Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(12):2116-2122. doi:10.7150/jca.24567 This issue Cite
Research Paper
Methylation patterns of RASA3 associated with clinicopathological factors in hepatocellular carcinoma
Hui Lin*1,2, Xiaoxiao Fan*1,2, LiFeng He1,2, Daizhan Zhou1,2,3,4 ![]()
1. Department of General Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China
2. Biomedical Research Center, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China.
3. Bio-X Center, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Shanghai Jiao Tong University, Shanghai, China.
4. Present address: Key Laboratory of Arrhythmias of the Ministry of Education of China, East Hospital, Tongji University School of Medicine; Institute of Medical Genetics, Tongji University, Shanghai, China.
* Hui Lin and Xiaoxiao Fan contributed equally for this work.
Received 2017-12-26; Accepted 2018-3-31; Published 2018-5-25
Abstract
Hepatocellular carcinoma (HCC) is the sixth most common tumor worldwide. The relationship between the gene methylation accumulation and HCC has been widely studied. In our study, we used the Sequenom EpiTYPER assay to investigate the methylation levels of the RASA3 in 164 HCC samples and paired adjacent non-cancerous tissues, and the association between methylation level and clinicopathological features. The methylation level of the RASA3 in HCC samples was found significantly lower than that in the adjacent non-cancerous tissues (P<0.0001). Moreover, the hypomethylation of RASA3 in HCC samples was connected with the presence of tumornecrosis (P=0.029) and alcohol intake (P=0.002). Furthermore, it was found that the expression of RASA3 was significantly decreased in tumor tissues (P=0.0053), which was also correlated with the methylation levels of RASA3 gene. Thus, RASA3 hypomethylation is a common feature in HCC, and may be a potential mechanism for HCC development, and serves as a useful biomarker for the early detection, especially in alcohol-associated HCCs.
Keywords: RASA3, Hepatocellular carcinoma, Methylation, clinicopathological factors
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common malignant tumor and the third leading reason of cancer-related death in the world [1]. The incidence and mortality rates of HCC were increasing in many parts of the world, particularly in Asia [2]. HCC usually has a poor prognosis due to its difficulty of early diagnosis, rapid infiltrating growth and high incidence of metastasis. Hence, the early detection and treatment of HCC has been a severe health problem worldwide.
It is well known that malignant transformation of the hepatocyte is a course of multiple steps associated with various genetic alterations [3]. During its development and progression, DNA methylation plays a dominant role by altering gene expression [4]. Some studies reported that the methylation of tumor suppressor genes was associated with hepatocarcinogenesis in HCC cell lines [5, 6]. The aberrant methylated genes have been reported to act as a useful biomarker for early detection, tumor progression or prognosis of HCC, which displays a promising clinical benefit [4, 7].
For being able to switch between two different conformations, an active GTP-bound state and inactive GDP-bound complex, RAS is thought to be a crucial oncogene in multiple human tumors. Besides genetic mutations and growth-factor-receptor activation, RAS can be activated continuously through the inactivation of RAS GTPase-activating proteins (RAS GAPs), which leads to tumorigenesis [8]. RASA3, a member of the family of GAPs, has been identified as a potential tumor suppressor gene, which correlated with some malignant tumors like colorectal cancer and bladder tumor [9, 10]. Moreover, subtelomeric deletion of chromosome 13 which takes away the RASA3 locus along with other genes, has been reported to be associated with head-and-neck squamous cell carcinoma [11]. Interestingly, belonging to the GAP1 subfamily as same as RASA3, RASAL is reported to be connected with some tumors including HCC through CpG methylation, resulting in aberrant RAS activation [12]. Therefore, we hypothesized that aberrant CpG methylation of RASA3 may constitute a mechanism by which aberrant RAS activation can lead to hepatocarcinogenesis.
In this study, we used the Sequenom EpiTYPER to analyze the methylation changes of the RASA3 between the HCC samples and adjacent non-cancerous samples. And we further analyzed the relationship between the methylation patterns of the RASA3 and clinicopathologic features and assessed if the methylation level of RASA3 could provide a potential biomarker for HCC.
Materials and methods
Patients and Tissue Samples
From January, 2012 to December, 2012, we collected HCC tissues and paired adjacent noncancerous tissues from 164 patients treated at the Shanghai Eastern Hepatobiliary Surgery Hospital. Their ages were from 22 to 76 years (51.5±10.3 years: means ± standard deviation) and the ratio of male-to-female was 139:25. Serum carcinoembryonic antigen (CEA), alpha-fetoprotein (AFP), and CA19-9 were detected by the electrochemiluminescence immunoassay using cobas E601 immunoassay analyzers (Roche Diagnostics, GmbH, Mannheim, Germany). All tissues we collected were defined as HCC by pathological diagnosis. The diagnosis of the HCC referred to morphology and immunohistochemistry provided by WHO [13]. We performed the immunohistochemistry on 4% formaldehyde fixed tissues. HCCs were made up of tumor cells that generally resemble hepatocytes morphologically. Immunohistochemically, we used Polyclonal antiserum to carcinoembryonic antigen (pCEA), Glypican 3, Hepatocyte Paraffin 1, and CD10 to discriminate HCC from other liver tumors. Grading of the HCCs was referred to the Edmondson-Steiner grading system [14], ranging from grade 1 to 4, based on nuclear and cellular atypia. Tumor sizes were measured in accordance to the size of the largest tumor for multiple masses and the largest dimension of solitary mass.
The clinicopathological features of the patients in our study are summarized in Table 1. 138 patients had hepatitis B and 4 patients had hepatitis C. Of the 164 patients, 34 patients had an alcohol consumption more than 50g/day. In addition, 124 patients presented with tumornecrosis and 54 patients had different types of liver cirrhosis.
The clinical characteristics of the HCC patients in this study
| Characters | Categories | NO. |
|---|---|---|
| SEX | Female | 25 |
| Male | 139 | |
| Hepatitis | NONE | 22 |
| HBV | 138 | |
| HCV | 4 | |
| Alcohol habit(≥50g/day) | NO | 130 |
| YES | 34 | |
| High Blood pressure | NO | 144 |
| YES | 14 | |
| Type 2 Diabetes | NO | 144 |
| YES | 13 | |
| Family History | NO | 149 |
| YES | 15 | |
| Age (mean± SD) | 51.5±10.3 | |
| AFP (mean± SE) | 29481.7±14940.5 | |
| CEA (mean± SD) | 2.83±1.67 | |
| CA19-9 (mean± SE) | 33.4±5.57 | |
| Diameter of tumor (mean± SD) | 6.81±3.94 | |
| Fibrous Capsule | NO | 56 |
| YES | 108 | |
| Satellite Tumor | NO | 120 |
| YES | 44 | |
| Tumornecrosis | NO | 40 |
| YES | 124 | |
| Microvascular invasion | NO | 107 |
| YES | 57 | |
| Liver cirrhosis | None | 107 |
| Micronodular cirrhosis | 30 | |
| Mixed macro-micro-nodular cirrhosis | 24 | |
| Macronodular cirrhosis | 3 | |
| Architectural patterns | Thin trabecular pattern | 16 |
| Thicker trabecular pattern | 118 | |
| Other types | 30 | |
| TNM stage | < III | 19 |
| III | 139 | |
| > III | 6 |
AFP, serum alpha-fetoprotein; CEA, carcino-embryonic antigen.
The collected tissue samples were frozen in liquid nitrogen immediately and stored at -80˚C until processing. With informed consent of patients, all the tissue samples and associated clinicopathological information were allowed to collect and all the study protocol were approved by the Clinical Research Ethics Boards of the Shanghai Eastern Hepatobiliary Surgery Hospital.
DNA Extraction and Bisulfite Conversion
We used the QIAamp DNA Mini Kit to extract genomic DNA from 25ug tissue samples according to the manufacturer's standard protocol (QIAGEN, Hilden, Germany). Genomic DNA (500ng) was modified through sodium bisulfite treatment following the protocol of the EpiTect Fast DNA Bisulfite Kit (QIAGEN, Hilden, Germany).
Sequenom analysis
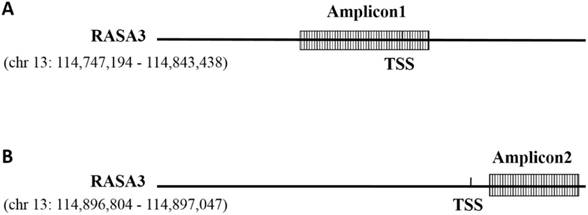
The methylation level of RASA3 was analyzed in Sequenom EpiTYPER assay. According to the UCSC genome database, RASA3 gene has several variants (chr13: 114747194 - 114898095; chr13: 114782713 - 114898095, chr13: 114747194 - 114843438, chr13: 114747194 - 114898095), there are two potential promotors around the two candidate TSS (Transcriptional Start Site) of RASA3 gene (chr13: 114843438 and chr13: 114898095 respectively). Two amplicons (Amplicon1) were selected for the methylation analysis. The first amplicon (Amplicon1) locates at chr13:114,843,368—114,843,766 (GRCh37/hg19), which locate at the promotor region in one variant of RASA3 (chr 13:114747194 - 114843438) (Fig. 1); the other amplicon (Amplicon2) locates at chr13: 114,896,804 - 114,897,047 (GRCh37/hg19), which comprise 6 CpG sites and locate at the promotor region in one variant of RASA3 (chr13:114747194-114898095) (Fig. 1). The primers were designed by using EpiDesigner (http://www.epidesigner.com/) with default setting (see Supplementaary Table 1).
Schematic representation of the amplicons of the RASA3 promoter. TSS: transcription initiation site. (A) Amplicon1 locates at the promotor region in one variant of RASA3 (chr13:114747194 - 114843438); (B) Amplicon2 locates at the promotor region in one variant of RASA3 (chr13:114747194 - 114898095).
Through PCR amplification, SAP cleanup, T Cleavage and conditioning of the hMC reaction products with Clean Resin steps, the products were transferred to a SpectroCHIP® array and analyzed on the MassARRAY® Analyzer 4 instrument.
Data analysis
We used the SPSS 19.0 to perform the statistical analyses. A p value <0.05 was considered statistically significant. The methylation level changes of RASA3 were analyzed by using paired t-test between HCC tissues and adjacent non-cancerous tissues. We detected the connections between methylation changes and clinical features by using independent-samples T-test. Paired t-test was also conducted to compare the mRNA expression difference between tumors and matched normal tissues.
Results
Methylation level of RASA3 in HCC and adjacent noncancerous tissues
The Amplicon 1 (chr13:114,843,368—114,843,766) cover 399bp and comprise 12 CpG sites, and 11 of them (CpG1, CpG2, CpG3, CpG4, CpG6, CpG7, CpG8, CpG9, CpG10, CpG11) were genotyped successfully by using Sequenom Technology (see supplementary Table 2). The Amplicon 2 (chr13: 114,896,804 - 114,897,047) cover 244bp and comprise 6 CpG sites, and all of them were genotyped successfully by using Sequenom Technology (see supplementary Table 3). By using paired t-test analysis, the methylation levels of the all 11 CpG sites in Amplicon 1 showed significant differences in methylation level between HCC and adjacent noncancerous tissues (P<0.0001, Table 2), but a marginal difference were detected in the CpG5.6 site in Amplicon 2 with hypermethylation in tumor tissues (P=0.048 Supplementary Table 4).
The comparison between methylation levels of the 11 CpG sites in 164 paired tissues
| CpGs | Group | mean | ⊿mean | P Value |
|---|---|---|---|---|
| CpG1 | Normal | 87.7% | 14.3% | <0.0001 |
| Tumor | 73.4% | |||
| CpG2.3 | Normal | 83.6% | 17.3% | <0.0001 |
| Tumor | 66.4% | |||
| CpG4 | Normal | 80.9% | 16.9% | <0.0001 |
| Tumor | 63.9% | |||
| CpG5.6 | Normal | 56.3% | 14.5% | <0.0001 |
| Tumor | 41.8% | |||
| CpG7.8 | Normal | 78.5% | 18.3% | <0.0001 |
| Tumor | 60.2% | |||
| CpG9 | Normal | 69.6% | 12.7% | <0.0001 |
| Tumor | 56.8% | |||
| CpG10 | Normal | 70.9% | 6.4% | <0.001 |
| Tumor | 64.6% | |||
| CpG11 | Normal | 59.1% | 7.2% | <0.005 |
| Tumor | 51.9% | |||
| Mean | Normal | 72.2% | 11.6% | <0.0001 |
| Tumor | 60.6% |
In the Amplicon 1, it showed significantly hypomethylated (P<0.0001) in HCC tissues compared with adjacent noncancerous tissues, with over 6% differences of methylation levels between paired tissues (from 6.4% to 17.3%). In addition, the methylation levels of the 11CpG sites were correlated with each other significantly (supplementary table 5).
RASA3 methylation associated with HCC risk factors and clinical pathological factors
To detect if hypomethylation of RASA3 is a potential biomarker of HCC, we analyzed the associations between the methylation levels and multiple clinicopathological factors. Meanwhile, to explore the integrated effect of designed CpG sites in this area, the mean value of methylation levels of 11 CpG sites was analyzed, which were correlated significantly with each other. We found that the methylation level of RASA3 in HCC samples was lower significantly in patients with tumornecrosis (P=0.029, Fig. 2A). Of the 123 patients with tumornecrosis, the mean methylation level of RASA3 was 59.01%, while of the 40 patients without tumornecrosis, the mean methylation level was 66.35%.
The association between methylation levels of RASA3 promoter in HCC and tumornecrosis (A), alcohol intake (B).
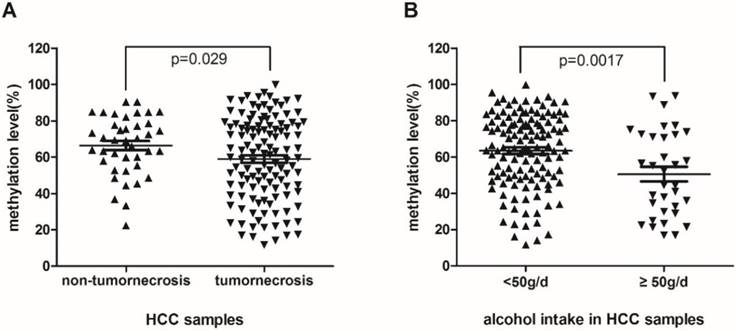
In addition, the methylation level of the RASA3 in alcohol-associated HCC samples was lower significantly than non-alcohol-related HCC (P=0.0017, Fig.2B). Of the 34 patients with alcohol habit (≥50g/day), the mean methylation level was 50.59%, which was lower than the mean methylation level of the other 129 patients (63.5%). Moreover, hypomethylation was also found in HCC samples with micronodular cirrhosis compared with the mixed macro-micro-nodular and macronodular cirrhosis HCC samples (P=0.043, supplementary Fig. 1). The mean methylation level of the former was 55.34% and the latter was 65.48%. For the other evaluated clinical factors, including age, high blood pressure, Type 2 Diabetes, family history, HBV, HCV, AFP, CEA, CA199, diameter of tumor, fibrous capsule, satellite tumor, microvascular invasion, architectural patterns, TNM stage, there was no significant association with the methylation level of the RASA3 in HCC.
The expression level of RASA3 in HCC and adjacent noncancerous tissues
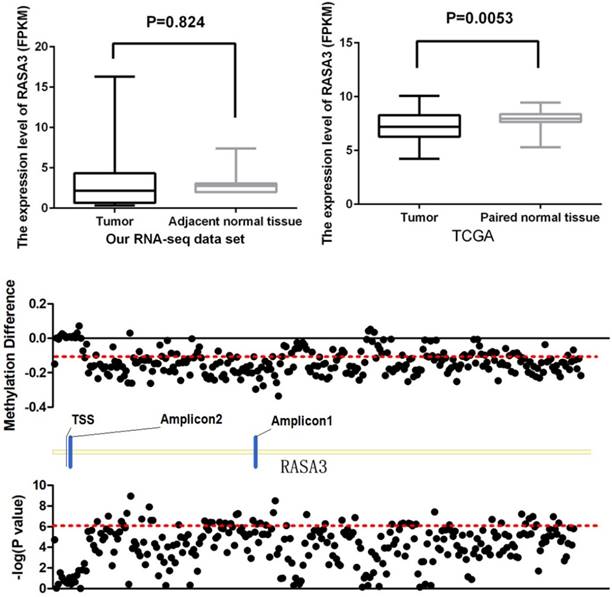
The RNA sequencing of 11 HCC tissue and their matched normal tissues had been performed in our institution as previously described [15]. In our RNA-seq data set, there was no significant difference between HCC samples and their paired normal tissues (P=0.824, Fig 3A), but the expression of RASA3 showed a trend to be higher in tumor tissues. Then, we proceeded to query RNA-seq data of fifty HCC tissues and paired adjacent noncancerous tissues from the Cancer Genome Atlas (TCGA) dataset. The expression was found significantly decreased in the tumor tissues (P=0.0053, Fig. 3B). From the TCGA data set, we also found most of CpG sites, successfully genotyped by the HumanMethylation450 BeadChip in RASA3 gene, were hypomethylated in HCC (Fig. 3C). The methylation in Amplicon 1 region were decreased significantly, but the methylation in Amplicon 2 region were slightly increased or had no changes, which were consistent with our results in present study.
The expression level of RASA3 in tumor tissues and their matched normal tissues. (A) Our RNA-seq data set (n=11), (B) The data from TCGA (n=50); (C) the methylation difference of 281 successfully genotyped CpG loci in RASA3 between tumor tissues and normal tissues from 50 paired samples in TCGA data set.
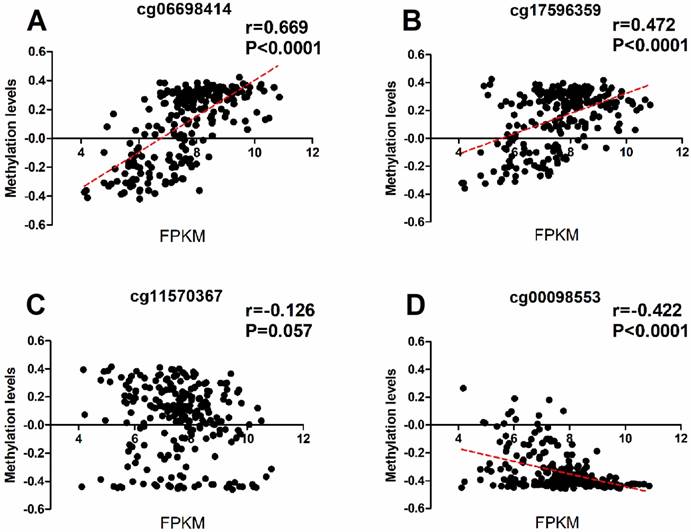
To investigate wheather the potiental regulation function of the methylations of RASA3, we downloaded the methylation and expression data of RASA3 in a total of 231 HCC samples from TCGA, and selected four CpG sites (cg06698414, cg17596359, cg11570367, and cg00098553; supplementary Table 2 and Supplementary Table 3) next to the two-amplicon regions in our study, and did a correlation analysis between methylation and gene expression. We found the cg06698414 (Chr13:114843024) and cg17596359 (Chr13:114844124) next to amplicon 1 were significantly hypomethylated in tumor tissues, and showed positive correlation with RASA3 expression (P<0.0001, Fig 4A, Fig 4B and Supplementary Table 6). We also found the cg00098553 (Chr13:114897502) next to amplicon 2 were slightly hypermethylated in tumor tissues, and showed negative correlation with RASA3 expression (P<0.0001, Fig 4C, Fig 4D and Supplementary Table 6). Thus, the methylations in RASA3 play very important roles in the regulation of gene expression.
The correlation between the methylation and expression data of RASA3 in four selected CpG sites closed to the amplicon studied. Cg06698141 and cg17596359 were next to the Amplicon 1 region, cg11570367 and cg00098553 were next to the Amplicon 2 region. (A), correlation analysis between the methylation levels of cg 06698414 and RASA3 gene expression. (B), correlation analysis between the methylation levels of cg 06698414 and RASA3 gene expression. (C), correlation analysis between the methylation levels of cg 06698414 and RASA3 gene expression. (D), correlation analysis between the methylation levels of cg 06698414 and RASA3 gene expression.
Discussion
DNA methylation is one of the most often investigated epigenetic mechanism which is involved in various tumors development [16]. Aberrant CpG island methylation of gene plays a significant role in inactivating tumor suppressor genes in human tumors progression including HCC [17]. In the present study, as a potential tumor suppressor gene, the methylation level of RASA3 gene were analyzed in HCCs. Our findings showed that the methylation levels of RASA3 in HCC tissues were significantly hypomethylated than adjacent noncancerous tissues (P<0.0001), and that there were over 6% differences of methylation level between paired tissues (from 6.4% to 17.3%), which indicated this epigenetic change might participate in the development of HCC carcinogenesis. RASA3 hypomethylation may activate RAS protein continuously to promote hepatocarcinogenesis through down-regulating RASA3 expression, which is different from the common viewpoint that hypermethylation of CpG islands tends to be associated with gene silencing. However, some studies have showed that hypermethylation of CpG islands in gene can be significantly associated with activation of some cancer-related genes, challenging the common view [18, 19]. Therefore, further studies should be done to explore the mechanisms between methylation status and the regulation of gene expression including RASA3.
Another important result of our study was that aberrant methylation level of RASA3 promoter was associated with major risk factors for HCC. HCC generally occurs on the basis of chronic inflammation that is secondary to the alcoholism or hepatitis virus infection, which increases the risks for hepatocarcinogenesis. In our study, methylation level of RASA3 was significantly lower in alcohol-associated HCC than in non-alcohol-related HCC indicating that alcohol intake may have a strong influence on RASA3 methylation levels in HCC. Some previous studies have reported a connection between alcohol consumption and aberrant methylation in HCC [20, 21]. Our findings further strengthen the notion that alcohol intake may impact DNA methylation levels in targeted genes, promoting tumor progression by means of an epigenetic mechanism. However, the precise mechanism by which alcohol can promote a reduction in methylation of RASA3 during HCC development remains unclear. Further studies should be done to detect whether alcohol consumption influences RASA3 directly or indirectly.
To our knowledge, abnormally methylated genes in cancer may serve as potential biomarkers for clinical use [22]. Tumor necrosis usually results from the lack of blood supply in the central portion of tumors associated with rapid tumor growth, which indicates worse prognosis and survival in HCC patients. Our results showed that the CpG islands methylation level of RASA3 in HCC samples was lower significantly in patients with tumornecrosis than HCC without tumornecrosis. It may be because RAS protein is activated by hypomethylation of RASA3, which contributed significantly to acceleration of tumor cell growth connected with tumor growth [8, 23]. Thereby the hypomethylation of RASA3 may serve as a prognosis biomarker for HCC. To facilitate clinical application, methylation levels of RASA3 should be further assessed in blood samples. In addition, a variety of methods such as serum AFP and radiographic test can be combined with DNA methylation test to improve its sensitivity and specificity for early diagnosis of HCC.
In summary, hypomethylation of RASA3 could be involved in the liver carcinogenesis, and associated with the presence of tumor necrosis and alcohol intake in HCC. Moreover, the methylation levels of RASA3 might discriminate HCC from liver cirrhosis or heavy drinkers. Thus, the methylation levels of RASA3 might be a promising epigenetic biomarker for HCC diagnosis and progression.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was funded by Medical Crossover Foundation of Shanghai Jiaotong University (No. YG2015MS42 and YG2014MS36), the National Nature Science Foundation of China (81121001, 81361120389, and 81421061) and the National Key Technology P&D Program (2016YFC1201701). Shanghai Key Laboratory of Psychotic Disorders (13dz2260500).
Author Contributions
Hui Lin and Xiaoxiao Fan contributed to the conception of the study, the data analyses and wrote the manuscript.
Xiaoxiao Fan and Lifeng He contributed to deal with Tissue Samples, DNA extraction and Bisulfite conversion.
Daizhan Zhou designed the experiments, collected the samples, performed the bioinformatics analysis, and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Murata S, Mine T, Sugihara F, Yasui D, Yamaguchi H, Ueda T. et al. Interventional treatment for unresectable hepatocellular carcinoma. World journal of gastroenterology: WJG. 2014;20:13453-65
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
3. De Minicis S, Marzioni M, Benedetti A, Svegliati-Baroni G. New insights in hepatocellular carcinoma: from bench to bedside. Annals of translational medicine. 2013;1:15
4. Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA. et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. The Journal of clinical investigation. 2007;117:2713-22
5. Huang J, Zhang YL, Teng XM, Lin Y, Zheng DL, Yang PY. et al. Down-regulation of SFRP1 as a putative tumor suppressor gene can contribute to human hepatocellular carcinoma. BMC cancer. 2007;7:126
6. Zhang C, Li H, Zhou G, Zhang Q, Zhang T, Li J. et al. Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J Pathol. 2007;212:134-42
7. Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology. 2012;56:994-1003
8. Downward J. Targeting RAS signalling pathways in cancer therapy. Nature reviews Cancer. 2003;3:11-22
9. Tang J, Li Y, Lyon K, Camps J, Dalton S, Ried T. et al. Cancer driver-passenger distinction via sporadic human and dog cancer comparison: a proof-of-principle study with colorectal cancer. Oncogene. 2014;33:814-22
10. Yao R, Yi Y, Grubbs CJ, Lubet RA, You M. Gene expression profiling of chemically induced rat bladder tumors. Neoplasia. 2007;9:207-21
11. Schurmans S, Polizzi S, Scoumanne A, Sayyed S, Molina-Ortiz P. The Ras/Rap GTPase activating protein RASA3: from gene structure to in vivo functions. Adv Biol Regul. 2015;57:153-61
12. Jin H, Wang X, Ying J, Wong AH, Cui Y, Srivastava G. et al. Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12353-8
13. SR H, LA A. World Health Organization classification of tumours. Pathology and genetics of tumours of the digestive system. Lyon: IARC Press. 2000
14. Edmondson HA, Henderson B, Benton B. Liver-cell adenomas associated with use of oral contraceptives. N Engl J Med. 1976;294:470-2
15. Zhang H, Ye J, Weng X, Liu F, He L, Zhou D. et al. Comparative transcriptome analysis reveals that the extracellular matrix receptor interaction contributes to the venous metastases of hepatocellular carcinoma. Cancer genetics. 2015;208:482-91
16. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33(Suppl):245-54
17. Baylin SB, Chen WY. Aberrant gene silencing in tumor progression: implications for control of cancer. Cold Spring Harb Symp Quant Biol. 2005;70:427-33
18. Bert SA, Robinson MD, Strbenac D, Statham AL, Song JZ, Hulf T. et al. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer cell. 2013;23:9-22
19. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T. et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14:534-42
20. Lambert MP, Paliwal A, Vaissiere T, Chemin I, Zoulim F, Tommasino M. et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. Journal of hepatology. 2011;54:705-15
21. Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV. et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PloS one. 2010;5:e9749
22. Liu WR, Shi YH, Peng YF, Fan J. Epigenetics of hepatocellular carcinoma: a new horizon. Chinese medical journal. 2012;125:2349-60
23. Qamra A, Xing M, Padmanabhan N, Kwok JJT, Zhang S, Xu C. et al. Epigenomic Promoter Alterations Amplify Gene Isoform and Immunogenic Diversity in Gastric Adenocarcinoma. Cancer discovery. 2017;7:630-51
Author contact
![]() Corresponding author: Daizhan Zhou, Phd, Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. 3 East Qingchun Rd, Hangzhou 310016, China. Tel: 86-13738055489; Fax: 86-571-86044817; Email: 3416252edu.cn/zhoudaizhancom
Corresponding author: Daizhan Zhou, Phd, Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. 3 East Qingchun Rd, Hangzhou 310016, China. Tel: 86-13738055489; Fax: 86-571-86044817; Email: 3416252edu.cn/zhoudaizhancom