Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(11):2012-2023. doi:10.7150/jca.24275 This issue Cite
Research Paper
SIRT1 promotes formation of breast cancer through modulating Akt activity
Xiaoxia Jin1,2#, Yingze Wei1,2#, Feng Xu3, Min Zhao2, Kui Dai2, Rong Shen2, Shuyun Yang2 ![]() , Nong Zhang1
, Nong Zhang1 ![]()
1. Department of Pathology, School of Basic Medical Sciences, Fudan University, Shanghai, China
2. Department of Pathology, Nantong Tumor Hospital, Nantong, Jiangsu, China
3. Department of Thoracic Surgery, Nantong Tumor Hospital, Nantong, Jiangsu, China
# Both authors contributed equally to this study.
Received 2017-12-9; Accepted 2018-3-15; Published 2018-4-30
Abstract
The silent information regulation factor 1 (sirtuin Type 1, SIRT1), as a kind of NAD+ dependent class III histone deacetylation enzyme, has been found to be involved in tumor proliferation, invasion, and metastasis. The roles of SIRTl in breast cancer is multifaceted depending on its substrate from upstream or downstream signaling pathway. In this study, we sought to make clear the regulating effects of SIRT1 in breast cancer cells, and to explore the underlying mechanisms through which SIRT1 regulates breast cancer. First, our results showed that SIRT1 was significantly up-regulated in breast cancer tissues and cells, which correlated with histological grade, tumor size, as well as lymph node metastasis. Then we established SIRT1-overexpressed and SIRT1- knockdown breast cancer cell lines to investigate the functions of SIRT1 in regulating colony formation, cell proliferation, cell cycle, cell apoptosis and migration. We found that overexpression of SIRT1 significantly promoted breast cancer growth both in vitro and in vivo, whereas knockdown of SIRT1 inhibited these phenotypes. Furthermore, SIRT1 was found to interact with Akt directly, consequently promoting the activity of Akt in breast cancer cells in vitro and positively correlating with expression of Akt, P-Akt, in breast cancer tissues in vivo. Down regulation the activity of Akt partially weakened the proliferative effect mediated by SIRT1. Taken together, our results demonstrated SIRT1's tumor promotion function and potential mechanisms in breast cancer, thus providing valuable therapeutic targets for breast cancer.
Keywords: SIRT1, Akt, cell proliferation, Breast cancer
Introduction
Breast cancer is the one of the most dangerous diseases and leads to the second highest incidence of cancer-rated deaths among women worldwide [1]. Development of breast cancer is associated with multiple etiologic factors, including hormonal disorders, inheritance, ionizing radiation, and unhealthy eating habits [2]. Breast cancer is normally treated by a combination of surgery with radiotherapy, endocrine therapy, and/or chemotherapy. Despite improvements in diagnosis and treatment, the 5-year survival rate of patients with breast cancer was still viewed as unsatisfactory in recent decades [3], which highlights the ongoing need to understand the mechanisms by which it progresses and to explore new therapeutic targets.
SIRTl is a mammalian homology of yeast Sir2 and the most studied sirtuins family member which functions as a histone deacetylase that has effects on not only histones but also nonhistones [4,5]. It plays important roles in cell survival, signal transduction, and cell apoptosis by deacetylating key cell signaling molecules and apoptotic related proteins, such as NF-kB, p53, Ku70, and HIFs [6-8]. Recent studies argued about the role of SIRT1 in cancer, because of its opposite effects as both a tumor activator or suppressor in various human cancers, including breast cancer. Indeed, there were significant differences found in the expression of SIRT1 with different tumors. For instance, Deng et al [9] found that the expression of SIRT1 was lower in prostate cancer, bladder cancer, ovarian cancer, and glioblastoma when compared with normal tissues. By contrast, others have found that, in the leukemia [10] and lung cancer [11], SIRT1 was significantly higher. To add to this debate, Huffman [12] found that the expression of SIRT1 was actually higher in prostate cancer when compared to control groups, a result which was converse to Deng's conclusions. These differences might be explainable based on tumor types or microenvironment, as well as the influence other factors upstream and downstream but they do underscore the need better to understand the role of SIRT1. Here, we further explored the role of SIRT1 in cell proliferation and progression in breast cancer, as well as its associated mechanisms.
The PI3K/Akt pathway plays important roles in many types of cancers. It serves as a tumor-inducing factor in that over-activation of Akt can cause uncontrolled cell proliferation and prevent programmed cell apoptosis [13]. Thus, inhibiting the activation of Akt might be a beneficial therapeutic strategy for breast cancer [14]. For these reasons, finding the underlying mechanisms of this pathway and searching for effective obstruction methods is essential. As we know, SIRT1 modulates a variety of cell signaling pathways through deacetylation of target proteins [15]. Lysine acetylation of Akt has been reported to be crucial for its stability and activity [16]. Recent studies also showed that SIRT1 deacetylated Akt and thus played an important role in cardiac hypertrophy. However whether SIRT1 interacts and deacetylates Akt in breast cancer and if this interaction is related to breast cancer cell biological functions is still unknown. It is possible that SIRT1 may facilitate progression of breast cancer by regulating Akt activity. With stable overexpressed or knocked down SIRT1 breast cancer cell lines, we demonstrate the causative effect of SIRT1 in regulating biological behaviors of breast cancer cells by modulating Akt activity both in vitro and in vivo, suggesting SIRT1 could be considered as a significant therapeutic target for breast cancer.
Materials and Methods
Cell culture and reagents
Four types breast cancer cell lines (BT549, T47D, MDA-MB-231, MCF7) and benign breast epithelial cell line (MCF-10A) were purchased from the Cell Culture Collection of the Chinese Academy of Sciences (Shanghai, China). BT549 and T47D cells were cultured in RPMI 1640 medium (RPMI1640, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco). MCF-7 and MDA-MB-231 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% FBS. MCF-10A cells was cultured in a 1:1 mixture of DMEM/F-12 medium supplemented with 10% FBS. All cell lines were grown at the 37℃ atmosphere contained 5% CO2.
Used antibodies included anti-SIRT1 (Abcam, Cambridge, MA, USA), anti- Akt and anti-phosphory-Akt (Ser473) (Cell Signaling Technology, Danvers, MA, USA), anti-GAPDH (Santa Cruz, CA, USA). Human AKT SiRNA was purchased from Santa Cruz. The PI3K/Akt inhibitor LY294002 was also obtained from Cell Signaling Technology.
Patient and samples
After diagnosed through histo-pathological and clinical evidences, all breast cancer samples from infiltrating ductal carcinoma patients used in this study were obtained from the Nantong Tumor Hospital between 2009 and 2012. The patients did not receive any anti-tumor therapy before surgery. All samples were pathologically diagnosed based on the World Health Organization (WHO) classification [17], and were referred to the protocol approved by the Ethical Committee of the Nantong Tumor Hospital.
Immunohistochemistry staining
The 5 μm-thickness paraffin-embedded tissue sections were deparaffinized by xylene, and then rehydrated in the graded ethanol solution. The slides were immersed in 0.1 M citric acid buffer for antigen retrieval, and boiled for 30 min at 100℃. After cooled to room temperature, the slides were incubated with the primary antibodies against SIRT1 (1:100), Akt (1:100) and P-Akt (Ser473) (1: 100) at 37℃ for 2 h. After the sections were washed with PBS buffer, the Dako Envision system/HRP (Dako Cytomation, Denmark) was used to color the sections. For evaluation standards by World Health Organization (WHO) classification, cells staining at the nucleus were divided groups into negative staining (-), low (<20% positive), medium (20%-50% positive) and high (>50% positive). Nucleus immunostaining >20% of tumor cells was determined as high expression. The expression of staining at cytoplasm was qualified by the percentage of positively-stained cells as negative staining (-), low (<25% positive), medium (25%-50% positive) and high (>50% positive) groups. The staining intensity scored as 0, negative; 1, weak; 2, moderate; and 3, strong. A score of 0-1 was as low expression, and a score of 2-3 was regarded as high expression.
Plasmids, lentivirus and transfection
To knockdown expression of SIRT1, the negative control small hairpin RNA (shcon,5'-TTCTCCGAACGTGTCACGT-3') and SIRT1 shRNA (sh SIRT1, 5'-GGGAATCCAAAGGATAATT-3', 5'-AATTATCCTTTGGATTCCC-3') were synthesized and cloned into lentivirus vectors (GV248) to construct GV248- shcon and GV248-shSIRT1 lentivirus by Genechem (Shanghai, China). For overexpression, SIRT1 plasmid (5'-CCGGATTGAAGAATGTTGG-3', 5'-ATCTGCTCCTTTGCCACTCT-3') was cloned into LV5 vector (named as LV5- Vector and LV5-SIRT1).
For lentivirus transfection, stable cell lines were generated in 24-well plates with serum-free medium. MDA-MB-231, MCF7 cells were transduced with SIRT1 lentivirus at the infection MOI≥90, also BT549 cells were transfected with shSIRT1 lentivirus at the infection MOI≥90 for 24 h. Then cells were cultured in the medium with 10% FBS and continuously cultured for 6 days followed by selection with G418 (Invitrogen) at 500 μg/ml. For human SiAkt transfection, about 2×105 cells were seeded in 24-well plates before transfection. Cells transfected SiAkt at a final concentration of 50 pM using Lipo3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the protocol.
Co-Immunoprecipitation (Co-IP) technique
In the present study, Co-IP technique was performed to detect the direct interrelation between SIRT1 and Akt. The protein A/G beads (ThermoFisher Scientific, MA, USA) were incubated with antibodies against SIRT1 and Akt at 4℃for 6 h, respectively. After washed with TBS three times, the beads which combined with antibodies were incubated with the cell lysates prepared from ultrasonic fragmentation at 4℃overnight. Next, the beads were also washed with TBS, centrifuged at 1,3000 rpm for 5 min. After discarding the supernatant, the equivoluminal SDS buffer was added into the beads. Finally, the beads were boiled for 5 min, and the relevant antibodies were used to detect the target proteins by Western blot analysis.
Western blot analysis
Cultured cells were lysed in RIPA buffer (Beyotime Biotechnology, Beijing, China) directly and determined the concentration by BCA Protein Assay Kit (Beyotime Biotechnology). About 30 μg total protein was separated by 10% SDS-PAGE and then transferred onto the PVDF membrane (Millipore, Danvers, MA, USA). After blocked by 5% skim milk, the membrane was incubated with antibodies against SIRT1 (1:1000), Akt (1:1000), P-Akt (Ser473) (1:1000), GAPDH (1:500) at 4℃overnight. The next day, the membrane was washed with TBS-T buffer and then incubated with appropriate secondary antibodies at 37℃ for 2 h. Finally, the samples were detected by the ECL system (ThermoFisher).
Immunofluorescence (IF) technique
Cells were fixed with 4% paraformaldehyde for 20 min, then permeated with 0.1% TritonX-100 for 15 min. After being washed with PBS, cells were blocked by 10% goat serum supplemented with 0.1% BSA (Beyotime Biotechnology) for 30 min. Following this, cells were incubated with the primary antibodies at 4℃ overnight, and washed with PBS three times. Then cells were incubated with the secondary antibodies conjugated to Alexa-488 and Alexa-555 fluorescence (Invitrogen). Lastly, nuclei were stained with Hochest (Beyotime Biotechnology) before being mounted on slides. Intracellular localization of target proteins was captured by a laser scanning confocal microscope (TCS-SP2, Leica, Germany).
CCK8 assay
Cell proliferation of breast cancer was tested by the Cell Counting Kit-8 (CCK8, DOJINDO, Japan). First, we seeded stably infected cells into a 96-well plate at the density of 3000 cells/well in medium with 100 μl final volume. Then 10 μl/well CCK-8 solution was added and incubated with cells for 2 hours at 37℃. Lastly, cell viability was measured at 450 nm absorbance by a microplate reader. All CCK8 experiments were repeated at least three times independently.
Cell cycle analysis
Stably infected cells were harvested and washed with PBS, fixed in 70% alcohol for at least 2 hours at 4℃. About 1×106 cells were detected using the cell cycle analysis kit (Beyotime Biotechnology). Cells were centrifuged at 1,000 g for 5 min. After discarding the supernatant, cells were incubated with 500 μl PI 20mg/ml and RNAase 50 μg/ml for 30 min. Each sample was analyzed by the flow cytometry (BD Bioscience, USA).
Colony formation
About 500 stably infected cells/well were seeded in 6-well plates, and cultured for 10-14 days. Cells were fixed with 4% paraformaldehyde for 20 min, stained with crystal violet (Beyotime Biotechnology) for 10 min. Only ≥50 cells regarded as positive colonies were counted and compared.
Cell apoptosis assay
Apoptotic cells were determined using flow cytometry with an Annexin-V- FITC kit (BD Bioscience). Stably infected cells were collected by centrifugation at 200×g for 5 min and resuspended in a binding buffer at room temperature at a density of 1×106 cell/ml. The cells (100 μl) were mixed with 5 μl of annexin-V-FITC and 5 μl of PI in a culture tube and incubated at room temperature in the dark for 15 min. After addition of 400μl binding buffer, the cells (~10,000 cells per assay) were then analyzed by using a dual-laser FACS VantageSE flow cytometer (Becton Dickinson, Mountain View, CA). The percentages of apoptotic cells for each sample were estimated.
Transwell migration assay
Cell migration was investigated by transwell migration assay using transwell inserts with a 8 μm pore filter (BD Bioscience). After trypsinizated, 4×104 cells supplemented with serum-free medium were seeded into the upper chamber while complete medium was added to the lower chamber. After incubated with 24 h, the cells were fixed with paraformaldehyde for 15min and stained with crystal violet (Beyotime Biotechnology) for 10 min. Next, the cells on the upper surface of the membrane were wiped off, and cells on the lower membrane were survied by Leica microscope. Migration ability of cells was evaluated by the average number of migrated cells from 4 random fields.
Xenograft experiments
Four-weeks female BALB/c nude mice were obtained from the Animal Facility of Nantong University (Nantong, China), and maintained in the pathogen-free condition. To determine the tumor growth of SIRT1 in vivo, the stably infected breast cancer cells were re-suspended with serum-free medium at the density of 1×106 cells/100 μl, then implanted 200 μl cell suspension into the flank of nude mice subcutaneously. About a week later, when the tumors were visible as large as soy bean, width and length of tumors were measured twice a week for 4 weeks. Finally, animals were euthanized, then tumors were picked up and weighed. The following formula was used to calculate the tumor volume: tumor size (mm3) = width2×length×0.522.
Statistical analysis
All data were presented as means±SEM from experiments repeated at least three times independently. Statistical analysis of significance was performed using GraphPad Prism 5.0 software. Data from individual groups were compared by Students' test, and values of p<0.05 was considered to have statistical significance.
Results
SIRT1 is moderately up-regulated in the human breast cancer tissues and cell lines
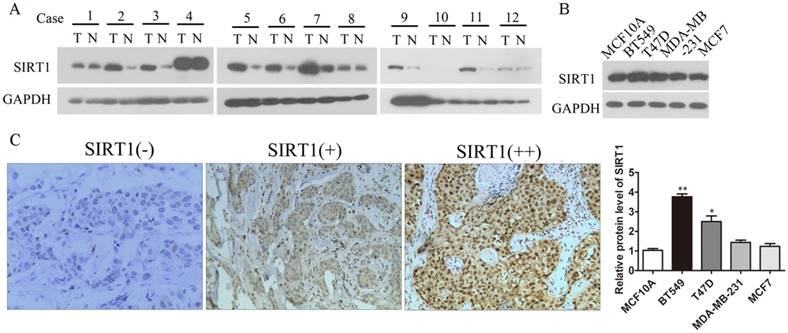
As SIRT1 was seen as having varying effects in previous studies, we investigated the expression of SIRT1 in breast cancer tissues and cell lines to explore the relationship between SIRT1 expressions and breast cancer development. Results showed that the protein level of SIRT1 was significantly increased in breast cancer tissues in comparison to adjacent normal breast tissues (Figure 1A). Also SIRT1 was up-regulated in breast cancer cell lines compared to normal breast epithelium cell line MCF10A, suggesting oncogenic activity in breast cancer (Figure 1B). Then IHC staining of SIRT1 was performed to detect SIRT1's role in tumors from breast cancer patients (n=100). IHC microscopy analysis revealed that SIRT1 was localized in both nucleus and cytoplasm of cancer cells (Figure 1C). Evaluated with standards described in Materials and Methods, the majority (74%, 74/100) of breast carcinoma samples was SIRT1-positive stained, whereas in non-cancerous samples, the percentage of SIRT1 positive staining plummeted to 28% (28/100) with the SIRT1-negative staining reaching 72% (72/100) (Table 1). At the same time, IHC staining of SIRT1 was statistically correlated with histological grade, tumor size, and lymph node metastasis (Table 2).
SIRT1 is up-regulated in human breast cancer tissues and cell lines. (A) Western blots analysis detected the expression of SIRT1 in breast cancer tissues (T) and normal counterparts (N) from 12 patients. (B) Western blots analysis tested the protein levels of SIRT1 in breast cancer cell lines and benign breast epithelial cell line (Data were presented as means±SEM. *p<0.05, **p<0.01. Student's t-test). (C) Representative IHC staining of SIRT1 on human breast cancerous tissue paraffin sections (n=100, Bar =100 μm).
| Group | n | SIRT1 expression | P | |
|---|---|---|---|---|
| Postive, n(%) | Negative, n(%) | |||
| adjacent normal breast tissues | 100 | 28(28%) | 72(72%) | <0.01 |
| breast cancer | 100 | 74(97%) | 26(3%) | |
| Group | n | SIRT1 expression | P | |
|---|---|---|---|---|
| Postive, n(%) | Negative, n(%) | |||
| Age | ||||
| ≤50 year | 60 | 41(68.3%) | 19(31.7%) | 0.131 |
| >50 year | 40 | 33(82.5%) | 7(17.5%) | |
| Histological grade | ||||
| Well (І) | 26 | 9(34.6%) | 17(65.4%) | 0.022 |
| Moderate (ΙΙ) | 23 | 20(87%) | 3(13%) | |
| Poor (ΙΙΙ) | 51 | 45(88.2%) | 6(11.7%) | |
| Tumor size(cm) | ||||
| ≤3.5 | 47 | 30(63.8%) | 17(36.2%) | 0.027 |
| >3.5 | 53 | 44(83%) | 9(17%) | |
| Lymph node metastasis | ||||
| no | 45 | 24(53.3%) | 21(46.7%) | 0.009 |
| yes | 55 | 50(90.9%) | 5(9.1%) | |
| Cancer embolus | ||||
| no | 40 | 30(75%) | 10(25%) | 0.061 |
| yes | 60 | 44(73.3%) | 16(26.7%) | |
Overexpression of SIRT1 promotes cell proliferation both in vitro and in vivo
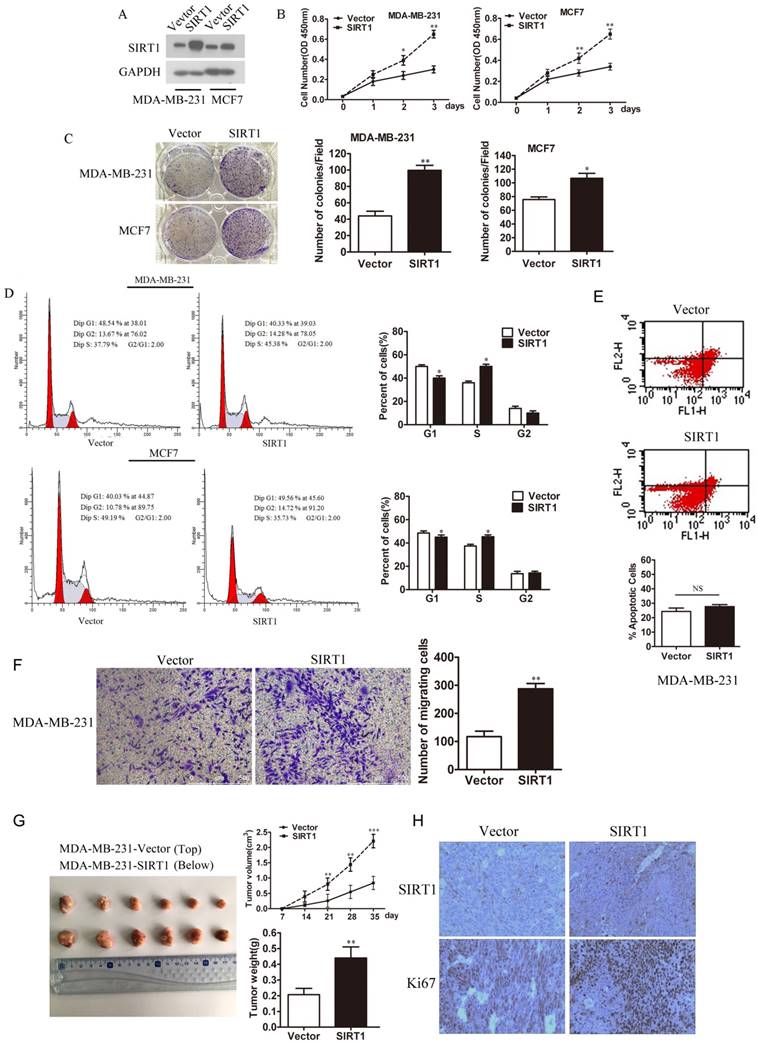
To understand the effect of SIRT1 on breast cancer, we established MDA-MB-231-SIRT1 and MCF7-SIRT1 cell lines that stably overexpressed SIRT1. Western blot analysis confirmed the higher expression levels of SIRT1 in these two cell lines over those vector cells (Figure 2A). Interestingly, overexpression of SIRT1 significantly promoted the cell viability and colony formation ability of breast cancer cells (Figure 2B and 2C). Meanwhile high-expression of SIRT1 facilitated transition from G1 to S, enhanced cell cycle progression in MDA-MB-231 and MCF7 cells (Figure 2D). Since P53 is well known substrate for SIRT1, which play important roles in cell apoptosis, Cell apoptosis was detected in MDA-MB-231 cells overexpressing SIRT1, using flow cytometry with an Annexin V FITC kit. The results showed that there were no significant differences between MDA-MB-231/SIRT1 cells and MDA-MB-231/vector cells (Figure 2E). To investigate whether SIRT1 regulate Metastasis of breast cancer cells, the transwell technique was used. The results showed overexpressing SIRT1 in MDA-MB-231cells promote cell metastasis (Figure 2F). In our in vivo study, xenograft nude mice models showed that overexpression of SIRT1 promoted breast cancer growth in vivo (Figure 2G). IHC staining analysis also demonstrated higher protein levels of Ki67 in tumors formed from MDA-MB-231-SIRT1 cells when compared with Vector cells (Figure 2H). All the above results demonstrated that overexpression of SIRT1 promoted cell growth of breast cancer both in vitro and in vivo.
Overexpression of SIRT1 promotes breast cancer cell growth in vitro and in vivo. (A) Western blot analysis of SIRT1 expression in MDA-MB- 231 and MCF-7 cell lines with or without stably overexpressed SIRT1. (B) Cell viability assay (C) Colony formation assay (D) Cell cycle analysis in MDA- MB-231 and MCF-7 cells with or without stably overexpressed SIRT1 (Data were presented as means±SEM. *p<0.05, **p<0.01, Student's t-test. Cell viability analysis with n=6; the rest analyzed with n=3). (E) Cell apoptosis (F) Cell migration ability in MDA-MB-231-SIRT1 and MDA-MB-231-Vector cells (Data were presented as means±SEM. **p<0.01, NS=no significance, Student's t-test. n=3). (G) In vivo tumor formation assay of MDA-MB-231-SIRT1 and MDA-MB-231-Vector breast cancer cells. At the end of the experiment, the tumors were dissected from mice. Volumes and weights of tumors between two groups were analyzed (Data were presented as means±SEM. **p<0.01, ***p< 0.001, Student's t test). (H) Representative IHC staining of SIRT1 and Ki67 in xenograft tumors between two groups (Bar=100 μm).
Knockdown of SIRT1 inhibits cell proliferation of breast cancer in vitro and in vivo
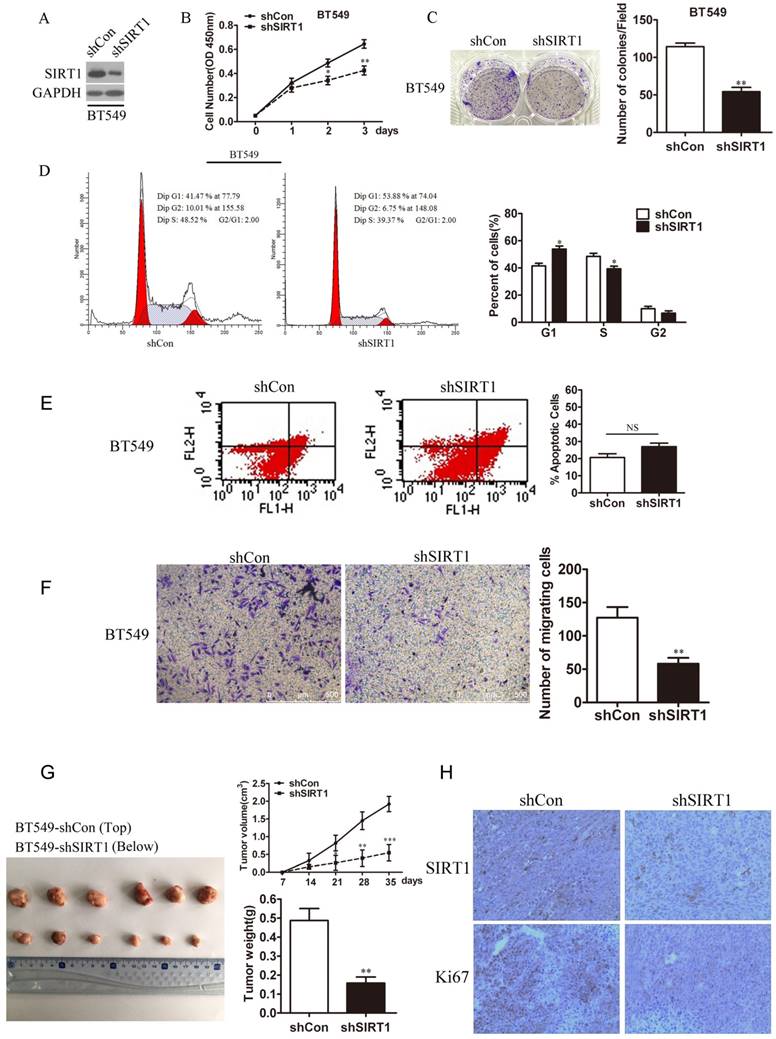
In the same way, we generated BT549-shSIRT1 cell line using shSIRT1 lentivirus to test breast cancer cell growth when SIRT1 was knocked down. Western blot analysis confirmed the SIRT1 protein level was knocked down in the cell line (Figure 3A). Down regulation of SIRT1 markedly inhibited breast cancer cell growth exhibited by attenuated ability in cell viability and colony formation (Figure 3B and 3C). In addition, after knocked down SIRT1, cell cycle progression was arrest via cell cycle analysis (Figure 3D). For cell apoptosis, there were no significant difference between BT549/sh SIRT1 cells and BT549/ shControl cells (Figure 3E). Knocking down SIRT1 in BT549 cells, the metastasis was dramatically decreased (Figure 3F). Consistently, in vivo bearing nude mice experiments revealed the inhibited effect of SIRT1 in tumor growth (Figure 3G). Also IHC analysis confirmed lower staining levels of Ki67 in tumors formed with BT549-shSIRT1 cells compared with control cell line (Figure 3H).
Knockdown of SIRT1 inhibits breast cancer growth in vitro and in vivo. (A) Western blot analysis was used to detect SIRT1 protein level in BT549-shSIRT1 cells and BT549-shCon cells. (B) Cell viability assay (C) Colony formation ability (D) Cell cycle analysis in BT549-shSIRT1 cells and BT549-shCon cells (Data were presented as means±SEM. *p<0.05, **p<0.01. Student's t-test. Cell viability analysis with n=6; the rest analyzed with n=3). (E) Cell apoptosis (F) Cell migration ability in BT549-shSIRT1 and BT549-shCon cells (Data were presented as means±SEM. **p<0.01, NS=no significance, Student's t-test. n=3). (G) In vivo tumor formation assay of BT549-shSIRT1 cells and BT549-shCon cells. The volumes and weights of tumors between two groups were analyzed. Photographed the dissected tumors from mice. (Data were presented as means± SEM. ***p<0.001, **p<0.01. Student's t test). (H) Representative IHC staining of SIRT1 and Ki67 in xenograft tumors between two groups (Bar =100 μm).
SIRT1 effects on breast cancer cell growth through modulating activity of Akt in vitro
To explore the molecular mechanisms of SIRT1 in breast cancer, we focused on some specific pathways. In previous studies, PI3K/Akt pathway has been found to confer tumorigenicity and promote tumor growth in various cancers. In addition, Akt and phosphorylated Akt were reported to be up-regulated in breast cancer and closely related to the development of the cancer.
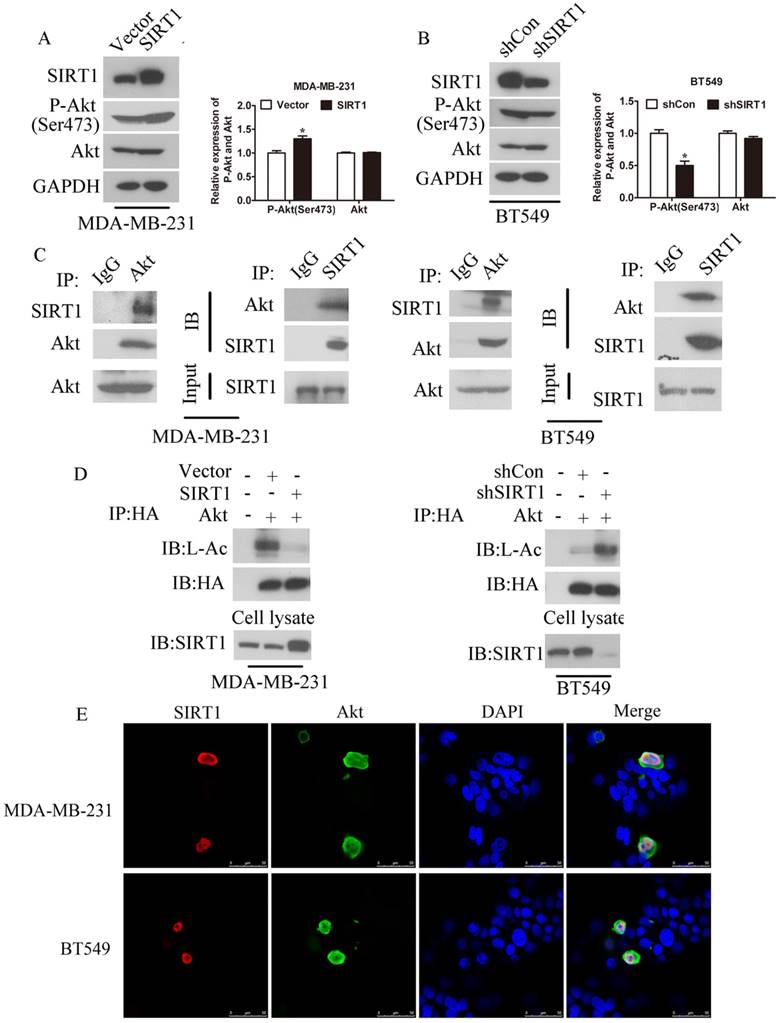
As SIRT1 obviously promoted breast cancer cell growth, we speculated that the effect of SIRT1 on breast cancer cells might be through the promotion of PI3K/Akt pathway. Thus we examined the expression of phosphorylated Akt in SIRT1 overexpressed breast cancer cell lines and Vector cells. Indeed, phosphorylation of Akt at Serine473 (P-Akt) was higher in breast cancer cells with high SIRT1 protein level than cancer cells with low SIRT1 level, but there is no significant difference in total Akt protein level. By contrast, knockdown of SIRT1 resulted in the decrease of P-Akt protein level (Figure 4A, S1 and 4B). These results implicated a positive correlation between the expression level of SIRT1 and P-Akt. Furthermore, the direct interrelation between SIRT1 and Akt was detected using Co-IP technology in MDA-MB-231 and BT549 breast cancer cells (Figure 4C). By the same way, we have observed the deacetylation of Akt was decreased after overexpression of SIRT1 in MDA-MB-231 cells while increased when SIRT1 was knocked down in BT549 cells (Figure 4D). We also performed the IF assay to investigate the cell location of these two molecules which showed both SIRT1 and Akt had co-location in cell nucleus (Figure 4E). These results confirmed that SIRT1 effected and deacetylated Akt directly in breast cancer cells in vitro.
SIRT1 effects on breast cancer cell growth through modulating activity of Akt in vitro. (A) Western blot analysis of P-Akt and Akt expression between MDA-MB-231-SIRT1 and MDA-MB-231-Vector breast cancer cells (Data were presented as means±SEM. **p<0.01. Student's t-test). (B) Western blot analysis of P-Akt and Akt expression between BT549-shSIRT1 cells and BT549-shCon cells (Data were presented as means±SEM. **p<0.01. Student's t-test). (C) Co-IP technique was performed to investigate the interrelation between SIRT1 and Akt in MDA-MB-231 cells and BT549 cells. (D) Deacetylation of Akt by SIRT1 was detected by Co-IP assay after overexpressing SIRT1 in MDA-MB-231 cells and knocking down SIRT1 in BT549 cells. (E) Co-localization of SIRT1 and Akt in MDA-MB-231 cells and BT549 cells was investigated by Co-IF technology. SIRT1 stained with red. Akt stained with green.
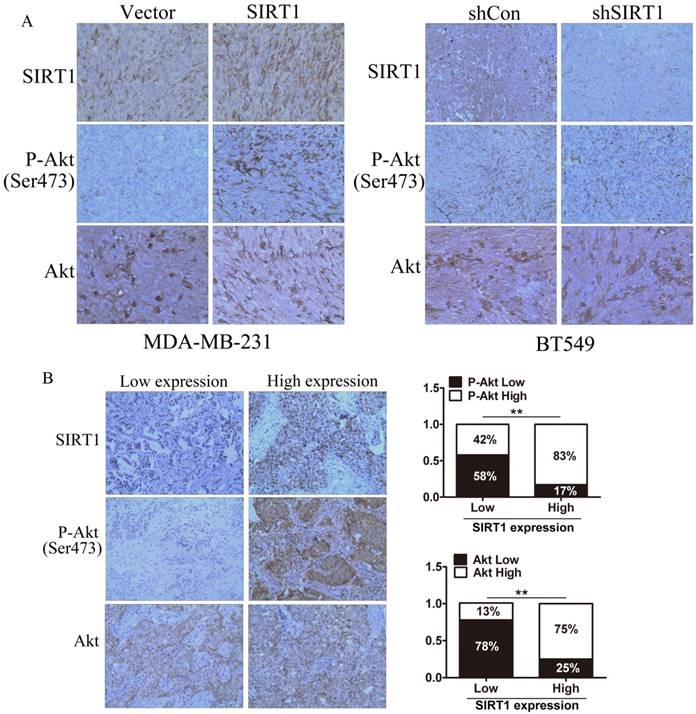
SIRT1 is correlated with expression of P-Akt in vivo
We investigated the expression of SIRT1, total Akt, and P-Akt as well as their correlation in subcutaneous implantation models and human breast cancer tissue samples by IHC staining analysis. The results demonstrated that the expression of P-Akt was higher in tissues formed from MDA-MB-231 cells with SIRT1 overexpression when compared to the Vector cells; BT549 cells with SIRT1 lower expression exhibited a lower P-Akt expression, while the expression of total Akt had no significant change in all groups (Figure 5A). In the samples from human breast cancer tissues with SIRT1 weak staining, there were 87% with low Akt expression and 58% with low P-Akt expression. Consistently, in the samples with SIRT1 strong staining, there were higher levels of Akt and P-Akt expression (75% and 87%, respectively). We noted that the protein level of SIRT1 was positively correlated with total Akt level (R=0.365, P=0.011) and P-Akt (R=0.497, P=0.028) in breast cancer tissues (Figure 5B). These results indicated expression of SIRT1 and Akt had positively correlation in breast cancer tissues in vivo.
SIRT1 is correlated with expression of P-Akt in vivo. (A) IHC staining of SIRT1, P-Akt, and Akt in tumors formed from xenograft (Bar=100 μm). (B) IHC analysis of SIRT1, P-Akt, and Akt in human breast cancer tissues. Left: representative photographs of IHC staining; right: low-high bar graph showing the correlation analysis (Bar=100 μm, **p<0.01).
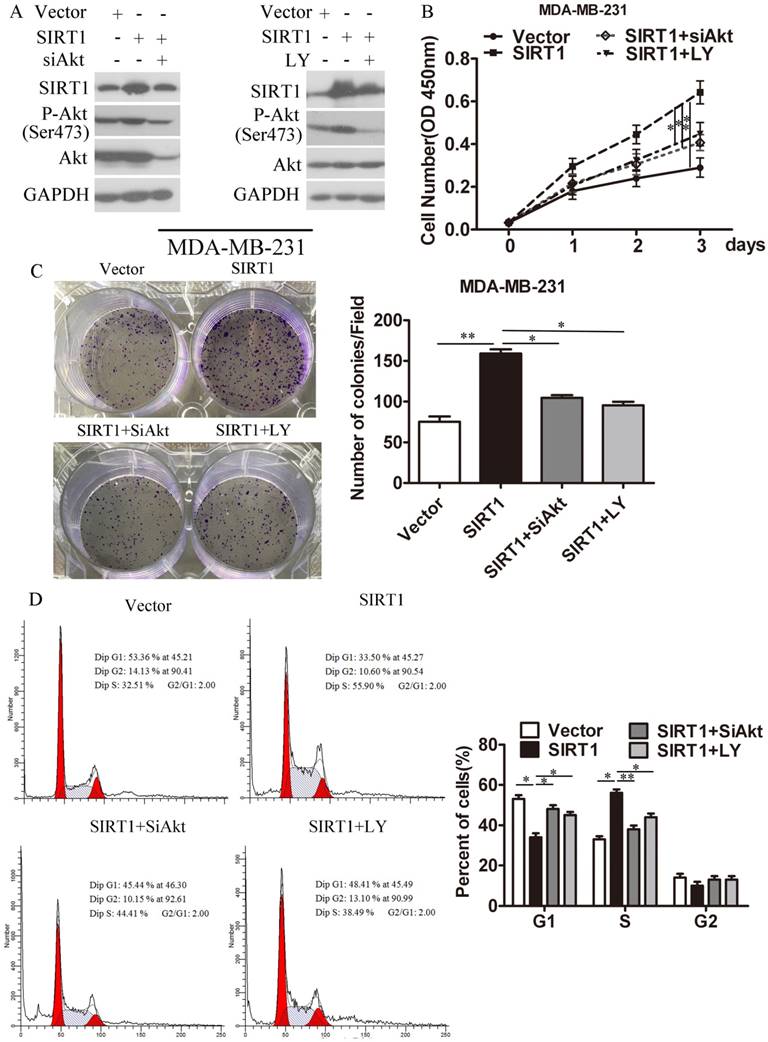
Reduction of Akt can partially eliminate the proliferative activity mediated by SIRT1
To further confirm the oncogenic role of SIRT1/Akt signaling pathway in breast cancer progression, we explored whether the effect of SIRT1 on breast cancer is dependent on the activity of Akt. We knocked down the Akt expression level by SiAkt or decreased the Akt activity with the PI3K/Akt inhibitor LY294002 in MDA-MB-231-SIRT1 and MCF-7-SIRT1 breast cancer cells (Figure 6A and S2A). CCK8 assays (Figure 6B and S2B) and cell colony formation analysis (Figure 6C and S2C) showed that ablation of both Akt and P-Akt could block SIRT1 mediated proliferation in breast cancer cells. Similarly, cell cycle analysis revealed that silencing of Akt and P-Akt hindered the ability of SIRT1 in boosting the breast cancer cells entry from G1 phase into S phase (Figure 6D and S2D).
Depletion of P-Akt and Akt in SIRT1-overexpressed cells abolished the oncogenic activity mediated by SIRT1. (A) Depletion of P-Akt and Akt in MDA-MB-231-SIRT1 cells with siAkt and PI3K/Akt pathway inhibitor LY294002. Protein levels of SIRT1, P-Akt and Akt were measured by Western blot analysis. (B) Cell viability analysis (C) Colony formation (D) Cell cycle distribution were evaluated (Data were presented as means±SEM. *p< 0.05, **p<0.01. Student's t-test. Cell viability analysis with n=6; the rest analyzed with n=3).
Discussion
Mammalian Sirtuins family members regulate numerous important cell function including cell survival, cell metabolism, proliferation, and differentiation [18]. SIRT1, as the most studied member of Sirtuins family, has been shown to have both pro- and anti-carcinogenic effects. On the one hand, SIRT1 presented as a tumor suppressor gene by maintaining genome stability of normal cells and promoting apoptosis of tumor cells [19]. On the other hand, SIRT1 created a conducive microenvironment for tumor cell survival and inhibited expression of tumor suppressor genes, thus was regarded as an oncogene to promote the development of tumorigenesis [20,21].
In breast cancer, a limited research has revealed that overexpression of SIRT1 was correlated with lymph node metastasis and the decrease of 5-year survival rate, indicating its oncogenic effect [22]. SIRT1 promoted cell survival through inactivation of the p53 pathway, which was also an essential for oncogenic signaling pathway of estrogen/estrogen receptor α (ERα) in breast cancer [4,5,23]. In addition, SIRT1 could promote cell migration by directly interacting and deacetylating cortactin, and then promoted the expression of multi-drug resistance-associated protein in tamoxifen-resistant breast cancer cells [24,25]. However, the mechanisms of SIRT1 induced breast cancer progression are little understood. In this study, we have uncovered that SIRT1 is significantly up-regulated in both breast cancer tissues and cells. Overexpression of SIRT1 promoted colony formation, cell proliferation, and cell cycle progression in breast cancer cells in vitro and accelerated tumor growth in vivo, whereas SIRT1 silencing inhibited these characteristics which were consistent with previous literatures.
CM et al [26] showed that Resveratrol (RES), as a Sirtuins activator, promotes cytotoxicity and pro-differentiation activity on breast cancer cells, which was in some conflict with our research. Indeed, RES activates SIRT3 in addition to SIRT1, and SIRT3 acts as a tumor suppressor inducing cancer cell death through the regulation of key proteins for malignant transformation [27]. Also, they showed the effects of RES on breast cancer cells were modulated not only by SIRT1/SIRT3, but also by mitochondrial complex inhibition. Most importantly, they found cytotoxic effects of RES, which is dependent on the increase of SIRT1 level, was only in high glucose containing medium, but not normal medium. We also detected the expression of SIRT1 in RES-treated breast cancer cells in our cultured conditions, however, there was no significant change in SIRT1 level (Figure S3). Thus, SIRT1 can function either as a tumor suppressor or as an oncogene, depending on the cellular background or environment cells are growing in.
In vitro studies, as SIRT1's regulation of p53 is well-known, we investigated cell apoptosis in MDA-MB-231/SIRT1 cells and BT549/shSIRT1 cells compared with the control groups using flow cytometry with an Annexin V FITC kit. The results showed that there were no significant differences between MDA-MB-231/SIRT1 cells and MDA-MB-231/Vector cells, as well as BT549/ shSIRT1 cells and BT549/shCon cells. We speculated that there might be other molecule signaling pathways to modulate SIRT1-mediated cell apoptosis besides p53, which compensates the effect of p53.
PI3K/Akt pathway is a key player in various types of malignant human tumors, such as breast cancer, lung cancer, melanoma, lymphoma and it significantly drives tumor development and biological processes including cell adhesion, growth, migration, invasion, and angiogenesis [28]. Recent literature suggested a crosstalk between SIRT1 and Akt signaling, Akt was deacetylated by SIRT1 which was necessary for the binding of Akt to PIP3 and for its membrane localization and activation in cardiac muscle [16]. In this study, we showed that up or down-regulation of SIRT1 made a difference on Akt activity, and SIRT1 directly interacted and deacetylated Akt in breast cancer cells. Ablation Akt activity could reverse high proliferation ability mediated by SIRT1. These results commonly revealed intricate connections between SIRT1 and Akt which demonstrated that the SIRT1/Akt signaling axis could significantly affect the development of breast cancer, suggesting its meaningful status in breast cancer diagnosis and treatment [29-31].
It is vital to recognize that SIRT1 promotes breast cancer progression through modulating Akt activity. Such molecular mechanisms underlying the progress of tumor by SIRT1 have not been reported before. As a result, controlling the expression of SIRT1 might be an alternative approach to manage PI3K/Akt pathway in breast cancer cells.
Abbreviations
SIRT1, Silent mating-type information regulation 2 homologue 1; Akt, Protein kinase B; P-Akt, phosphorylation of Protein kinase B; NAD+, nicotinamide adenine dinucleotide.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81270810).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Akram M, Iqbal M, Daniyal M. et al. Awareness and current knowledge of breast cancer. Biol Res. 2017;50(1):33
2. Kolahdooz F, Jang SL, Corriveau A. et al. Knowledge, attitudes, and behaviours towards cancer screening in indigenous populations: a systematic review. Lancet Oncol. 2014;15(11):504-16
3. Zhang XH, Hao S, Gao B. et al. A network meta-analysis for toxicity of eight chemotherapy regimens in the treatment of metastatic/advanced breast cancer. Oncotarget. 2016;7(51):84533-43
4. Vaziri H, Dessain SK, Ng Eaton E. et al. hSIR2(SIRT1) functions as an NAD-Dependent p53 Deacetylase. Cell. 2001;107(2):149-59
5. Luo J, Nikolaev AY, Imai S. et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107(2):137-48
6. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev of Biochem. 2004;73:417-35
7. Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene. 2014;33(13):1609-20
8. Zhao W, Kruse JP, Tang Y. et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451(7178):587-90
9. Deng CX. SIRT1, Is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5(2):147-52
10. Chen W, Bhatia R. Roles of SIRT1 in leukemogenesis. Curr Opin Hematol. 2013;20(4):308-13
11. Han L, Liang XH, Chen LX, et a1. SIRT1 is highly expressed in brain metastasis tissues of non-small cell lung cancer (NSCLC) and in positive regulation of NSCLC cell migration. Int J Clin Exp Pathol. 2013;6(11):2357-65
12. Huffman DM, Grizzle WE, Bamman MM, et a1. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67(14):6612-8
13. Riggio M, Polo ML, Blaustein M. et al. PI3K/AKT pathway regulates phosphorylation of steroid receptors, hormone independence and tumor differentiation in breast cancer. Carcinogenesis. 2012;33(3):509-18
14. Lee MS, Jeong MH, Lee HW. et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun. 2015;6:7769
15. Pinton G, Zonca S, Manente AG. et al. SIRT1 at the crossroads of AKT1 and ERβ in malignant pleural mesothelioma cells. Oncotarget. 2016;7(12):14366-79
16. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt Signaling by Sirtuins. Circ Res. 2014;114(2):368-78
17. Wan G, Tian L, Yu Y. et al. Overexpression of Pofut1 and activated Notch1 may be associated with poor prognosis in breast cancer. Biochem Biophys Res Commun. 2017;491(1):104-11
18. Hwang BJ, Madabushi A, Jin J. et al. Histone/protein deacetylase SIRT1 is an anticancer therpeutic target. Am J Cancer Res. 2014;4(3):211-21
19. Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol. 2009;185(2):203-11
20. Wang RH, Sengupta K, Li C. et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14(4):312-23
21. Wood JG, Rogina B, Lavu S. et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoan. Nature. 2004;430(7000):686-9
22. Nin V, Escande C, Chini CC. et al. Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem. 2012;287(28):23489-501
23. Moore RL, Faller DV. SIRT1 represses estrogen-signaling, ligand- independent ERα-mediated transcription, and cell proliferation in estrogen- responsive breast cells. J Endocrinol. 2013;216(3):273-85
24. Jin MS, Hyun CL, Park IA. et al. SIRT1 induces tumor invasion by targeting epithelial mesenchymal transition-related pathway and is a prognostic marker in triple negative breast cancer. Tumour Biol. 2016;37(4):4743-53
25. Chung SY, Jung YY, Park IA. et al. Oncogenic role of SIRT1 associated with tumor invasion, lymph node metastasis, and poor disease-free survival in triple negative breast cancer. Clin Exp Metastasis. 2016;33(2):179-85
26. Deus CM, Serafim TL, Magalhães-Novais S. et al. Sirtuin 1-dependent resveratrol cytotoxicity and pro-differentiation activity on breast cancer cells. Arch Toxicol. 2017;91(3):1261-78
27. Chen Y, Fu LL, Wen X. et al. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047
28. Massihnia D, Galvano A, Fanale D. et al. Triple negative breast cancer: shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget. 2016;7(37):60712-22
29. Sarma P, Bag I, Ramaiah MJ. et al. Bisindole-PBD regulates breast cancer cell proliferation via SIRT-p53 axis. Cancer Biol Ther. 2015;16:1486-501
30. Whitlock NC, Baek SJ. The anticancer effects of resveratrol: modulation of transcription factors. Nutr Cancer. 2012;64(4):493-502
31. Oh WK, Cho KB, Hien TT. et al. Amurensin G, a potent natural SIRT1 inhibitor, rescues doxorubicin responsiveness via down-regulation of multidrug resistance 1. Mol Pharmacol. 2010;78(5):855-64
Author contact
![]() Corresponding author: Shuyun Yang. Department of Pathology, Nantong Tumor Hospital. 30 North Tongyang Road, Pingchao, Nantong, Jiangsu, 226361, China. Tel: +86 0513 86729733. E-mail: shuyun_yangcom; Nong Zhang. Department of Pathology, School of Basic Medical Sciences, Fudan University. 138 Yixueyuan Road, Xuhui District, Shanghai, 200030, China. Tel: +86 021 54237825. E-mail: 14111010068edu.cn
Corresponding author: Shuyun Yang. Department of Pathology, Nantong Tumor Hospital. 30 North Tongyang Road, Pingchao, Nantong, Jiangsu, 226361, China. Tel: +86 0513 86729733. E-mail: shuyun_yangcom; Nong Zhang. Department of Pathology, School of Basic Medical Sciences, Fudan University. 138 Yixueyuan Road, Xuhui District, Shanghai, 200030, China. Tel: +86 021 54237825. E-mail: 14111010068edu.cn