Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(15):2589-2602. doi:10.7150/jca.23023 This issue Cite
Research Paper
Subtyping Of Triple Negative Breast Carcinoma On The Basis Of RTK Expression
Harald Hessel1, Manuela Poignée-Heger2, Sabine Lohmann2, Bianca Hirscher3, Andrea Herold2, Gerald Assmann1,4, Jan Budczies5, Karl Sotlar1,6, Thomas Kirchner1 ![]()
1. Institute of Pathology, Faculty of Medicine, LMU Munich, Germany
2. Roche Diagnostics GmbH, Penzberg, Germany
3. Roche Diagnostics International AG, Rotkreuz, Switzerland
4. Pathologiepraxis München, Germany
5. Institute of Pathology, Charité University Hospital, Berlin, Germany
6. University Institute of Pathology, University Hospital Salzburg, Paracelsus Medical University, Austria
Received 2017-9-26; Accepted 2018-4-16; Published 2018-6-23
Abstract
Background: "Triple-negative breast cancers" (TNBC) comprise a heterogeneous group of about 15% of invasive BCs lacking the expression of estrogen and progesterone receptors (ER, PR) and the expression of HER2 (ERBB2) and are therefore no established candidates for targeted treatment options in BC, i.e., endocrine and anti-HER2 therapy. The aim of the present study was to use gene expression profiling and immunohistochemical (IHC) characterization to identify receptor tyrosine kinase (RTK) profiles that would allow patient stratification for the purposes of target-oriented personalized tumor therapy in TNBC.
Methods: Twenty-nine cases of TNBC selected according to routine diagnostic IHC/cytogenetic criteria were examined by reverse transcription polymerase chain reaction (RT-PCR). RTK mRNA expression profiles were generated for a total of 31 tumor-relevant biomarkers, mainly belonging to the IGF- and EGF-receptor families but also including biomarkers related to downstream signaling. Protein expression of selected biomarkers was investigated by IHC.
Results: Hierarchical cluster analysis revealed a dichotomous differentiation pattern amongst TNBCs. A significant difference in gene expression was observed for 16 of the 31 RTK-associated tumor relevant biomarkers between the two newly identified TNBC subgroups. The findings were verified at the posttranslational level by the IHC data. The RTKs HER4, IGF-1R and IGF-2R and the hormone receptors ER and PR below the IHC detection limit play a central role in the differentiation of the two TNBC subgroups. Observed survival was reported as Kaplan-Meier estimates and point towards an improved survival of patients with RTK-high with superior three-year survival rate of 100% compared to RTK-low gene signatures with superior three-year survival rate of 60% (log-rank test, p-value = 0.022).
Conclusion: Gene-expression and IHC analysis of the EGF and IGF receptor families and biomarkers associated with downstream signaling point to the existence of two distinct TNBC subtypes. The RTKs HER4, IGF-1R, IGF-2R and the hormone receptors ER and PR appear to be of particular importance here. Based on survival analysis the differentiation of TNBC with RTK-high and RTK-low gene signatures seems to be of prognostic relevance. Additionally, correlation analysis of the relationship between RTKs and ER suggests co-regulatory mechanisms that may have potential significance in new therapeutic approaches.
Keywords: triple-negative breast cancer, subtyping, personalized tumor therapy, Real-Time RT-PCR, receptor tyrosine kinase (RTK), RTK-high gene signature, RTK-low gene signature
Introduction
About one in eight women will be diagnosed with breast cancer during their lifetime, making this the most frequent malignant diseases in women [1]. Figures published by the WHO indicate that, worldwide, about 1,000,000 women develop breast cancer every year. Breast cancer comprises a heterogeneous group of malignant diseases that can be differentiated primarily on the basis of histopathological criteria into several distinct sub-groups of varying incidences [2].
Intense research efforts in recent years have provided insight into a large number of the complex molecular mechanisms involved in the pathogenesis of breast cancer [3]. Current attempts to develop individualized tumor therapies are aimed at identifying and treating further distinct subtypes on the basis of therapeutically relevant surrogate markers [4, 5, 6, 7, 8]. In addition to the classical histopathological parameters, the IHC characterization of hormone receptor status and HER2 as well as when indicated the cytogenetic detection of HER2 gene amplification represent the first important parameters upon which individualized tumor-specific therapy for breast carcinoma can be based. About 85% of all breast cancer patients currently receive such individualized therapy, which is aimed at the inhibition of HER2 (Trastuzumab, Lapatinib) and estrogen receptors (Tamoxifen, Letrozol) and is now considered standard treatment [9].
Thus, breast carcinoma can be roughly divided into tumors that are positive for estrogen/progesterone receptors and/or HER2 (triple-positive) which are suited to established, personalized therapy and those that are negative for these markers (triple-negative) without such benefit.
Triple-negative tumors are primarily defined, therefore, by an absence of surrogate markers of relevance for individualized therapy. As has been shown in numerous studies, triple-negative tumors vary in tumor progression, the development of therapeutic resistance, and the clinical course [10, 11, 12, 13, 14, 15], which indicates that they comprise a heterogeneous group. Further characterization and subtyping of triple-negative tumors on the basis of therapeutically relevant biomarkers is therefore strongly necessary.
To this end, a number of international microarray and NGS studies have produced data on the characterization of triple-negative tumors at the molecular level [13, 16, 17, 18]. In addition, further attempts to subtype these tumors have differentiated between basal-like and myoepithelial tumors [19, 20, 21] the findings being primarily of prognostic relevance. However, little therapeutic strategies targeting basal-like tumor cell specific surrogate markers have yet been developed.
The search for new approaches to individualized therapy during recent years has revealed biomarkers with predictive relevance for a large number of malignant diseases, which have been used in the development of individualized tumor therapy modalities in clinical trials. Various RTK families are of particular importance here. The treatment success achieved with trastuzumab (Herceptin) in HER2+ breast cancer has stimulated a search for other therapeutic targets including other members of elements of the epidermal growth factor receptor (EGFR) family.
The concept of growth factor receptor based anti-tumor therapy has been investigated and developed in recent years, especially in relation to EGFR. This has produced a series of EGFR inhibitors that have been approved for the treatment of epithelial tumors. In addition to the therapeutic monoclonal antibodies cetuximab and panitumumab, which are of great importance in the treatment of colorectal carcinoma, antineoplastic effects have been demonstrated for the receptor tyrosine kinase inhibitors (RTKI) gefitinib (Iressa) and erlotinib (Rarceva) in non-small cell lung cancer and for lapatinib (Tykerb) in metastatic breast cancer [22, 23, 24].
In addition to the representatives of the EGF receptor family, insulin-like growth factor receptor 1 (IGF-1R) is a further promising candidate for the development of individual therapeutic approaches in oncology. Various specific therapeutic monoclonal antibodies and RTKI against IGF-1R, growth factor antagonists and IGF-1R are currently undergoing clinical trials [25, 26, 27].
The aim of the present study was, by means of gene expression analysis and IHC investigations, to identify various RTK of the EGFR and IGFR families that would enable patient stratification for the purposes of target-oriented individualized tumor therapy in TNBC. In addition, correlation analysis was undertaken to shed light on possible interactions amongst the various RTK and provide a basis for innovative approaches to individualized tumor therapy.
Materials and Methods
Patients and tumors
A total of 29 triple-negative invasive ductal breast carcinomas, which had been diagnosed in the course of routine histopathological investigation at the Institute of Pathology, Ludwig-Maximilians-Universität Munich during the years 2003 to 2005, were selected for investigation. All tumors were re-determined for negative hormone receptor and negative HER2 status according to up-to-date histopathological guidelines and recommendations [28, 29]. ER and PR negativity was defined as positive staining in <1% of tumor cells. HER2 negativity was defined as an IHC score of 0 or 1+; those with an immunoreactive score of 2+ underwent HER2-specific FISH analysis to exclude HER2 gene amplification. Further selection criteria included the high-risk parameters of high tumor grade (grade 3) and low age (cut-off of ≤ 55 years) at primary manifestation of disease (average age 41 years; x = 41.3 years; s = 7.8 years) at the time of diagnosis. Tumor size was documented in relation to the patient specific chemotherapeutic treatment regimen before adjuvant respectively before neoadjuvant chemotherapeutic treatment. The mean tumor size was 3.3 cm (x = 3.3 cm, s = 3.5 cm). Nineteen patients received adjuvant chemotherapy and 9 patients neoadjuvant chemotherapy. Within the observation time of this study recurrence rate was 24% for patients treated with adjuvant chemotherapy and 63% for patients with neoadjuvant treatment. None of the patiens with neoadjuvant chemotherapy showed a pathological complete response (pCR). Tumors in 6 patients showed an infiltration of lymph or blood vessels (lymphangiosis-, hemangiosis carcinomatosa) at the time of diagnosis and one patient had distant metastases. Details of the diagnostically relevant parameters in the study group are given in Table 1.
Diagnostically relevant parameters in the study group
| Case | Age | Grade | Estrogen | Progesterone | HER2/ neu | Lymph nodes | Tumor size (cm) | Lymphangiosis carcinomatosa / Angiosis carcinomatosa | Neoadjuvant chemo-therapy | Adjuvant chemo-therapy | Distant metastases |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 42 | 3 | 0 | 0 | 0 | 0 | 2.3 | x | |||
| 2 | 31 | 3 | 0 | 0 | 1+ | 1 | 1.6 | x | x | ||
| 3 | 43 | 3 | 0 | 0 | 0 | 1 | 1.5 | x | |||
| 4 | 42 | 3 | 0 | 0 | 0 | 0 | 1.1 | x | |||
| 5 | 53 | 3 | 0 | 0 | 0 | 0 | 1.3 | x | |||
| 6 | 38 | 3 | 0 | 0 | 1+ | 0 | 1.6 | x | |||
| 7 | 45 | 3 | 0 | 0 | 0 | 1 | 15 | x | x | ||
| 8 | 48 | 3 | 0 | 0 | 1+ | 0 | 2.5 | x | x | ||
| 9 | 28 | 3 | 0 | 0 | 1+ | 0 | 1.9 | x | |||
| 10 | 42 | 3 | 0 | 0 | 0 | 0 | nd | x | x | ||
| 11 | 34 | 3 | 0 | 0 | 0 | nd | 3.3 | x | |||
| 12 | 31 | 3 | 0 | 0 | 0 | 1 | 3.6 | x | |||
| 13 | 47 | 3 | 0 | 0 | 0 | nd | nd | nd | nd | ||
| 14 | 53 | 3 | 0 | 0 | 0 | 0 | 4.3 | x | |||
| 15 | 34 | 3 | 0 | 0 | 0 | 1 | 2.2 | x | |||
| 16 | 51 | 3 | 0 | 0 | 0 | 0 | 0.7 | x | |||
| 17 | 40 | 3 | 0 | 0 | 0 | 0 | 2.2 | x | |||
| 18 | 49 | 3 | 0 | 0 | 0 | nd | 0.9 | x | |||
| 19 | 53 | 3 | 0 | 0 | 0 | 1 | 2.5 | x | x | ||
| 20 | 51 | 3 | 0 | 0 | 2+ | 0 | 5 | x | |||
| 21 | 35 | 3 | 0 | 0 | 2+ | 1 | 3 | x | x | ||
| 22 | 49 | 3 | 0 | 0 | 1+ | 0 | 2.1 | x | |||
| 23 | 43 | 3 | 0 | 0 | 1+ | 1 | 2 | x | x | ||
| 24 | 35 | 3 | 0 | 0 | 0 | 0 | 2 | x | |||
| 25 | 33 | 3 | 0 | 0 | 2+ | 0 | 2.6 | x | |||
| 26 | 41 | 3 | 0 | 0 | 1+ | 0 | 10 | x | |||
| 27 | 32 | 3 | 0 | 0 | 0 | 0 | 2.5 | x | |||
| 28 | 30 | 3 | 0 | 0 | 0 | 1 | 2 | x | |||
| 29 | 46 | 3 | 0 | 0 | 0 | 0 | 5 | x |
Material investigated
The investigations were performed on formalin-fixed, paraffin-embedded tissue (FFPET) processed for routine histopathological diagnostic investigation. All analyses were performed at resection specimens.
Deparaffinisation and RNA extraction
For each case, nine 4-µm FFPET whole sections comprising more than 70% tumor were cut and deparaffinized according to standard protocols. Total RNA was isolated by means of a commercial RNA isolation kit (High-Pure RNA Isolation Kit, Roche). RNA samples were analyzed photometric for their qualitative and quantitative content (NanoDrop, Thermo Scientific, USA). The minimum RNA content required for gene expression analysis was 300 ng/µl (total volume: 45 µl). An adequate amount of total RNA could be isolated from a total of 29 of the 31 triple-negative tumors that were initially selected for investigation.
cDNA synthesis
cDNA synthesis was performed by primer-specific reverse transcription of total RNA according to a standard protocol (Transkriptor First Strand cDNA Synthesis Kit, Roche). In brief, 3 µl RNA (50 ng/µl), 1 µl sequence-specific primer (1 µM), 9 µl distilled water, 4 µl 5X buffer, 2 µl nucleotide mix (each 10 mM), 0.5 µl protector RNase inhibitor (40 U/µl) and 0.5 µl transcriptor reverse transcriptase (20 U/µl) per reaction were mixed, preincubated for 10 minutes at 95°C and incubated for one hour at 50°C.
Per 96-well plate a standard dilution series (dilution factor 1:5), calibrator, negative control and 29 tumor samples were processed. A total of 34 biomarkers, including representatives of the EGFR and IGF-1R families and biomarkers related to downstream signaling, and five housekeeping genes (HKG) were analyzed in separate runs.
PCR Amplification
Gene expression data were obtained using sequence-specific detection probes (hydrolysis probes). With the help of the Universal ProbeLibrary Software [30], primer-specific sequences and target-specific hydrolysis probes for 34 biomarkers and 5 HKG were determined on the basis of freely accessible data banks (Table 2).
Biomarkers / Universal ProbeLibrary Assays
| Biomarker | Primersequence (5' -> 3'-direction) | Probe No. | Reverse Primer RT |
|---|---|---|---|
| EGFR Fwd | cat gtc gat gga ctt cca ga | 44 | ttc tcc tct cct gca cc |
| EGFR Rev1 | ggg aca gct tgg atc aca ct | ||
| HER2 Fwd | tgg ctc agt gac ctg ttt tg | 75 | tca ggt ttc aca ccg ct |
| HER2 Rev1 | ggt cct tat agt ggg cac agg | ||
| Her3 Fwd | cac aat gcc gac ctc tcc | 86 | tgg gca atg gta gag tag ag |
| Her3 Rev1 | cac gag gac ata gcc tgt ca | ||
| Her4 Fwd | ttc cac ttt acc aca aca tgc ta | 78 | aca gca aat gtc aga ccc |
| Her4 Rev1 | cag aat gaa gag ccc acc a | ||
| TGF-alpha Fwd | tgc tgc cac tca gaa aca gt | 63 | agt gct gtc ctg aag aag c |
| TGF-alpha Rev1 | atc tgc cac ag tcc acc tg | ||
| EGF Fwd | tgg ttg tgg ttc atc cat tg | 68 | tca cag cct ccg ttt tga ta |
| EGF Rev1 | tca cag cct ccg ttt tga ta | ||
| beta-cellulin Fwd | act gca tca aag gga gat gc | 49 | acc ttg ctc caa tgt agc |
| beta-cellulin Rev1 | tcc aat gta gcc ttc atc aca | ||
| Amphiregulin Fwd | cgg aga atg caa ata tat aga gca c | 38 | tgt caa tca tgc tgt gag ttt |
| Amphiregulin Rev1 | cac cga aat att ctt gct gac a | ||
| HB-EGF Fwd | tgg ggc ttc tca tgt tta gg | 55 | ccg att cct tga gca ca |
| HB-EGF Rev1 | tgc cca act tca ctt tct ctt c | ||
| NRG-Common Fwd | gat cag caa att agg aaa tga cag | 53 | tct gaa gac aca tat gct cct |
| NRG-Common Rev1 | ggc ata cca gtg atg atc tcg | ||
| NRG-GGF2 Fwd | gct gcc act act gct gct g | 70 | cgg gga cga gta gca cac |
| NRG-GGF2 Rev1 | cgg gga cga gta gca cac | ||
| NRG-SMDFsecSet Fwd | aga acg ccc aag tca gca | 5 | atg gct tgt ccc agt g |
| NRG-SMDF secSetRev1 | ttg tcc cag tgg tgg atg ta | ||
| NRG2 Fwd | ttc gca tca aat atg gca ac | 39 | gac ggt gtc ctt ccc c |
| NRG2 Rev1 | ggc ctc gca gac ata ctc c | ||
| NRG3 Fwd | gcc agt ctg tca aac acc ac | 26 | cca cct agc cta ctt cgg |
| NRG3 Rev1 | atg gag cat gcc act tct tt | ||
| NRG4 Fwd | ggg gct ttg tta tgt gat acc t | 23 | ctg gag cct ggg aga a |
| NRG4 Rev1 | cct gta tag ttt tca acg cac ct | ||
| BCL2 Fwd | agg tgc atc tgg tga tgt ga | 26 | cac tcc aac ccc cga tct |
| BCL2 Rev1 | cac tcc aac ccc cga tct | ||
| G6PDH secSet Fwd | aga gac cgt gga tgc tga ag | 65 | ctc cgc act gct gac a |
| G6PDH secSet Rev1 | tga gga cct ccg tca gat g | ||
| beta-Aktin Fwd | att ggc aat gag cgg ttc | 11 | atg tcc acg tca cac ttc at |
| beta-Atkin Rev | gga tgc cac agg act cca t | ||
| ER-Fwd | aac cag tgc acc att gat aaa a | 69 | att ctc cct cct ctt cgg |
| ER-Rev | tcc tct tcg gtc ttt tcg tat c | ||
| PGR-Fwd | ttt aag agg gca atg gaa gg | 11 | ttt ttc tgc gga ttt tat caa |
| PGR-Rev | cgg att tta tca acg atg cag | ||
| PTEN-Fwd | gca caa gag gcc cta gat ttc | 60 | aat aca cat agc gcc tct ga |
| PTEN-Rev | cgc ctc tga ctg gga ata gt | ||
| PIK3CA-Fwd | cga gat cct ctc tct gaa atc ac | 2 | gaa ttt cgg gga tag tta cac aa |
| PIK3CA-Rev | gaa ttt cgg gga tag tta cac aa | ||
| PIK3R1 Iso 1-Fwd | aat gaa cga cag cct gca c | 16 | cag tac cat tca gca tct tgt aa |
| PIK3R1 Iso 1-Rev | ccg ttg ttg gct aca gta gta gg | ||
| AKT common-Fwd | ggc tca ccc agt gac aac tc | 41 | agc agc ttc agg tac tca aa |
| AKT common-Rev | act caa act cgt tca tgg tca c | ||
| SRCcom-Fwd | cga gaa agt gag acc acg aa | 34 | gtt gag gcc ctt ggc |
| SRCcom-Rev | ttg gcg ttg tcg aag tca | ||
| SCUBE2-Fwd | gct gcc atc cac agt aca ag | 17 | gac agt gtc ctc tcg ctc caa |
| SCUBE2-Rev | gac agt gtc ctc tcg ctc caa | ||
| Ki-67-Fwd | tga att tcc aag aaa aat acg tga | 36 | agc ttt ctc atc agg gtc a |
| Ki-67-Rev | tca ggg tca gaa gag aag cta ga | ||
| Cyclin B1-Fwd | cgc ctg agc cta ttt tgg t | 34 | aga aag cct gac aca ggt c |
| Cyclin B1-Rev | gca cat cca gat gtt tcc att | ||
| HNF3α-Fwd | agg gct gga tgg ttg tat tg | 1 | atg ttg ctg acc ggg a |
| HNF3α-Rev | acc ggg acg gag gag tag | ||
| Survivin com-Fwd | ctt gaa agt ggc acc aga gg | 29 | cct cac ttc tca cct ggt aa |
| Survivin com-Rev | caa aaa tga gcc ccc aaa a | ||
| TRIP13-Fwd | ctc atg cgc tgt atg tcc a | 65 | gca agc ttc ttt ctc tct tca |
| TRIP13-Rev | gtc cac tgc cag aga cag g | ||
| HPRT-Fwd | tga cct tga ttt att ttg cat acc | 73 | aat gtg atg gcc tcc ca |
| HPRT-Rev | cga gca aga cgt tca gtc ct | ||
| PBGD common-Fwd | agc tat gaa gga tgg gca ac | 25 | gcc tgc atg gtc tct tgt a |
| PBGD common-Rev | ttg tat gct atc tga gcc gtc ta | ||
| PPP1CA common-Fwd | cct ata aga tca agt acc ccg aga | 15 | cag ttg aag cag tca gtg aa |
| PPP1CA common-Rev | gat gtt gta gcg tct ctt gca c | ||
| IGF1 Fwd | tgt gga gac agg ggc ttt ta | 67 | cac tca tcc acg atg cct |
| IGF1 Rev | atc cac gat gcc tgt ctg a | ||
| IGF2 Fwd | gct ggc aga gga gtg tcc | 10 | cat tgg gat tcc cat tgg |
| IGF2 Rev | gat tcc cat tgg tgt ctg ga | ||
| IGF-1R Fwd | ttc agc gct gct gat gtg | 10 | caa gtt ccc ggc tca tg |
| IGF-1R Rev | ggc tca tgg tga tct tct cc | ||
| IGF-2R Fwd | cag acc aca tat acc acg agg a | 1 | agt tga gag aaa aga tgg ggt |
| IGF-2R Rev | aga aaa gat ggg gtg gct gt |
For quantitative PCR, 3.5 µl cDNA (50ng/µl) were added to 16.5 µl PCR Master Mix according to the manufacturer´s instructions (LightCycler® 480 Probes Master, Roche). For all targets, uniform ThermoCycler running conditions were used. In brief, initial preincubation at 95°C for 5 minutes was followed by 45 cycles of 95°C for 10 seconds, 60°C for 20 seconds and 72°C for 1 second. Gene expression profiles were generated on the LightCycler® 480 Real-Time PCR System (Roche).
A standard curve, consisting of diluted human reference RNA (Universal Human Reference RNA, Stratagene), was included for every RT reaction and subsequent RT-PCR. For a run to be accepted there had to be a valid negative control and an amplification efficiency of between 1.9 and 2.1. Expression data were normalized to five averaged HKG. All experiments were performed in duplicate and the results averaged. Calibrator based calculations were used to determine relative gene expression profiles [31].
Immunohistochemical analysis
IHC investigation for various surrogate markers was performed to substantiate the results of gene expression analysis and to identify basal-like breast carcinomas. Markers investigated included the RTK EGFR, HER2 and IGF-1R, the hormone receptors ER and PR, and the basal-like marker CK5/6 [32]. Whole FFPET sections cut at 3 µm were stained with a Ventana Benchmark XT autostainer (Ventana Medical Systems). Details of the antibodies and methods employed are given in Table 3.
Antibodies employed in the study
| Antigen | Source | Order No. | Dilution | Method / System | Pretreatment | Incubation |
|---|---|---|---|---|---|---|
| EGFR | Ventana/Roche | 790-2988 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 20 min, Protease | 32 min |
| HER2 | Ventana/Roche | 800-2996 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 30min, CC1 | 16 min |
| IGF-1R | Ventana/Roche | 790-4346 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 60 min, CC1 | 28 min |
| ER | Ventana/Roche | 790-4325 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 60 min, CC1 | 28 min |
| PR | Ventana/Roche | 790-4296 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 60 min, CC1 | 28 min |
| CK 5/6 | Ventana/Roche | 760-4253 | ready to use | Ventana /3,3`-diamino-benzidine kit1 | 60 min, CC1 | 32 min |
1 (XT Ultra View DAB kit, Ventana Medical Systems, Tucson, Arizona)
Slides were counterstained with haematoxylin (Ventana). System and isotype controls were included. IHC investigation for ER and PR and for HER2 was performed according to up-to-date histopathological guidelines and recommendations [28, 29].
Immunostaining for EGFR and IGF-1R was evaluated semiquantitatively with a scoring system similar to that established for HER2 (0: no membrane-specific staining, 1+: weak, incomplete cell membrane staining in <10% of cells, 2+: weak or moderate staining of the complete cell membrane in > 10% of cells, 3+: strong, complete membrane staining in > 10% of cells. Immunostaining for CK 5/6 was evaluated according to the criteria of Dabbs et al. [33] (0: no staining, R: single cells stained, 1+: 5-30% of cells stained, 2+: >30-60% of cells stained, 3+: > 60% of cells stained). The scores 0 and R were considered negative, 1+ to 3+ positive.
Statistical analysis
Statistical evaluation was performed with the help of the statistics software SPSS (Chicago, USA) and R [34]. The Kolmogorov Smirnov test was used to test for normal distribution. The Mann-Whitney U test and Student's t-test were used to test for significant differences in gene expression between the postulated subtypes RTK-high and RTK-low. T-test for equality of means and Levene´s test for equality of variances was applied to demonstrate the statistic independence of tumor samples with adjuvant and neoadjuvant chemotherapeutic treatment in context to the gene-expression analyses. The influence of clinical parameters on the gene expression profile was investigated by multivariate analysis on the basis of the Chi-square test and the Mann-Whitney U test. Correlation between variables was observed with Pearson respectively Spearman rank correlation analyses. Hierarchical cluster analysis was performed to identify subtypes within the study cohort. The 3-year survival rate was analyzed with the Kaplan-Meier curve.
Ethics approval and consent to participate
Specimens and data were anonymized, and the need for consent was waived by the institutional ethics committee of the Medical Faculty of the Ludwig Maximilians University of Munich.
Results
Biomarker detection efficiencies within RT-PCR analyses
In the present study 34 tumor associated biomarkers relevant for EGFR and IGF-1R families and related biomarkers of the downstream signaling as well as 5 HKG were analyzed. Expression profiles for all tumor samples could be obtained for 31 of the biomarkers and all five HKG.
Differentiation of triple-negative tumors into two distinct subtypes on the basis of gene expression profiling of RTK associated biomarkers
Hierarchical cluster analysis revealed a dichotomous differentiation pattern amongst the triple-negative tumors (Figure 1).
Pearson correlation - triple-negative breast cancer
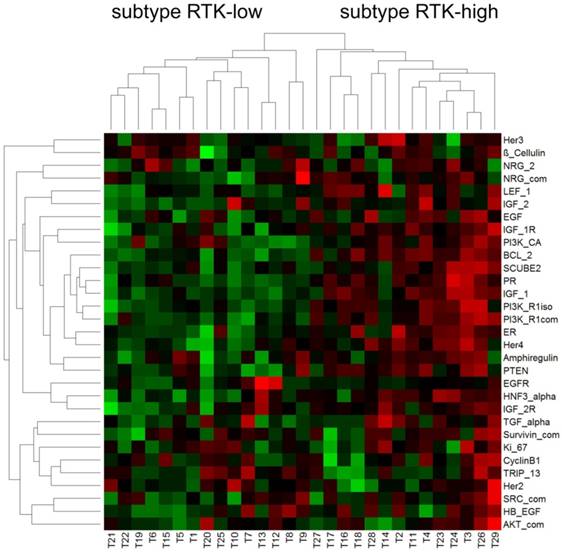
The two subtypes were characterized by low (subtype RTK-low; N=15) and high (subtype RTK-high; N=14) gene expression of the biomarkers investigated. The corresponding relative gene expression values of each biomarker are shown in Table 4.
Differential biomarker expression in triple-negative tumors of the subtypes RTK-low and RTK-high
| Biomarker | p | TNBC subtype | Mean | 95 % CI | Biomarker | p | TNBC subtype | Mean | 95 % CI |
|---|---|---|---|---|---|---|---|---|---|
| PR | 0.000 | RTK-low | 2.789 | 1.802 - 3.776 | Amphiregulin | 0.24 | RTK-low | 2.880 | 0.840 - 4.920 |
| RTK-high | 17.582 | 5.256 - 29.908 | RTK-high | 4.610 | 2.122 - 7.278 | ||||
| HER4 | 0.000 | RTK-low | 1.510 | 0.827 - 2.373 | NRG-com | 0.408 | RTK-low | 16.995 | -16.498 - 50.488 |
| RTK-high | 10.550 | 3.887 - 17.213 | RTK-high | 3.390 | 1.669 -5.111 | ||||
| EGF | 0.043 | RTK-low | 3.728 | 1.153 - 6.303 | Ki-67 | 0.407 | RTK-low | 1.390 | 1.114 - 1.665 |
| RTK-high | 18.825 | 4.383 - 33.267 | RTK-high | 1.628 | 1.069 - 2.186 | ||||
| PI3K-R1iso | 0.000 | RTK-low | 4.494 | 3.659 - 5.329 | SRC-com | 0.254 | RTK-low | 17.375 | 13.012 - 21.738 |
| RTK-high | 12.392 | 8.023 - 16.762 | RTK-high | 22.269 | 14.142 - 30.397 | ||||
| SCUBE2 | 0.003 | RTK-low | 5.696 | 3.953 - 7.439 | Survivin-com | 0.119 | RTK-low | 0.262 | 0.185 - 0.339 |
| RTK-high | 22.497 | 9.788 - 35.206 | RTK-high | 0.3981 | 0.228 - 0.568 | ||||
| BCL2 | 0.000 | RTK-low | 10.077 | 7.813 - 12.340 | TRIP13 | 0.382 | RTK-low | 1.214 | 0.948 - 1.479 |
| RTK-high | 34.702 | 26.705 - 42.699 | RTK-high | 1.035 | 0.690 - 1.381 | ||||
| IGF-1R | 0.001 | RTK-low | 1.424 | 0.876 - 1.971 | Cyclin B1 | 0.425 | RTK-low | 0.92 | 0.738 - 1.102 |
| RTK-high | 4.332 | 2.159 - 6.504 | RTK-high | 1.082 | 0.696 - 1.467 | ||||
| IGF-2R | 0.015 | RTK-low | 2.545 | 1.827 - 3.263 | HER2 | 0.794 | RTK-low | 6.353 | 5.173 - 7.533 |
| RTK-high | 3.493 | 2.980 - 4.007 | RTK-high | 6.093 | 4.299 - 7.888 | ||||
| IGF2 | 0.004 | RTK-low | 0.376 | 0.224 - 0.976 | HER3 | 0.798 | RTK-low | 6.324 | 5.258 - 7.389 |
| RTK-high | 0.473 | 0.069-1.015 | RTK-high | 8.650 | 5.070 - 12.231 | ||||
| HNF3-α | 0.001 | RTK-low | 0.142 | 0.025-0.308 | β-Cellulin | 0.889 | RTK-low | 63.354 | 37.120 - 89.588 |
| RTK-high | 0.306 | 0.135 - 0.477 | RTK-high | 60.973 | 35.728 - 86.218 | ||||
| IGF1 | 0.000 | RTK-low | 31.943 | 25.742 - 38.143 | HBEGF | 0.446 | RTK-low | 3.408 | 1.849 - 4.968 |
| RTK-high | 113.493 | 73.942 - 153.044 | RTK-high | 4.189 | 2.689 - 5.688 | ||||
| ER | 0.000 | RTK-low | 0.833 | 0.520 - 1.146 | NRG-2 | 0.727 | RTK-low | 5.416 | 1.366 - 9.467 |
| RTK-high | 2.554 | 1.452 - 3.656 | RTK-high | 4.651 | 2.572 - 6.730 | ||||
| PTEN | 0.000 | RTK-low | 3.704 | 2.278 - 5.129 | AKT-com | 0.311 | RTK-low | 4.606 | 3.578 - 5.633 |
| RTK-high | 6.786 | 4.883 - 8.689 | RTK-high | 5.675 | 3.640 - 7.710 | ||||
| Pi3K-CA | 0.043 | RTK-low | 5.415 | 3.521 - 7.310 | EGFR | 0.257 | RTK-low | 23.781 | -12.782 - 60.344 |
| RTK-high | 8.481 | 6.383 - 10.579 | RTK-high | 3.645 | 2.668 - 4.621 | ||||
| LEF1 | 0.003 | RTK-low | 0.871 | 0.684 - 0.106 | TGF-α | 0.219 | RTK-low | 3.587 | 1.112 - 6.062 |
| RTK-high | 0.2561 | 0.131 - 0.381 | RTK-high | 5.865 | 2.817 - 8.913 | ||||
| PI3K R1 iso | 0.000 | RTK-low | 4.494 | 3.659 - 5.329 | |||||
| RTK-high | 12.392 | 8.023 - 16.762 |
Further statistical analysis confirmed significant differences in gene expression between the subgroups RTK-low and RTK-high for 16 of the 31 biomarkers evaluated (Table 4). Of particular interest were significant differences for 3 of the 6 analyzed RTK (HER4, p≤ 0.001; IGF-1R, p < 0,001; IGF-2R, p = 0.015; Figure 2).
Differential gene expression by the triple-negative subtypes RTK-low und RTK-high in relation to PR, ER, HER4, IGF-1R and IGF-2R
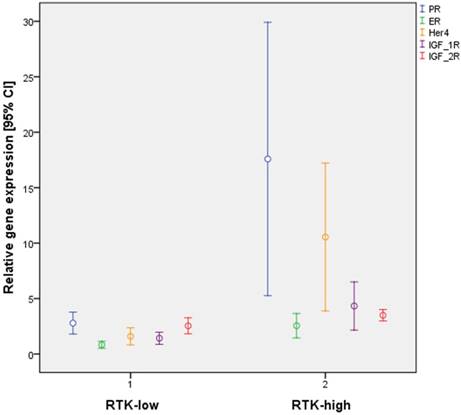
Interestingly, significant differences in gene expression patterns for ER (p = 0.001) and PR (p < 0.001) were also found between the two subtypes (Figure 2). By contrast, all cases of triple-negative tumors, by definition lacked IHC staining for ER and PR.
Correlation analysis was performed to determine whether the RTK and hormone receptors investigated are subject to functional interaction (Table 5).
Correlation coefficients for RTK, ER and PR gene expression
| ER | PR | EGFR | egfr | HER3 | HER4 | IGF-1R | IGF-2R | |
|---|---|---|---|---|---|---|---|---|
| ER | 1 | |||||||
| PR | 0.654** | 1 | ||||||
| EGFR | 0.034 | 0.001 | 1 | |||||
| HER2 | 0.057 | -0.081 | 0.099 | 1 | ||||
| HER3 | -0.109 | 0.078 | 0.023 | 0.000 | 1 | |||
| HER4 | 0.675** | 0.623** | -0.021 | -0.105 | 0.222 | 1 | ||
| IGF-1R | 0.419* | 0.555** | -0.056 | 0.112 | 0.206 | 0.394* | 1 | |
| IGF-2R | 0.293 | 0.284 | 0.519** | -0.034 | 0.354 | 0.369* | 0.264 | 1 |
** p<0.005; * p<0.05
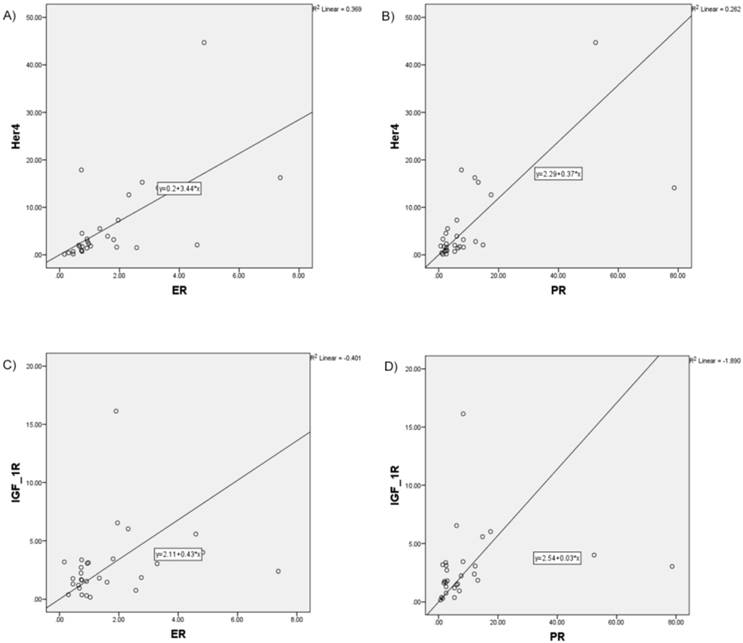
Increased correlation, in particular for HER4 with ER (r = 0.675; p≤0.001), HER4 with PR (r=0.623, p≤0.001), as well as IGF-1R with ER (r = 0.419; p=0.024) and IGF-1R with PR (r=0.555; p=0.002) was found (Figure 3).
Increased correlation between analyzed RTK and hormone receptors pictured in scatter plot charts A) HER4 and ER, B) HER4 and PR, C) IGF-1R and ER, D) IGF-1R and PR
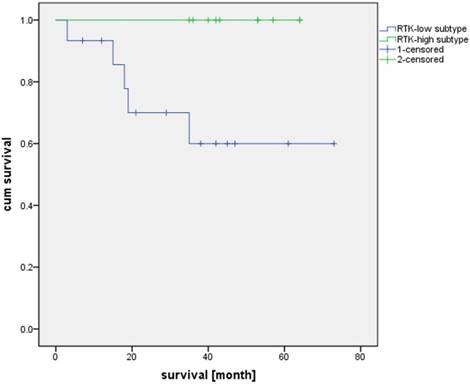
Additionally, we compared the patient survival based on the RTK-high and RTK-low gene signature. Kaplan-Meier analysis demonstrated superior three-year survival in the RTK-high group as opposed to the patients in the RTK-low group (three-year survival rate of 100% v. 60%; log-rank test, p-value = 0.022; Figure 4).
Overall survival of patients with RTK-high and RTK-low gene signature
In summary, two subtypes of TNBC could be identified on the basis of gene expression profiles of 16 biomarkers of the EGF and IGF receptor families and biomarkers of their downstream signaling as well as the hormone receptors ER and PR (Table 4). Of special interest are the RTK HER4 and IGF-1R, as well as the hormone receptors ER and PR, for which the findings of correlation analysis indicate functional interaction in cell signaling.
Comparison of IHC and gene expression findings
In order to determine to what extent the differences in gene expression between the subtypes RTK-high and RTK-low as detected by RT-PCR are also reflected at the protein level, IHC investigations were carried out for certain key proteins from the biomarker panel (IGF-1R and EGFR: study findings, HER2, ER and PR: findings of routine diagnostic work-up). For IGF-1R and EGFR the IHC findings were consistent with the gene expression data. Accordingly, differences in IGF-1R expression at both transcriptional and translational levels were found between the subtypes RTK-high and RTK-low (IHC: p Mann-Whitney U test = 0.005, RT-PCR: p Mann-Whitney U test < 0.001; r Spearman-Rho = 0.706, p ≤ 0.001). The expression profile of EGFR in both subtypes of triple-negative tumors could also be confirmed at the IHC level (IHC: p Mann-Whitney U test = 0.377, RT-PCR: p t-test = 0.257; r Spearman-Rho = 0.311, p = 0.100).
As shown in Table 1, each case in the study group exhibited a triple negative phenotype according to established histopathological criteria. However, there were highly significant differences in the gene expression profiles for both ER (p Mann-Whitney U test = 0.001) and PR (p Mann-Whitney U test ≤ 0.001) amongst the triple-negative tumors in relation to the postulated subtypes RTK-high and RTK-low. As with the IHC findings, no significant difference was found for HER2 at the transcriptional level between the postulated subtypes RTK-high and RTK-low (p t-test = 0.794; r Spearman-Rho = -0.087, p = 0.655).
Immunohistochemical characterization of basal-like subtypes
In relation to the much debated question as to the extent to which triple-negative tumors correspond to the basal-like breast cancer subtype, and to determine to what extent one of the two triple-negative subtypes defined in the study correlates with the basal-like subtype, IHC staining for the basal-like tumor associated biomarker CK5/6 and EGFR was performed. The IHC analyses revealed a basal-like phenotype within 79% of the samples. Statistical analysis did show no significant difference between the triple-negative subtypes defined in this study on the basis of gene expression analyses and the basal-like and non-basal-like TNBC subtypes (p Pearson Chi-Square test = 0.080).
Comparison of clinical parameters and genetic expression of tumor-related biomarkers
By means of multivariate analysis the variables nodal status, age, tumor size and angiosis carcinomatosa were tested for a possible relationship to biomarker expression and subtype (RTK-high and RTK-low). The Chi-square test and Mann-Whitney U test failed to show a significant relationship between any of the clinicopathological parameters and the subtypes.
T-test for equality of means and Levene's test for equality of variances demonstrated the statistic independence of tumor samples with adjuvant and neoadjuvant chemotherapeutic treatment in context to the RTK gene-expression analyses (Table 6). None of the changes was significant after Bonferroni correction.
Statistic independence of tumor samples with adjuvant and neoadjuvant chemotherapeutic treatment in context to the RTK gene-expression analyses
| Biomarker | Fold Change (neoadjuvant versus adjuvant) | p-value (Welch) | p-value (Levene) | Biomarker | Fold Change (neoadjuvant versus adjuvant) | p-value (Welch) | p-value (Levene) |
|---|---|---|---|---|---|---|---|
| PR | -1.847 | 0.269 | 0.212 | PI3K_R1iso | -1.589 | 0.078 | 0.607 |
| ER | -1.145 | 0.755 | 0.254 | AKT_com | 1.176 | 0.325 | 0.478 |
| EGFR | -1.022 | 0.960 | 0.158 | SRC_com | -0.114 | 0.511 | 0.780 |
| HER2 | 1.135 | 0.393 | 0.834 | SCUBE2 | -0.135 | 0.515 | 0.222 |
| Her3 | -1.009 | 0.970 | 0.777 | BCL2 | -1.712 | 0.081 | 0.915 |
| Her4 | -1.170 | 0.804 | 0.376 | IGF-1R | -1.454 | 0.390 | 0.690 |
| TGF_alpha | -1.079 | 0.817 | 0.943 | IGF-2R | -1.403 | 0.090 | 0.250 |
| EGF | -2.634 | 0.150 | 0.604 | IGF1 | -1.557 | 0.234 | 0.477 |
| ß_Cellulin | -1.125 | 0.826 | 0.279 | IGF2 | -1.297 | 0.729 | 0.605 |
| Amphiregulin | -1.945 | 0.259 | 0.482 | HNF3-alpha | -1.842 | 0.318 | 0.046 |
| HB_EGF | 1.183 | 0.573 | 0.924 | Survivin com | -1.600 | 0.117 | 0.765 |
| NRG_com | -1.506 | 0.531 | 0.962 | TRIP 13 | 1.044 | 0.794 | 0.256 |
| NRG_2 | 0.140 | 0.348 | 0.153 | CyclinB1 | -1.159 | 0.386 | 0.151 |
| PTEN | -1.546 | 0.102 | 0.881 | Ki-67 | 1.037 | 0.840 | 0.161 |
| PI3K_CA | -1.242 | 0.385 | 0.274 | LEF1 | -1.855 | 0.041 | 0.973 |
| PI3K_R1com | -1.558 | 0.082 | 0.233 |
Discussion
Characterization and subtyping of triple-negative tumors based on therapeutically relevant RTK
The search for individualized treatment strategies in recent years has revealed biomarkers with predictive relevance for a large number of malignancies. This information has been applied to the development of individualized tumor therapies. The various RTK are of special importance here, particularly representatives of the EGF and IGF receptor families which, in the case of overexpression, represent target proteins for individualized tumor therapy for an increasing number of tumor entities.
In the case of breast cancer, too, gene expression data and IHC findings have characterized patients for whom target-specific therapy is suitable. A number of clinical trials are concerned with the question as to what extent inhibition of EGFR or IGF-1R by specific monoclonal antibodies or RTKI can benefit patients with advanced breast cancer [35, 36, 37, 38, 39]. The combination of chemotherapy with EGFR inhibition appears so far to have mixed results. Adequate results of an individual therapeutic IGF-1R inhibition are not yet available.
At the same time, it has been shown in recent years that despite the importance of single tumor-relevant biomarkers, the neoplastic behaviour of tumors is influenced by numerous interactions in downstream signaling. The crosstalk of various overexpressed RTK, hormonal transcriptional regulation (ER) or mutational, constitutive activation in downstream signaling (EGFR, KRAS) represent multifactorial mechanisms involved in tumor growth. Because of the complexity of the situation, more recent clinical trials have focused increasingly on combination treatment strategies in narrowly defined patient groups. The characterization of breast tumors using a panel of tumor-specific surrogate markers not only contributes to a better understanding of functional mechanisms involved in tumor differentiation, growth and invasion, but also represents a basis for new combination treatment strategies. In addition, exact subtyping of breast cancer cases such as luminal A, luminal B, normal breast-like, ERBB2-overexpressing and basal-like molecular subtypes of breast cancer is still used for optimal patient selection in clinical trials.
In the study presented, we examined gene expression levels of various RTK and related biomarkers in TNBC without individualized treatment options with the aim of defining heterogeneous expression patterns and possible TNBC subtypes. Hierarchical cluster analysis was able to identify a panel of biomarkers that allows two distinct subtypes of TNBC to be distinguished. Further analysis of the genetic cluster emphasizes in particular the significance of the two RTK IGF-1R and HER4, as well as the hormone receptors ER and PR. Cases with overexpression of these markers described above (RTK-high) represent a potentially interesting group for clinical investigations. In addition, the possibility to identify those breast cancer patients who are potential responders for personalized medicine targeting IGF-1R or ER is of particular significance.
Differentiation of triple-negative tumors into basal-like positive and negative subtypes
Several microarray-based studies have demonstrated a differentiation of breast cancer into at least 5 different subtypes [40, 41, 42, 43]. Selected gene sets for around 500 biomarkers enable subtyping of breast carcinoma into luminal A, luminal B, normal breast-like, ERBB2-overexpressing and basal-like tumors. Tumors with the basal-like phenotype which are characterized by, among other features, by the absence of IHC ER and HER2 expression, are of particular clinical interest. Scientific studies employing IHC techniques and gene expression analysis have shown that up to 15% of all breast cancers exhibit the basal-like subtype [32, 33]. Numerous studies have attempted to equate triple-negative tumors with basal-like breast cancer, and indeed an accumulation of the basal-like phenotype amongst triple-negative tumors has been documented. Nevertheless, it has been shown that the two are not identical with each other [44]. Comparative analysis has shown that, although triple-negative tumors (immunohistochemically defined) often show a basal-like phenotype, and tumors diagnosed as basal-like breast cancer (on the basis of gene expression) are predominantly triple-negative, the two categories are up to 30% discordant [19, 45].
Methods for the identification of the basal-like phenotype rely mainly on the detection of expression of proliferation-specific biomarkers by gene expression analysis and immuno-histochemical techniques. Our study utilized IHC staining for CK 5/6. Eighty-one percent of the tumor samples were found to express a basal-like phenotype, a prevalence consistent with figures quoted in the literature.
In our study, comparison of the subtypes RTK-high and RTK-low and the group classed as CK 5/6 positive revealed no significant correlation. Therefore, on the basis of our findings it can be assumed that this is a newly defined and independent gene expression signature that permits further subtyping of TNBC into RTK-high and RTK-low genotypes.
Gene expression of ER in triple-negative breast cancer
IHC analysis of the hormone receptor status, is one of the standardized clinical investigations used in the diagnostic work-up of patients with breast cancer for treatment prediction. Selection of the cases for our study was based upon the findings of routine histopathological investigations. By definition, all exhibited a negative (<1%) IHC score for ER and PR. By contrast, our gene expression analysis revealed over-expression of ER and PR in some cases, there being a highly significant difference between the subtypes RTK-high and RTK-low. The discrepancy may be due to the increased sensitivity of RT-PCR. Differences in regulation of protein expression at the translational level are also conceivable.
The results of correlation analysis pertaining to IGF-1R, HER4 and ER in tumors with the RTK-high genotype suggest that these receptors interact with each other through regulatory mechanisms. Hence, further in-vivo investigations, especially into the potential of new immunotherapeutic approaches using combined immunotherapeutic approaches in TNBC patients with the RTK-high genotype, would be of great interest [46, 47, 48].
Crosstalk between IGF-1R and ER signaling
Recent studies have shown that there is crosstalk in cell signaling between RTK and ER. Various different trans-activating mechanisms with stimulating effects on the downstream signaling of adjacent RTK and ER were generally found [49, 50].
IGF-1R is one of the most important representatives of the IGF receptor family and, like ER, is associated with neoplastic properties when overexpressed [51, 52, 53].
In our study we found increased expression of IGF-1R and ER in TNBC of the RTK-high subtype. In addition, strong correlation between IGF-1R and ER was seen, which suggests functional interaction between the two receptors. As shown in recent studies, IGF-1R and ER are able to trans-activate each other and lead to activation or enhancement of downstream signaling [52]. In addition, in vivo experiments have demonstrated their combined significance in tumorigenesis [54].
In studies of IGF-1R-mediated ER activation [55, 56, 57] it has been shown that tyrosine kinases involved in IGF-1R downstream signaling, such as MAPK, RSK and AKT, are able to phosphorylate and activate ER at the AF-1 domain (Serin118, Serin167). Furthermore, in vivo experiments showed that IGF-1 is able to increase the expression of PR, an estrogen response element.
In addition to IGF-1R-mediated ER phosphorylation and activation, effects of ER on IGF-1R signaling have also been demonstrated [47]. ER is able, by means of various different regulatory mechanisms, to impact on IGF-1R downstream signaling. Studies have shown that anti-estrogens like tamoxifen can inhibit IGF-mediated growth [58]. Thus, it has been demonstrated that estrogens are able to regulate IGF-1R at the transcriptional level and to transactivate IGF-1R by phosphorylation. ER belongs to the steroid hormone receptor family and functions primarily as a transcription factor. IGF-1R is one of the most important estrogen response elements [58].
Besides the ability of ER to inactivate inhibitory elements of IGFR signaling, such as IGFBP-3, binding of ER to Shc with subsequent phosphorylation of MAPK have also been described. In addition to the singular importance of hormone receptors as a therapeutic target, several studies have demonstrated the existence of complex regulatory mechanisms and interactions in downstream signaling between ER/PR and growth factors [59, 60]. Several clinical trials concerning the combined use of IGF-1R and ER-inhibitors are under investigation [61, 62].
Crosstalk between HER4 and ER signaling
The c-erbB-4 gene (HER4) belongs to the EGFR family. Little is known about the functional significance of HER4 in the pathogenesis of breast cancer. In normal breast tissue, its functions include the differentiation of myocardial cells and mammary epithelial cells and the development of the central nervous system [63, 64]. Compared to the most important representatives of the EGFR family, EGFR and HER2, the prognostic impact of HER4 expression in breast cancer are basically unclear and controversially discussed [65, 66]. Unlike the remaining members of the EGF receptor family, which act primarily as mitogenic effectors in breast cancer cells, HER4 appears to have a large number of different functions in normal and neoplastic breast tissue. With regard to the overexpression of HER4, experimental studies have shown both oncogenic and tumor suppressive functions [67, 68]. Other investigations have also shown that HER4 is generally associated with differentiated and less aggressive tumors, and that it correlates with a better prognosis and a longer disease-free interval [66, 69, 70]. In addition, it has been shown that breast cancer patients who exhibit coexpression of HER4 and ER have a lower recurrence rate [71] and a higher survival rate [72] than those who express ER alone.
Interestingly, several in vivo and in vitro studies have shown a strong correlation and crosstalk between HER4 and ER expression in breast cancer [68, 73, 74]. Intense efforts to elucidate mechanism of crosstalk between HER4 and ER could identify an autocrine HER4/ER signaling pathway where the factor HER4 intracellular domain (4ICD), a cleavage product of HER4, acts as transcriptional coregulator of ER [73, 75, 76, 77, 78].
Our results point to an increased rate of coexpression of HER4 and ER in triple-negative tumors with the RTK-high phenotype. The coexpression of HER4 and ER suggests the existence of crosstalk in downstream-signaling within defined subpopulations of breast cancer patients.
ER / RTK crosstalk- development of intrinsic and acquired resistance
Antihormonal therapy (tamoxifen, anastrozole) and HER2 inhibition (trastuzumab, lapatinib) represent important cornerstones in personalized therapy for the treatment of breast cancer in women with overexpression of the corresponding receptors. Further promising targets for individualized therapy the RTK EGFR and IGF-1R and targeting the PI3K-Akt pathway are currently subject of several studies and clinical trials [79, 80, 81, 82]. However, follow-up studies did show the development of resistance in some patients receiving antihormonal therapy and/or RTK inhibitors.
Studies have demonstrated intrinsic or acquired resistance to treatment with tamoxifen in 20-30% of hormone-receptor-positive breast cancer patients [83]. Although the mechanisms involved have not yet been clarified in detail, it appears that, in addition to coregulatory and epigenetic mechanisms, transactivating processes between RTK and ER could be responsible [80, 84]. Further investigations have revealed complex co-operation of genomic and non-genomic/fast ER signaling and their crosstalk with growth factor receptors, such as EGFR, HER2, HER4 or IGF-1R, in the development of endocrine resistance [85, 86, 87]. In brief, activated cytoplasmic and membrane-bound ER show the ability to stimulate RTK, either directly via adaptor proteins like Shc, or indirectly by nuclear ER through increased release of RTK-specific ligands, for example HB-EGF. Following subsequent activation of the downstream signaling kinases MAPK and AKT, phosphorylation and thus activation of nuclear ER occurs, which leads to increased gene expression, including the expression of RTK-related genes. Thus, innovative treatment strategies aim at inhibition of both RTK and ER.
Current indications for hormone receptor therapy are based on standardized IHC findings of routine diagnostic investigations. According to the histopathological guidelines and recommendations [28], antihormonal treatment is indicated when more than 1% of the tumor cells are ER and/or PR positive. Large overviews of randomized clinical trials have confirmed the therapeutic value of antihormonal treatment only in immunreactive ER-positive breast cancers.
Our investigations demonstrated a significant increase in ER gene expression level in a newly characterized subtype (RTK-high) of TNBC. In addition, correlation analysis suggests functional interaction between IGF-1R, HER4 and ER. It is not yet clear to what extent antihormonal therapy would be effective in tumors in which hormone receptor status is negative by immunohistochemistry but gene expression is elevated. However, on the basis of crosstalk between ER and RTK, it is conceivable that a combination of inhibition of both ER and corresponding RTK, like IGF-1R or HER4, could be of benefit.
Conclusions
RTK-associated gene expression profiles generated in this study revealed dichotomous differentiation within the triple-negative study group. IHC analysis verified this at the posttranslational level. Correlation analysis of the biomarkers investigated suggests a functional connection between IGF-1R, IGF-2R, HER4, PR and ER. Based on survival analysis the differentiation of triple-negative tumors with RTK-high and RTK-low gene signature seems to be from prognostic relevance.
Abbreviations
TNBC: Triple-negative breast cancers; ER: estrogen receptor; PR: progesterone receptors; IHC: immunohistochemical; RTK: receptor tyrosine kinase; RT-PCR: reverse transcription polymerase chain reaction; EGFR: epidermal growth factor receptor; RTKI: receptor tyrosine kinase inhibitors; IFF-1R: insulin-like growth factor receptor 1; pCR: pathological complete response; FFPET: formalin-fixed, paraffin-embedded tissue; HKG: housekeeping genes; 4ICD: HER4 intracellular domain.
Acknowledgements
We thank Annegret Schäfer of the IHC laboratory of the Institute of Pathology of the Ludwig Maximilians University of Munich for their dedication and contributions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. Cancer Facts & Figures 2014. https:// www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014.
2. Lakhani S, Ellis I, Schnitt S. et al. WHO Classification of Tumours of the Breast. 4rd ed. Lyon: IARC Press. 2012
3. Ellis MJ, Perou CM. The Genomic Landscape of Breast Cancer as a Therapeutic Roadmap. Cancer Discovery. 2013;3:27-34
4. Tessari A, Palmieri D, Di Cosimo S. Overview of diagnostic/targeted treatment combinations in personalized medicine for breast cancer patients. Pharmgenomics Pers Med. 2013;7:1-19
5. Mayer IA, Abramson VG, Lehmann BD. et al. New strategies for triple-negative breast cancer-deciphering the heterogeneity. Clinical Cancer Research. 2014;20:782-790
6. Kos Z, Dabbs DJ. Biomarker assessment and molecular testing for prognostication in breast cancer. Histopathology. 2016;68:70-85
7. Yadav BS, Chanana P, Jhamb S. Biomarkers in triple negative breast cancer: A review. World J Clin Oncol. 2015;6:252-263
8. Zhang JF, Liu J, Wang Y. et al. Novel therapeutic strategies for patients with triple-negative breast cancer. Onco Targets Ther. 2016;21:6519-6528
9. Russell CA. Personalized medicine for breast cancer: it is a new day!. Am J Surg. 2014;207:321-325
10. Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232:142-150
11. Braicu C, Chiorean R, Irimie A. et al. Novel insight into triple-negative breast cancers, the emerging role of angiogenesis, and antiangiogenic therapy. Expert Rev Mol Med. 2016;18:e18
12. Ishitha G, Manipadam MT, Backianathan S. et al. Clinicopathological Study of Triple Negative Breast Cancers. J Clin Diagn Res. 2016;10:EC05-EC09
13. Lehmann BD, Jovanović B, Chen X. et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11:e0157368
14. Engebraaten O, Vollan HK, Børresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183:1064-1074
15. Chiorean R, Braicu C, Berindan-Neagoe I. Another review on triple negative breast cancer. Are we on the right way towards the exit from the labyrinth? Breast. 2013;22:1026-1033
16. Yau C, Esserman L, Moore DH. et al. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Research. 2010;12:R85
17. Chen Xi, Li J, Gray WH. et al. TNBC type: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Informatics. 2012;11:147-156
18. Weisman PS, Ng CK, Brogi E. et al. Genetic alterations of triple negative breast cancer by targeted next-generation sequencing and correlation with tumor morphology. Mod Pathol. 2016;29:476-488
19. Kreike B, van Kouwenhove M, Horlings H. et al. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Research. 2007;9:R65
20. Sotiriou C, Soek-Ying N, McShane LM. et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. PNAS. 2003;100:10393-10398
21. Rakha EA, Ellis IO. Triple-negative/basal-like cancer: review. Pathology. 2009;41:40-47
22. Aifa S, Rebai A. ErbB antagonists patenting: “playing chess with cancer”. Recent Pat Biotechnol. 2008;2:181-187
23. Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77:400-410
24. Irshad S, Ellis P, Tutt A. Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr Opin Oncol. 2011;23:566-577
25. Golan T, Javle M. Targeting the Insulin Growth Factor Pathway in Gastrointestinal Cancers. Oncology. 2011;25:518-526
26. Gradishar WJ, Yardley DA, Layman R. et al. Clinical and Translational Results of a Phase II, Randomized Trial of an Anti-IGF-1R (Cixutumumab) in Women with Breast Cancer That Progressed on Endocrine Therapy. Clin Cancer Res. 2016;22:301-309
27. Lin EH, Lenz HJ, Saleh MN. et al. A randomized, phase II study of the anti-insulin-like growth factor receptor type 1 (IGF-1R) monoclonal antibody robatumumab (SCH 717454) in patients with advanced colorectal cancer. Cancer Med. 2014;3:988-997
28. College of American Pathologists (CAP). Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2931060/.
29. Wolff AC, Hammond ME, Hicks DG. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer. American Society of Clinical Oncology. College of American Pathologists. Clinical practice guideline update. Arch Pathol Lab Med. 2014;138:241-256
30. The Universal ProbeLibrary. https://www.lifescience.roche.com/products/universal-probe-library.
31. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45
32. Nielsen TO, Hsu FD Jensen K. et al. Immunohistochemical and Clinical Characterization of the Basal-Like Subtype of Invasive Breast Carcinoma. Clin. Cancer Res. 2004;10:5367-5374
33. Dabbs DJ, Chivukula M, Carter G. et al. Basal phenotype of ductal carcinoma in situ: recognition and immunohistologic profile. Mod Pathol. 2006;19:1506-1511
34. The R Project for Statistical Computing. https://www.r-project.org.
35. Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol. 2013;4:30-37
36. Haisa M. The type 1 insulin-like growth factor receptor signalling system and targeted tyrosine kinase inhibition in cancer. J Int Med Res. 2013;41:253-264
37. Gelmon K, Dent R, Mackey JR. et al. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol. 2012:23 2223-2234
38. Xue M, Cao X, Zhong Y. et al. Insulin-like Growth Factor-1 Receptor (IGF-1R) Kinase Inhibitors in Cancer Therapy: Advances and Perspectives. Curr Pharm Des. 2012;18:2901-2913
39. Schwartz GK, Dickson MA, LoRusso PM. et al. Preclinical and first-in-human phase I studies of KW-2450, an oral tyrosine kinase inhibitor with insulin-like growth factor receptor-1/insulin receptor selectivity. Cancer Sci. 2016;107:499-506
40. Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molecular Oncology. 2011;5:5-23
41. Perou CM, Sorlie T, Eisen MB. et al. Molecular portraits of human breast tumors. Nature. 2000;406:747-752
42. Sorlie T, Perou CM, Tibshirani R. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869-10874
43. Sorlie T, Tibshirani R, Parker J. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418-8423
44. Irvin WJ Jr, Carey LA. What is triple-negative breast cancer? Eur J Cancer. 2008;44:2799-2805
45. Bertucci F, Finetti P, Cervera N. et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123:236-240
46. Chen HX, Sharon E. IGF-1R as an anti-cancer target-trials and tribulations. Chin J Cancer. 2013;32:242-252
47. Yu Z, Gao W, Jiang E. et al. Interaction between IGF-IR and ER induced by E2 and IGF-I. PLoS One. 2013;8:e62642
48. Chakraborty AK, Welsh A, Digiovanna MP. Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and pro-apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast Cancer Res Treat. 2010;120:327-335
49. Zabransky DJ, Park BH. Estrogen Receptor and Receptor Tyrosine Kinase Signaling: Use of Combinatorial Hormone and Epidermal Growth Factor Receptor/Human Epidermal Growth Factor Receptor 2-Targeted Therapies for Breast Cancer. J Clin Oncol. 2014;32:1084-1086
50. Heskamp S, Boerman OC, Molkenboer-Kuenen JD. et al. Upregulation of IGF-1R expression during neoadjuvant therapy predicts poor outcome in breast cancer patients. PLoS One. 2015;10:e0117745
51. Bartella V, De Marco P, Malaguarnera R. et al. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cellular Signalling. 2012;24:1515-1521
52. Liu C, Zhang Z, Tang H. et al. Crosstalk between IGF-1R and Other Tumor Promoting Pathways. Curr Pharm Des. 2014;20:2912-2921
53. Christopoulos PF, Msaouel P, Koutsilieris M. The role of the insulin-like growth factor-1 system in breast cancer. Mol Cancer. 2015;14:43-56
54. Bradley LM, Gierthy JF, Pentecost BT. Role of the insulin-like growth factor system on an estrogen-dependent cancer phenotype in the MCF-7 human breast cancer cell line. J Steroid Biochem Mol Biol. 2008;109:185-196
55. Tremblay GB, Tremblay A, Copeland NG. et al. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol. 1997;11:353-365
56. Simoncini T, Hafezi-Moghadam A, Brazil DP. et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538-541
57. Becker MA, Ibrahim YH, Cui X. et al. The IGF pathway regulates ERα through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol. 2011;25:516-528
58. Fagan DH, Yee D. Crosstalk between IGF-1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:423-429
59. Ribeiro JR, Freiman RN. Estrogen signaling crosstalk: Implications for endocrine resistance in ovarian cancer. J Steroid Biochem Mol Biol. 2014;143:160-173
60. Engels CC, de Glas NA, Sajet A. et al. The influence of insulin-like Growth Factor-1-Receptor expression and endocrine treatment on clinical outcome of postmenopausal hormone receptor positive breast cancer patients: A Dutch TEAM substudy analysis. Mol Oncol. 2016;10:509-516
61. Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials-early lessons. J Mammary Gland Biol Neoplasia. 2008;13:471-483
62. Fagan DH, Uselman RR, Sachdev D. et al. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor (IGF1R): implications for breast cancer treatment. Cancer Res. 2012;72:3372-3380
63. Long W, Wagner K-U, Lloyd KCK. Impaired differentiation and lactational failure in ErbB4-deficient mammary glands identify ERBB4 as an obligate mediator of Stat5. Development. 2003;130:5257-5268
64. Tidcombe H, Jackson-Fisher A, Mathers K. et al. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc Natl Acad Sci USA. 2003;100:8281-8286
65. Fujiwara S, Hung M, Yamamoto-Ibusuk CM. et al. The localization of HER4 intracellular domain and expression of its alternately-spliced isoforms have prognostic significance in ER+ HER2- breast cancer. Oncotarget. 2014;5:3919-3930
66. Machleidt A, Buchholz S, Diermeier-Daucher S. et al. The prognostic value of Her4 receptor isoform expression in triple-negative and Her2 positive breast cancer patients. BMC Cancer. 2013;13:437-446
67. Gullick WJ. c-erbB-4/HER4: friend or foe? J Pathol. 2003;200:279-281
68. Junttila TT, Sundvall M, Lundin M. et al. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005;65:1384-1393
69. Portier BP, Minca EC, Wang Z. et al. HER4 expression status correlates with improved outcome in both neoadjuvant and adjuvant Trastuzumab treated invasive breast carcinoma. Oncotarget. 2013;4:1662-1672
70. Wang J, Yin J, Yang Q. et al. Human epidermal growth factor receptor 4 (HER4) is a favorable prognostic marker of breast cancer: a systematic review and meta-analysis. Oncotarget. 2016;7:76693-76703
71. Barnes NL, Khavari S, Boland GP. et al. Absence of HER4 expression predicts recurrence of ductal carcinoma in situ of the breast. Clin Cancer Res. 2005;11:2163-2168
72. Witton CJ, Reeves JR, Going JJ. et al. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290-297
73. Rokicki J, Das PM, Giltnane JM. et al. The ERalpha coactivator, HER4/4ICD, regulates progesterone receptor expression in normal and malignant breast epithelium. Mol Cancer. 2010;9:150-154
74. Pawlowski V1, Révillion F, Hebbar M. et al. Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000;6:4217-4225
75. Thor AD, Edgerton SM, Jones FE. Subcellular localization of the HER4 intracellular domain, 4ICD, identifies distinct prognostic outcomes for breast cancer patients. Am J Pathol. 2009;175:1802-1809
76. Jones FE. HER4 intracellular domain (4ICD) activity in the developing mammary gland and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:247-258
77. Naresh A, Thor AD, Edgerton SM. et al. The HER4/4ICD estrogen receptor coactivator and BH3-only protein is an effector of tamoxifen-induced apoptosis. Cancer Res. 2008;68:6387-6395
78. Zhu Y, Sullivan LL, Nair SS. et al. Coregulation of estrogen receptor by estrogen-inducible ERBB4/HER4 establishes a growth promoting autocrine signal in breast cancer. Cancer Res. 2006;66:7991-7998
79. Nabholtz JM, Chalabi N, Radosevic-Robin N. et al. Multicentric neoadjuvant pilot Phase II study of cetuximab combined with docetaxel in operable triple negative breast cancer. Int J Cancer. 2016;138:2274-2280
80. Engels CC, de Glas NA, Sajet A. et al. The influence of insulin-like Growth Factor-1-Receptor expression and endocrine treatment on clinical outcome of postmenopausal hormone receptor positive breast cancer patients: A Dutch TEAM substudy analysis. Mol Oncol. 2016;10:509-516
81. Iams WT, Lovly CM. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin Cancer Res. 2015;21:4270-4277
82. Massacesi C, Di Tomaso E, Urban P. et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203-210
83. Ali S, Rasool M, Chaoudhry H. et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation. 2016;12:135-139
84. Higgins MJ, Stearns V. Understanding resistance to tamoxifen in hormone receptor-positive breast cancer. Clinical Chemistry. 2009;55:1453-1455
85. Arpino G, Wiechmann L, Osborne CK. et al. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine Reviews. 2009;29:217-233
86. Koutras AK, Fountzilas G, Kalogeras KT. et al. The upgraded role of HER3 and HER4 receptors in breast cancer. Crit Rev Oncol Hematol. 2010;74:73-78
87. Osborne CK, Schiff R. Mechanism of Endocrine Resistance in Breast Cancer. Annu Rev Med. 2011;62:233-247
Author contact
![]() Corresponding author: Dr. Harald Hessel, Institute of Pathology, Faculty of Medicine, LMU Munich, Thalkirchner Strasse 36, 80337 München, Germany. harald.hesseluni-muenchen.de; Phone: +49 89 2180 73680; Fax: +49 89 2180 73742
Corresponding author: Dr. Harald Hessel, Institute of Pathology, Faculty of Medicine, LMU Munich, Thalkirchner Strasse 36, 80337 München, Germany. harald.hesseluni-muenchen.de; Phone: +49 89 2180 73680; Fax: +49 89 2180 73742