.001).
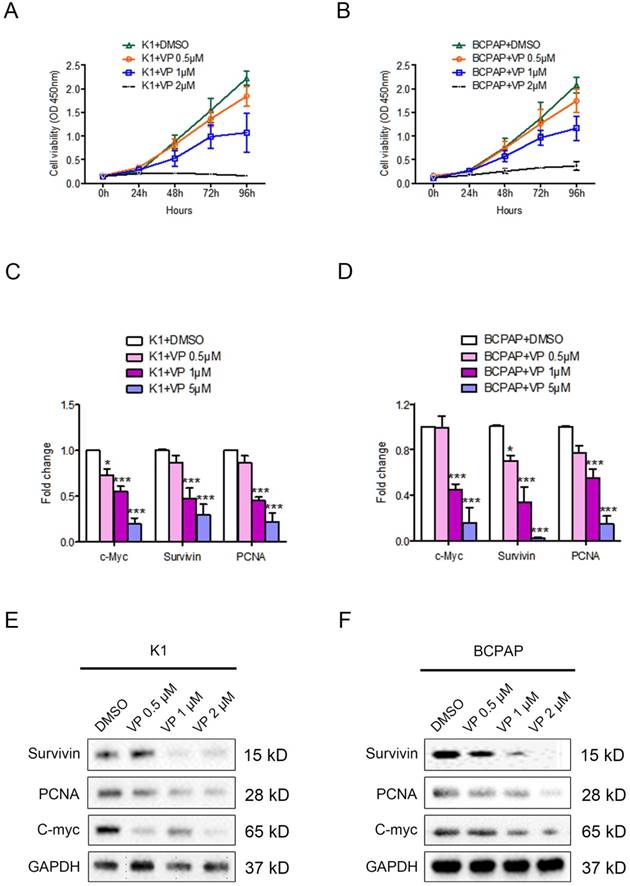
The proliferation related gene expression of survivin, PCNA, and c-Myc were decreased in verteporfin-treated K1 and BCPAP cells (Fig. 1C and 1D). Western blotting analysis showed the expression level of protein (survivin, PCNA, and c-Myc) in both K1 and BCPAP cells were downregulated after the treatment of verteporfin (Fig. 1E and 1F), which was also in a dose-dependent manner. 1 μM and 5μM verteporfin treatment for 72h seems to be more effective than 0.5 μM in both K1 and BCPAP cells. The results revealed that verteporfin could significantly inhibit the cell proliferation of papillary thyroid cancer cell.
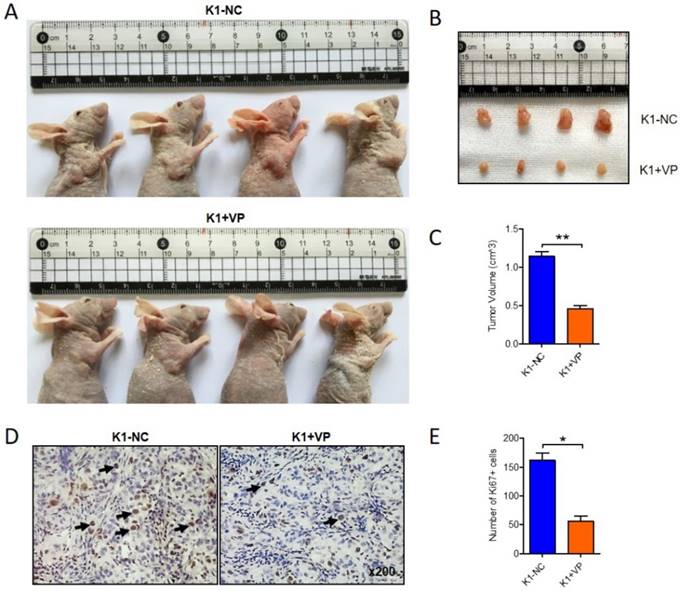
To further determine the anticancer effect of verteporfin on papillary thyroid cancer in vivo, 1x107 K1 cells were subcutaneously interjected into nude mice. After 14 days, the transplanted xenograft tumors were constructed. verteporfin (100mg/kg) was intraperitoneally injected into the mouse in verteporfin-treated group (K1+VP) every 3 day, while mouse in vehicle-group treated with PBS. Animals were sacrificed after five doses of verteporfin and tumor volumes were measured (Fig. 2A). The verteporfin-treated mice generated tumors were dramatically smaller than the tumors present in vehicle-group mice (Fig. 2B and C). The tumors were formalin-fixed and paraffin-embedded and tumor tissue sections were analyzed by IHC. The positive number of Ki67+ cells revealed verteporfin treatment significantly suppressed the proliferation of the implanted tumors (Fig. 2D and E). In a word, the data indicated that VP can inhibit proliferation and growth of papillary thyroid cancer in vitro and in vivo.
Verteporfin suppressed tumor growth of papillary thyroid cancer in vivo. 1x107 K1 cells were subcutaneously injected into nude mice, VP (100mg/kg) was intraperitoneally injected every 3 days when transplanted xenograft tumors were constructed. After five doses of VP, tumor volumes were measured. (A-B) Four representative tumors from each group (n=4) are shown. (C) Volume comparison for tumors from K1-VP group and K1-NC group. (D) IHC analysis of Ki67 in formalin-fixed, paraffin-embedded tumors from K1-VP group and K1-NC group (Original magnification ×200; Arrow marked Ki67 positive cells). (E) Number of Ki67+ cells in tumors from K1-VP group and K1-NC group. (*P<0.05, **P<0.01).
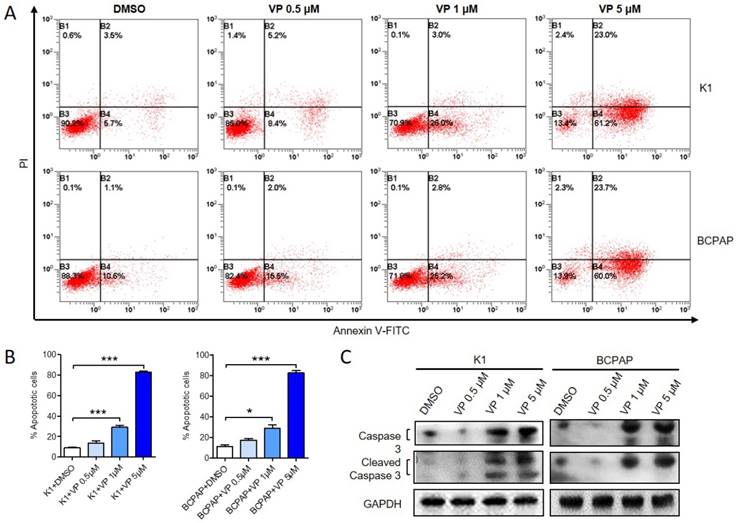
Verteporfin induces apoptosis of papillary thyroid cancer cells
Next, we examined whether verteporfin could induce apoptosis of K1 and BCPAP cells. After treated with 0.5 μM, 1 μM or 5 μM verteporfin or DMSO, apoptotic cells were analyzed by flow cytometry (Fig. 3A and B). Compared with DMSO-treated group (8.87 ± 1.53% of K1 and 11.17 ± 2.59% of BCPAP), 0.5 μM verteporfin induced about 13.8 ± 1.53% in K1 and 17.47 ± 2.59% in BCPAP (P > 0.05), while 1 μM and 5μM verteporfin induced about 30.93 ± 3.63% and 80.66 ± 2.03% in K1 (P < 0.05), 28.6 ± 6.391% and 85.32 ± 3.822% in BCPAP (P < 0.05), respectively. The results showed that the apoptosis rate of K1 and BCPAP increased when the concentration of verteporfin increased. Moreover, both caspase 3 and cleaved-caspase 3 were activated in a dose-dependent manner in verteporfin-treated K1 and BCPAP cells (Fig. 3C).
Verteporfin induces PTC cell apoptosis in vitro. K1 and BCPAP were treated with 0.5 μM, 1 μM or 5 μM verteporfin or DMSO. (A) Cell apoptosis was examined by flow cytometry. UL, necrotic cells; UR, terminal apoptotic cells; LR, early apoptotic cells. (B) Apoptotic cells of 0.5 μM, 1 μM or 5 μM verteporfin-treated group were quantified to DMSO group. The results are presented as the mean ± SEM from three independent experiments. (C) Caspase 3 and cleaved-caspase 3 expression were determined by WB. (*P<0.05, ***P<0.001).
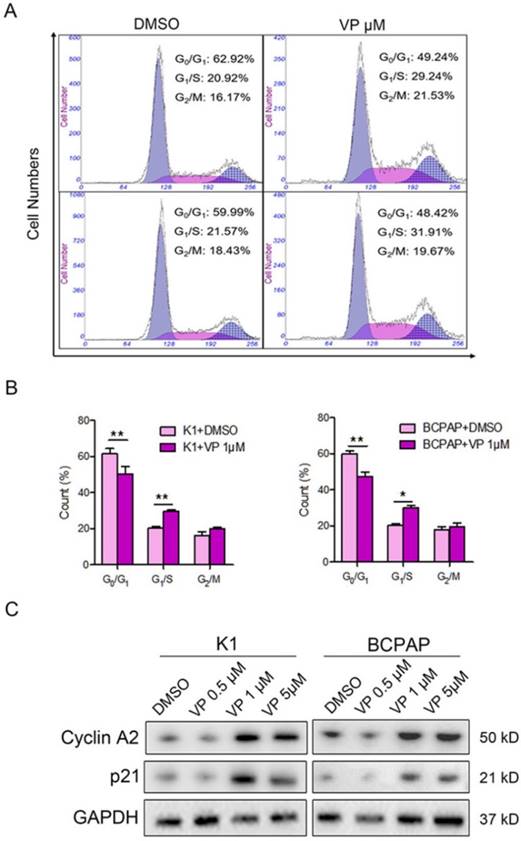
Verteporfin arrests G1/S phase of papillary thyroid cancer cells through cyclin A2 and p21
The above data suggested that 1 μM verteporfin could significantly inhibit proliferation and induce apoptosis of K1 and BCPAP cells. Thus, we further verify whether 1 μM verteporfin affected cell cycle progression. Firstly, K1 and BCPAP cells were treated with 1 μM for 48 h, and then cell cycle was accessed using flow cytometry assay. As shown in Fig. 4A and B, the treated K1 cells had a low percentage of G0/G1 phase (50.3%) and high percentage of G1/S phase (29.63%) compared with control group (61.4% and 20.24%), while the treated BCPAP cells also had a low percentage of G0/G1 phase (47.37%) and a high percentage of G1/S phase (30.19%) compared with control group (59.76% and 20.46%), respectively. G2/M phase of treated K1 and BCPAP cells were 19.95% and 19.31%, control group had 16.05% and 17.8% G2/M phase, respectively. It suggested that both K1 and BCPAP cells were arrested in G1/S phase by 1 μM verteporfin. Next, we examined the expression of cyclin A2 and p21 in verteporfin-treated K1 and BCPAP cells, which have a key role in the cell cycle. Western blotting analysis data showed that the expression of cyclin A2 and p21 were enhanced after verteporfin treatment, exhibiting a dose-dependent manner (Fig. 4C).
Cell cycle inhibition of PTC cells with verteporfin treatment. K1 and BCPAP were treated with 0.5 μM, 1 μM or 5 μM verteporfin or DMSO. (A) Cell cycle distributions of the treated cells were analyzed by flow cytometry. (B) Cells of G0/G1, G1/S, G2/M phase were quantified in treated cells. The results are presented as the mean ± SEM from three independent experiments. (C) Expression of cyclin A2 and p21 were detected in the treated cells using WB. (*P<0.05, **P<0.01).
Verteporfin inhibits proliferation and cell cycle of papillary thyroid cancer cells through ERK1/2 signalling pathway
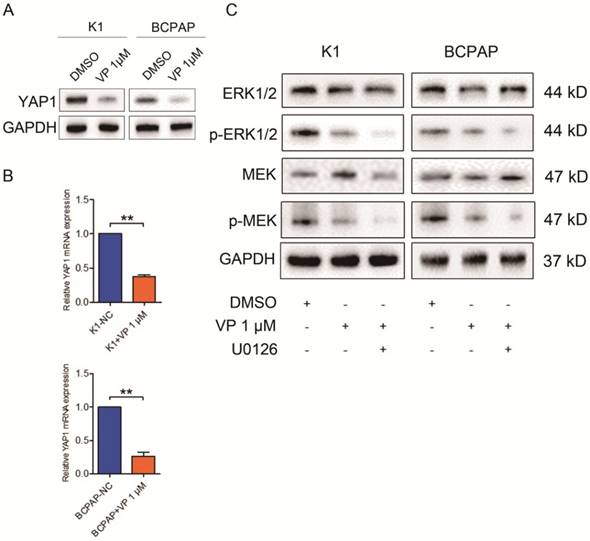
Verteporfin is known as a YAP1 inhibitor, independent of light activation [10]. Here, we accessed the expression level of YAP1 in treated K1 and BCPAP cells. As shown in Fig. 5A and B, both mRNA and protein level of YAP1 in K1 and BCPAP cells dramatically decreased after 1 μM verteporfin treatment. To further explore the underlying signaling involved in the inhibitory effect of verteporfin on thyroid cancer cells, we treated the cells with U0126, a MEK inhibitor, and examined the expression of protein involved in ERK1/2 signaling pathway. The results revealed that the total ERK1/2 and MEK did not change with the treatment of U126, while phosphorylated ERK1/2 and MEK significantly declined in 1 μM verteporfin treated K1 and BCPAP cells. These data suggested that verteporfin exhibit inhibitory effect on PTC cells partly through ERK1/2 signaling pathway.
Verteporfin-induced proliferation suppression and cell cycle arrest via ERK1/2 and MEK. 1 μM verteporfin treatment decreased YAP1 expression in both protein (A) and gene level (B). qPCR results were normalized to β-actin mRNA expression. (C) Treatment of U0126 suppressed the expression of phosphorylated ERK1/2 and MEK in verteporfin-treated K1 and BCPAP cells. (**P<0.01). All the experiments were performed in triplicate.
Discussion
Verteporfin is a second-generation photosensitizer that has been reported to be a candidate of photodynamic therapy for the management of vertebral breast metastases [5] and locally advanced pancreatic cancer [6]. However, there are rare reports about the effect of verteporfin on papillary thyroid cancer and the underlying mechanism. In this study, we displayed verteporfin exhibited inhibitory effect on proliferation and cell cycle, and induced apoptosis of thyroid cancer cells in vitro, and it could also suppress thyroid cancer xenograft growth in vivo. Our finding indicated that verteporfin may be a therapeutic target to treat thyroid cancer.
After the first preliminary clinical study reported for the treatment of skin cancer [11], accumulating evidences have proved that verteporfin is a promising anti-tumor drug against multiple types of solid cancers. Liu-Chittenden Yi et al. group have identified that verteporfin as a small molecule can inhibit the interactions of YAP-TEAD and prevents YAP1-induced oncogenic growth by binding to YAP1 and changing its conformation, thereby abrogating its interaction with TEAD2 [12]. In our previous study, we covered that YAP1 played a vital role as an oncogene on the tumorigenicity of PTC [9]. To explore the effect of verteporfin on PTC, we complement the data and confirm its inhibitory effect on PTC oncogenesis in current study. YAP1 expression level in PTC cells decreased with the treatment of 1μM verteporfin, which make us speculate that the inhibitory effect of verteporfin may result from its disrupting YAP1's function in PTC.
Previous researches showed that MAPK signaling pathway involves in cell proliferation of multiple tumor [13-15], including thyroid cancer. Previous study has detected that verteporfin could block RAS-induced MAPK signaling pathway in ATC cell lines [16]. In our previous study, we also found that YAP1 was involved in PTC oncogenesis through MAPK signaling pathway [9]. Herein, exposure to MEK inhibitor U0126 could also partly suppressed phosphorylated ERK1/2 and MEK expression in verteporfin-treated PTC cells, further complement with previous study. However, warranting further underlying mechanism investigation is still needed to figure out how verteporfin influence the YAP1-ERK/MAPK crosstalk in the process of PTC.
In conclusion, we for the first time reveal verteporfin inhibits the proliferation and cell cycle of papillary thyroid cancer cells partially via ERK1/2 signaling pathway. Taken together, verteporfin may be an encouraging candidate for the therapeutic management of papillary thyroid cancer.
Methods
Cell culture and treatments
Papillary thyroid cancer cell lines BCPAP and K1 were grown in RPMI1640 media supplemented with 10% heat-inactivated FBS (Invitrogen, Carlsbad, CA, USA) and 1% penicillin-streptomycin (10,000 units penicillin and 10 mg streptomycin) at 37°C with 5% CO2. Verteporfin (VP) was dissolved in DMSO (100 mg/mL) and stored at -20°C. BCPAP and K1 cells were treated with dimethyl sulfoxide or VP with a dose of 0.5μM, 1μM or 5μM and U0126 (10 μM) (Sigma Chemical Co., St. Louis, MO).
Total RNA extraction and RT-PCR
We extracted total RNA of cultured cells with TRIzol Reagent (Invitrogen, Inc.). 1μg of total RNA was utilized for cDNA synthesis in a PrimeScriptTM RT reagent kit (Takara Bio, Inc., Japan). qRT-PCR was performed with SYBR Premix Ex TaqTM kit (Takara Bio, Inc., Japan) in triplicate according to the manufacturer's instructions. The primers of target genes were synthesized by Sangon Company (Sangon Biotech Co., Ltd., Shanghai, China) (Table 1). The house keeping gene β-actin was used as endogenous control for mRNA assays. Threshold cycle (Ct) values were analyzed using the comparative Ct (2-ΔΔCt) method. The expression of target genes was calculated by normalizing to β-actin and relative to the control.
qRT-PCR primers of target genes
| Target genes | Forward sequence (5'to3') | Reverse sequence (5'to3') |
|---|---|---|
| YAP1 | TAGCCCTGCGTAGCCAGTTA | TCATGCTTAGTCCACTGTCTGT |
| c-Myc | GGAGGCTATTCTGCCCATTT | CGAGGTCATAGTTCCTTGTTGGT |
| survivin | CACTTTCTTCGCAGTTTCCT | GACCACCGCATCTCTACATTC |
| PCNA | AAACTAGCTAGACTTTCCTC | TCACGCCCATGGCC AGGTTG |
| β-actin | TGACGTGGACATCCGCAAAG | CTGGAAGGTGGACAGCGAGG |
Western blotting
Cell lysates from 1 × 106 cultured cells were obtained in a mixture of ProteoJET Mammalian Cell Lysis Reagent (Fermentas, Inc.). A total of 20 μg protein extracted from each sample was separated by 10 % SDS-PAGE. After blocked in 5 % nonfat milk, the target protein was incubated with antibody either against human YAP1 (1:1000; abcam), c-Myc, PCNA (Proliferating cell nuclear antigen, PCNA), survivin, ERK1/2, MEK, phosphorylatd-ERK1/2 and MEK (1:1000; Cell Signaling Technology), or GAPDH (1:5000; Abcam); then incubated with goat anti-rabbit or goat anti-mouse IgG (1:5000 for both; Cell Signaling Technology); and detected using enhanced chemiluminescence reagents (Thermo Fisher Scientific). Target bands were detected using the Alpha Imager (Alpha Innotech, San Leandro, CA, USA).
CCK8 proliferation assays
Cell viability was examined using the Cell Counting Kit-8 (CCK8; Dojindo Molecular Technologies, Gaithersburg, Maryland, USA). In brief, BCPAP and K1 cells were treated with DMSO or VP with a dose of 0.5μM, 1μM or 5μM. The treated cells were seeded into 96-well plates at 4×103 cells/well. An aliquot of 10 μl CCK-8 solution was added into each well, and then was incubated for 4 h at 37 °C. At the indicated time points, the absorbance at 450 nm was measured using a spectrophotometer. For each group, data from five wells were pooled.
Apoptosis assays
Apoptotic cells were accessed by flow cytometric analysis of Annexin V-FITC/PI staining (BD Biosciences, Mississauga, ON). The cells were cultivated in different concentrations of VP for 48 h, and then trypsined and washed. Resuspended cells were stained with 5 μl Annexin V-FITC and 10 μl PI at room temperature for 15 min (Biovision, CA, USA). After incubation, the cells were then analyzed by flow cytometry immediately. Apoptotic cells were marked based on Annexin V expression (Annexin V+/PI- and Annexin V+/PI+), quantified and compared to the controls from each group. Data from independent triplicate experiments were calculated to analyze the number of apoptotic cells.
Cell cycle analysis
In order to synchronize the cell cycle, cells were harvested in ice-cold PBS, and fixed in ice-cold 70% ethanol. The fixed cells were then resuspended in cold PBS. RNAase (2μg/ml) was added and incubated at 37°C for 30 min, followed by cultivation in 400 μl propidium iodide (PI, 10 μg/ml) for 40 min at room temperature. The DNA content was determined by a FACS Calibur flow cytometer and the cells in different phase of G0, G1, S, M were analyzed. All experiments were performed in triplicates.
Animal experiments
Five weeks old BALB/c nude mice (4 mice/group) were obtained from the Shanghai experimental animal center (Shanghai, China). Briefly, 1x107 K1 cells were subcutaneously injected into the right back skin area of BALB/c nude mice. The size of tumors was measured by vernier caliper once a week. After 14 days, the tumors were constructed, mice were administered intraperitoneally with VP at a dose of 100 mg/kg every 3 day whereas control mice were injected with equivalent PBS. After 5 dose of VP, mice were sacrificed, and tumor tissues were collected, photographed and weighted. Paraffin-embedded tissues were sectioned for immunohistochemistry of Ki-67 (Santa Cruz Biotechnology). Animal experiments have been pre-approved by the Shanghai Cancer Center, Fudan University and all the procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Immunohistochemical staining
Immunohistochemical staining was performed according to procedures as described previously. In brief, formalin-fixed and paraffin-embedded tissue sections were deparaffinized in xylene and hydrated through descending concentrations of ethanol before being placed in blocking solution to inhibit endogenous peroxidase activity. The slides were incubated with primary antibody at 4°C overnight. A HRP-conjugated rabbit or mouse secondary antibody was added for 60 min at room temperature, followed by 3,3′-diaminobenzidine (DAB) development. Slides were fixed and images obtained with the Olympus IX71 inverted microscope using the DP2-BSW Olympus image acquisition software system. The results were confirmed by two experienced pathologists who were blinded to the clinic pathologic data of the patients. We took 5 visual field per section and number of Ki67 positive cells in each field were calculated to analyze the average of positive Ki67 cells in each group.
Statistics
Statistical analysis was performed with GraphPad Prism 5.1. For comparison among the groups, Student's t test, one-way ANOVA and two-way ANOVA were performed, and P<0.05 was defined as statistically significant. The data and error bars report the mean ± SEM. All the experiments were repeated at least three times.
Abbreviations
PTC: papillary thyroid carcinoma; PCNA: proliferating cell nuclear antigen.
Acknowledgements
The authors would like to thank University of Colorado Cancer Center Cell Bank for providing PTC cell line K1.
Funding
This work was partly supported by funds from The Project Sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (to Tian LIAO), the National Natural Science Foundation of China (81272934, 81572622, 81772854 to Qing-Hai JI, 81702753 to Tian LIAO), the Shanghai Science and Technology Commission Western Guide project (14411962402 to Duan-Shu LI), the Shanghai municipal planning commission of science and research fund for young scholar (20154Y0050 to Jun Xiang).
Contributions
T.L., W.J.W. and J.Q.H performed all experiments, B.M and Y.M.C performed statistical analysis, D.W., N.Q. and R.L.S prepared figures, G.H.S, Y.X.Z., D.S.L and Q.H.J designed the experiments, T.L. wrote the manuscript. All authors were involved in writing the paper and had final approval of the submitted manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lubitz CC, Sosa JA. The changing landscape of papillary thyroid cancer: Epidemiology, management, and the implications for patients. Cancer. 2016
2. Dai X. et al. YAP activates the Hippo pathway in a negative feedback loop. Cell Res. 2015;25:1175-8
3. Yu FX. et al. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355-71
4. Varelas X. et al. Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol. 2012;22:88-96
5. Akens MK. et al. Defining the therapeutic window of vertebral photodynamic therapy in a murine pre-clinical model of breast cancer metastasis using the photosensitizer BPD-MA (Verteporfin). Breast Cancer Res Treat. 2010;119:325-33
6. Huggett MT. et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110:1698-704
7. Celli JP. et al. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg Med. 2011;43:565-74
8. Corvaisier M. et al. Regulation of cellular quiescence by YAP/TAZ and Cyclin E1 in colon cancer cells: Implication in chemoresistance and cancer relapse. Oncotarget. 2016
9. Liao T, Wen D, Ma B, Hu JQ, Qu N, Shi RL. et al. Yes-associated protein 1 promotes papillary thyroid cancer cell proliferation by activating the ERK/MAPK signaling pathway. Oncotarget. 2016
10. Zhang H. et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8:ra98
11. Michels S, Schmidt-Erfurth U. Photodynamic therapy with verteporfin: a new treatment in ophthalmology. Semin Ophthalmol. 2001;16:201-6
12. Liu-Chittenden Y. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300-5
13. Han J. et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015;138:668-75
14. Chen DL. et al. EDAG-1 promotes proliferation and invasion of human thyroid cancer cells by activating MAPK/Erk and AKT signal pathways. Cancer Biol Ther. 2016;17:414-21
15. Gong Y. et al. Phosphorylation of myofibrillogenesis regulator-1 activates the MAPK signaling pathway and induces proliferation and migration in human breast cancer MCF7 cells. FEBS Lett. 2014;588:2903-10
16. Garcia-Rendueles ME. et al. NF2 Loss Promotes Oncogenic RAS-Induced Thyroid Cancers via YAP-Dependent Transactivation of RAS Proteins and Sensitizes Them to MEK Inhibition. Cancer Discov. 2015;5:1178-93
Author contact
![]() Corresponding author: Qing-Hai JI, Department of Head and Neck Surgery, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China. Phone: 0086-021-64175590, fax: 0086-021-64175590. E-mail: ji_qh2015com
Corresponding author: Qing-Hai JI, Department of Head and Neck Surgery, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China. Phone: 0086-021-64175590, fax: 0086-021-64175590. E-mail: ji_qh2015com