Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(16):2807-2816. doi:10.7150/jca.25680 This issue Cite
Research Paper
MT1G is Silenced by DNA Methylation and Contributes to the Pathogenesis of Hepatocellular Carcinoma
Ju-deng Zeng1*, Ning Zhang2*, Gui-jun Zhao3*, Li-xia Xu2, Yang Yang1, Xiao-yi Xu1, Meng-ke Chen1, Hui-yun Wang1 ![]() , Steven Xiao-feng Zheng1,4
, Steven Xiao-feng Zheng1,4 ![]() , Xiao-xing Li1
, Xiao-xing Li1 ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-Sen University Cancer Center, Guangzhou, Guangdong, China
2. Department of Gastroenterology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong, China
3. Department of Gastroenterology and Hepatology, Inner Mongolia People's Hospital, Hohhot, Inner Mongolia Autonomous Region, China
4. Rutgers Cancer Institute of New Jersey and Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, New Brunswick, NJ, USA
* These authors have equally contributed to this work.
Received 2018-2-23; Accepted 2018-6-2; Published 2018-7-16
Abstract
Using genome-wide screening and TCGA-based data analysis, we identified a DNA methylation-related gene named metallothionein-1G (MT1G), which may play an important role in hepatocellular carcinoma (HCC). In this study, we found that MT1G expression was silenced in 4/6 HCC cell lines and negatively related to aberrant promoter hypermethylation. Its mRNA level was restored with demethylation treatment. Moreover, MT1G downregulation at both the transcriptional and protein level was also detected in 8 pairs of clinical HCC samples compared with its expression in adjacent normal tissues. Ectopic expression of MT1G in silenced HCC cell lines inhibited colony formation, suppressed cell migration and invasion, and repressed xenograft tumor growth in nude mice. In contrast, knockdown of MT1G by short hairpin RNA showed the opposite effect on cell proliferation and the malignant phenotype. Moreover, our data showed that MT1G suppressed tumor invasion and metastasis mainly through regulating the expression of proteins in the matrix metalloproteinase family (MMP) and modulating the epithelial-mesenchymal transition (EMT) process. To our surprise, the data from TCGA showed that hypermethylation of MT1G is associated with good survival of HCC patients. In conclusion, our study demonstrated that MT1G acts as a tumor suppressor gene in HCC development, but its clinical potential in HCC requires further evaluation.
Keywords: MT1G, Hepatocellular Carcinoma, Tumor Suppressor Gene, Promoter Hypermethylation
Introduction
Hepatocellular carcinoma (HCC) is the fifth most prevalent cancer and the third most frequent cause of cancer-related death in the world [1]. In most cases, HCC develops within an established background of chronic liver injury. Major risk factors for HCC include chronic HBV or HCV infection, alcoholic and probable non-alcoholic hepatitis [2]. For early-stage HCC patients, Liver resection and transplantation are the best choice and are associated with significantly prolonged survival time [3, 4]. Although there are several therapeutic regiments for advanced HCC, including local ablation [5], transarterial chemoembolization (TACE) [6, 7] and targeted molecular therapy [8], the prognosis of patients is still very poor. Therefore, early diagnosis is a major method to reduce the mortality of HCC. However, early-stage HCC is currently difficult to detect; even the most advanced imaging methods are unable to efficiently identify the precancerous lesions [9]. In the genomic era, many molecular markers related to tumorigenesis have been discovered. Biomarker detection is expected to become an effective tool for diagnosing HCC and determining individualized therapeutic regimens [10].
Metallothioneins (MTs) are a group of low-molecular-weight cysteine-rich proteins that play a key role in removing toxic heavy metals and scavenging free radicals to protect cells against oxidative stress[11]. Generally, mammalian metallothionein isoforms can be divided into four groups, called MT1, MT2, MT3 and MT4 [12]. Most studies focus on MT1, the most widely expressed isoform in humans and at least eight members have been reported (MT1A, 1B, 1E, 1F, 1G, 1H, 1M, 1X) [13, 14]. The liver is the major organ involved in metabolism, which is accompanied by a large number of free radicals [15], and MTs play important roles in maintaining intracellular redox homeostasis [16]. Unsurprisingly, the aberrant expression of MTs play a part in hepatocarcinogenesis because of the associated excessive oxidative damage. In fact, previous studies have shown that MT1G, a well-known isoform of MT1, was downregulated in most HCC specimens compared to its expression in adjacent non-tumor tissues and was frequently silenced by promoter hypermethylation. In addition, the expression and promoter methylation of MT1G is expected to be a biomarker to predict the prognosis of HCC patients [17, 18]. However, some researchers believe that MT is a multi-functional and paradoxical protein. On one hand, MT protects normal cells from toxic metals and free radical damage. On the other hand, under disease conditions, MT can protect cancer cells against the therapies based on generating oxidative damage [19-21].
In this study, we integrate TCGA data analysis and experimental results to systematically evaluate the biological role of MT1G in hepatocarcinogenesis and its potential as an independent prognostic factor in HCC.
Materials and Methods
Cell lines and cell culture
In this study, 6 human HCC cell lines (HepG2, Hep3B, Bel7402, SMMC7721, MHCC97L and MHCC97H) and 1 normal immortalized liver cell line LO2 were used for the detection of MT1G methylation status and expression. HepG2 and Hep3B were obtained from ATCC. Bel7402, SMMC7721, MHCC97L, MHCC97H and LO2 were purchased from the cell resource center of Shanghai Institutes for Biological Science. HepG2, Hep3B, MHCC97L, MHCC97H and LO2 were cultured in DMEM (Gibco, USA) supplemented with 10% FBS (BI, USA); Bel7402 and SMMC-7721 were cultured in RPMI-1640 medium (Gibco, USA) supplemented with 10% FBS (BI, USA). All cell lines were grown at 37˚C and 5% CO2.
Clinical samples
Eight paired biopsy specimens from primary tumor and adjacent non-tumor sites from patients with HCC were collected from the Inner Mongolia People's Hospital, Hohhot, China. The study protocol was approved by the Clinical Research Ethics Committee of the Inner Mongolia People's Hospital, Hohhot, China.
DNA extraction and bisulfite conversion
Genomic DNA from HCC cell lines and tumor tissues were isolated by using TIAN-amp Genomic DNA Kit (TIANGEN BIOTECH, Beijing, DP304). Sodium metabisulfite was used to modify 2 μg of DNA with the EZ DNA Methylation-Gold Kit (Zymo, USA, D5006).
Methylation-specific PCR and bisulfite genomic sequencing
Methylation-specific PCR was performed to amplify the bisulfite-modified DNA, and two primer pairs were designed to identify methylation and unmethylation sites respectively. The PCR products were sequenced and analyzed by FinchTV. MT1G-M-sense: 5'-TGGTAGGGTGAGAGAAGTCGTATAC-3'; MT1G-M-anti sense: 5'-TTTTTATAATCAAAAAACGAAACCG-3'; MT1G-U-sense: 5'-GGTAGGGTGAGAGAAGTTGTATATG-3'; MT1G-U-anti sense: 5'-TTTTATAATCAAAAAACAAAACCAAA-3'; MT1G-B-sense: 5'-GGAAGTTGGGAAGGTTTGGTAGG-3'; MT1G-B-anti sense: 5'-AACATCCCAAAACACAAAACCC-3'.
Semi-quantitative RT-PCR and real-time quantitative PCR
Trizol reagent (Ambion, USA, 15596018) was used to extract total RNA from HCC cells and tumor tissues. cDNA was synthesized using GoScript Reverse Transcription Kit (Promega, USA, A5001). Semi-quantitative RT-PCR and real-time quantitative PCR were performed to detect the relative MT1G expressions, the primers used in this experiment were listed as follow: MT1G-RT-sense: 5'-CTTCTCGCTTGGGAACTCTA-3'; MT1G-RT-anti-sense: 5'-AGGGGTCAAGATTGTAGCAAA-3'.
Demethylating treatment with 5-aza-dC
HCC cells were grown in 60mm dishes and then treated with 10 μM 5-aza-dC (Selleck, USA, S1200) for 72 h and harvested for subsequent analysis.
Western blot assay and antibody
RIPA lysate was used to isolate total protein from cells and measuring the concentration with a BCA protein quantitation kit (ThermoFisher, USA). Then, 30 µg protein of each sample was separated in a 10% SDS-PAGE gel, transferred onto a polyvinylidene Fluoride (PVDF) membrane. The non-specific binding sites on membrane were sealed with 5% milk, and then incubated with a primary antibody (UC1MT; Abcam, ab12228) and secondary antibody. The signals were detected using the enhanced chemiluminescence method.
Colony formation assay
0.25% trypsin was used to digest HCC cells and cell suspensions were diluted to 1X103 cells/ml. Then, 50 cells were planked in each well of a 96-well plate and cultured in an incubator at 37 ℃ for 12-14 days. Colonies were counted after they were stained with crystal violet.
Transwell migration and invasion assay
HCC cells were digested into a single-cell suspension, and the concentration was adjusted to 1X106 cells/ml with serum-free medium. Then, 100 μL cells were planked into upper chambers of transwell migration chambers (migration assay) or Matrigel invasion chambers (invasion assay), and medium supplemented with 10% FBS was added to the lower chambers. The chambers were incubated for 6-8 h (migration assay) or 18-24 h (invasion assay) in an incubator with 5% CO2 at 37 ˚C. The upper surface of the chambers was carefully wiped with a cotton swab to remove the residual cells. Cells that adhered to the lower surface were fixed with 4% paraformaldehyde and stained with crystal violet. Five random fields were counted using an optical microscope.
In vivo tumorigenicity
SMMC7721 cells transfected with MT1G or vector and MHCC97H cells transfected with shMT1G or shVector were digested, and the concentration was adjusted to 1X107 cells/ml. A 200-μL cell suspension was injected into the dorsal right flank of 6-week-old BALB/c-nude mice with six mice in each group. The tumor weights and volumes were measured each week. After 6 weeks of observation, mice were sacrificed, and tumors were collected, photograph, and embedded in paraffin for HE staining. Animal care and all experimental procedures were approved by the Animal Ethics Committee of the Inner Mongolia People's Hospital.
Lentiviral transfection
Lentiviruses containing MT1G CDS or short hairpin RNA targeting MT1G were purchased from GenePharma (Shanghai) and were used to infect HCC cell lines according to the manufacturer's instruction. Puromycin was used to select stable transfected cells.
Databases and statistical analysis
MT1G mRNA expression data (TCGA_LIHC_exp_HiSeqV2-2015-02-24; genomic-Matrix) were download from the UCSC Cancer Browser database (https://genome-cancer.ucsc.edu/proj/site/hgHeatmap/). Both clinical data (lihc_tcga.tar; data_bcr_clinical_data_patient) and MT1G methylation data (lihc_tcga.tar; data_methylation_hm450) were downloaded from the cBioPortal database (http://www.cbioportal.org/study?id=lihc_tcga#summary). All the above data originated from TCGA data from the same cohort. SPSS 20.0 (IBM, USA) and GraphPad Prism 6.0 (GraphPad Software, USA) were applied for plotting and statistical analysis. All experimental data were displayed as the means±SD. Comparisons between groups were assessed using student's unpaired t-test (two-tailed). Pearson's correlation analysis were performed to analyzed the correlation between MT1G mRNA expression and methylation. The correlation between clinical features and MT1G methylation levels was analyzed by Pearson's chi-square test. Kaplan-Meier survival curve and log-rank test were used to evaluate the overall survival and disease-free survival time corresponding to MT1G mRNA expression and methylation levels. Differences were considered statistically significant when the p-value was less than 0.05.
Results
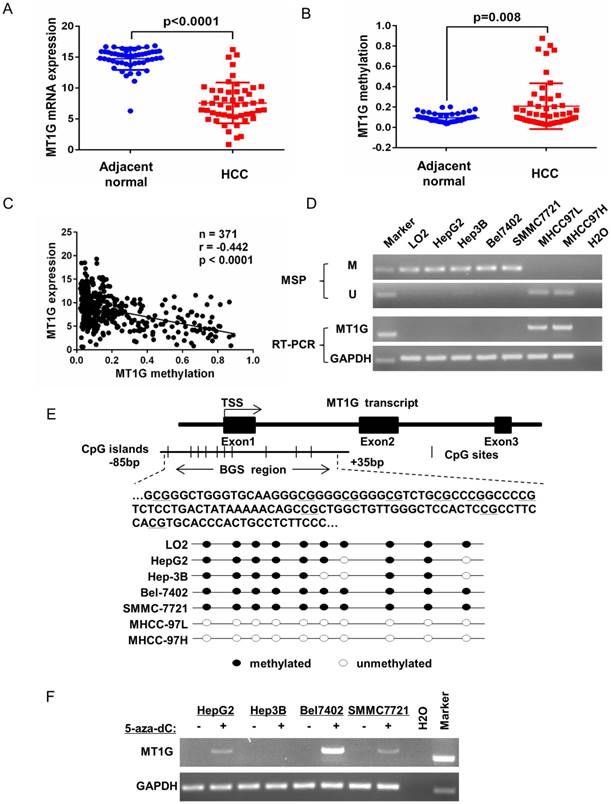
MT1G is silenced by promoter methylation in HCC
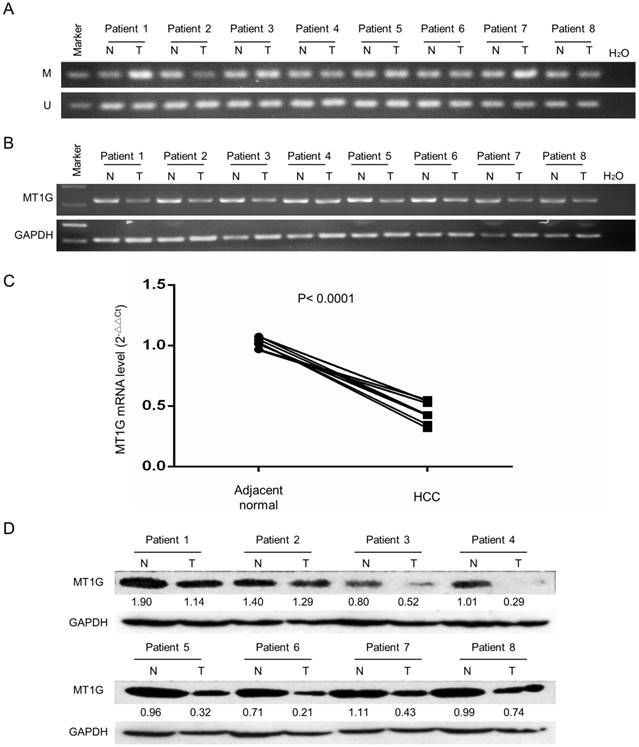
To evaluate MT1G expression and methylation status in HCC, we first analyzed TCGA data obtained from the Cancer Browser and CBioPortal databases. The results showed that MT1G mRNA expression is significantly downregulated in HCC accompanied with reciprocal promoter hypermethylation in 50 pairs of HCC tumor and adjacent non-tumor tissues (Figure 1A, B). In addition, a remarkable negative correlation between MT1G expression and methylation was observed in 371 HCC samples (r = -0.442, P < 0.0001, Figure 1C). The associations between clinical features and MT1G methylation levels are listed in Table 1. On the other hand, using methylation-specific PCR (MSP) and bisulfite genomic sequencing (BGS), most (4/6) of the HCC cell lines were found to be hypermethylated in the promoter region of MT1G (Figure 1D, E). Moreover, RT-PCR results showed that MT1G was only expressed in unmethylated cell lines (Figure 1D). MT1G-silenced cell lines were treated with the demethylation agent 5-Aza-dC, which was able to partially recover the mRNA expression (Figure 1F). We further performed MSP, RT-PCR and western blot to detect MT1G methylation, mRNA and protein expression in paired tumor samples and adjacent non-tumor tissues from 8 HCC patients. As shown in Figure 2A, the methylation of MT1G was higher in tumor samples in half of the paired samples. Both the MT1G mRNA and protein levels were dramatically downregulated in all of the tested HCC tissues compared with the levels in the paired adjacent non-tumor tissues (Figure 2B, C, D). According to these results, MT1G was silenced in both HCC samples and cell lines partially due to promoter hypermethylation.
MT1G silencing is closely linked to DNA methylation. A) MT1G mRNA expression and B) methylation levels in HCC samples compared with the expression in their adjacent non-tumor tissues were analyzed in two genomic screening datasets (mRNA expression dataset: TCGA_LIHC_exp_HiSeqV2-2015-02-24 obtained from Cancer Browser; methylation levels dataset: lihc_tcga.tar obtained from CBioPortal; student's unpaired t-test, n = 50). C) The correlation between MT1G mRNA expression and methylation status was analyzed in HCC samples of the above two datasets (r = -0.4442, n = 371, p-value < 0.0001). D) The methylation status and mRNA expression levels in 1 normal immortalized liver cell line LO2 and 6 HCC cell lines (HepG2, Hep3B, Bel7402, SMMC7721, MHCC97L and MHCC97H) were measured by MSP (M: methylation, U: unmethylation) and semi-quantitative RT-PCR. E) The methylation status was further confirmed by bisulfite genomic sequencing. F) MT1G mRNA expression was partially restored after treatment with the demethylation reagent 5-Aza-dC in 4 HCC cell lines.
Correlation between MT1G methylation levels and clinicopathological features in liver cancer data from TCGA
| Low methylation | High methylation | ||||
|---|---|---|---|---|---|
| Variable | (n = 127) | % | (n = 125) | % | p-value |
| Age | |||||
| < 60 | 58 | 47.5% | 64 | 52.5% | 0.380 |
| ≥ 60 | 69 | 53.1% | 61 | 46.9% | |
| Gender | |||||
| Male | 99 | 55.6% | 79 | 44.4% | 0.010 |
| Female | 28 | 37.8% | 46 | 62.2% | |
| Family history | |||||
| of cancer | |||||
| Yes | 42 | 48.8% | 44 | 51.2% | 0.722 |
| No | 85 | 51.2% | 81 | 48.8% | |
| Alcohol consumption | |||||
| Yes | 45 | 50.6% | 44 | 49.4% | 0.969 |
| No | 82 | 50.3% | 81 | 49.7% | |
| HBV/HCV infection | |||||
| Yes | 59 | 50.9% | 57 | 49.1% | 0.891 |
| No | 68 | 50.0% | 68 | 50.0% | |
| Non-alcoholic fatty liver disease | |||||
| Yes | 4 | 26.7% | 11 | 73.3% | 0.058 |
| No | 123 | 51.9% | 114 | 48.1% | |
| Tumor stage | |||||
| I & II | 91 | 49.7% | 92 | 50.3% | 0.729 |
| III & IV | 36 | 52.2% | 33 | 47.8% | |
| Recurrence | |||||
| Yes | 72 | 51.8% | 67 | 48.2% | 0.622 |
| No | 55 | 48.7% | 58 | 51.3% |
MT1G expression is significantly downregulated in clinical specimens of HCC. A) MT1G methylation status was detected by MSP in 8 HCC tumor specimens and was compared with the status in their adjacent non-tumor tissues. B) MT1G mRNA expression levels in paired samples were measured by semi-quantitative RT-PCR. C) MT1G mRNA expression levels in paired samples were measured by real-time PCR. D) MT1G protein expression levels in paired samples were measured by western blot.
MT1G overexpression inhibits cell proliferation and invasiveness
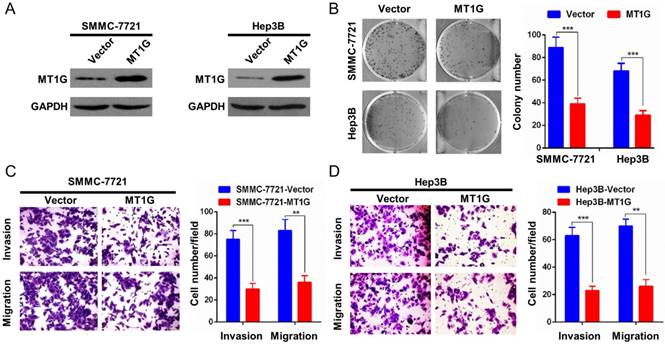
The remarkable downregulation of MT1G mediated by promoter methylation in HCC samples indicated that MT1G may be a potential tumor suppressor gene. To evaluate the role of MT1G in HCC proliferation, we ectopically expressed MT1G in SMMC7721 and Hep3B cells (Figure 3A), in which MT1G expression was silenced by promoter methylation. SMMC7721 and Hep3B cells overexpressing MT1G exhibited significantly fewer and smaller colonies than the vector-transfected control cells in the colony formation assay (Figure 3B). Moreover, to further assess the effect of MT1G in cell invasion and migration, we performed a transwell migration assay and Matrigel invasion assay. The results showed that the invaded and migrated cell numbers of cells transfected with MT1G were significantly lower than those of control cells, suggesting that MT1G suppresses the malignant phenotype of HCC (Figure 3C, D).
Overexpression of MT1G inhibits cell colony formation, invasion and migration. A) The ectopic expression efficiency of MT1G in SMMC7721 and Hep3B cells was confirmed by western blot. B) The colony formation assay showed that the number of colonies was lower in MT1G-overexpressed cell lines than in control groups. C) Matrigel invasion assay and D) transwell migration assay were used to evaluate the cell invasion and migration abilities. The results showed that invaded or migrated cell numbers were significantly lower in MT1G-overexpressed cell lines than in the control groups.
Knockdown of MT1G promotes cell proliferation and a malignant phenotype
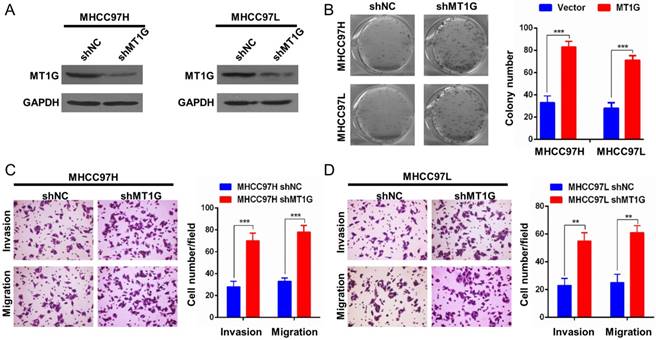
To further confirm the role of MT1G in HCC development, we performed a knockdown assay with MT1G short hairpin RNA in two MT1G-expressing cell lines, MHCC97L and MHCC97H (Figure 4A). Contrary to MT1G overexpression, knockdown of MT1G markedly promoted colony formation (Figure 4B). In addition, an enhanced migration and invasion phenotype of HCC cell lines was observed with reduced MT1G expression in transwell migration and Matrigel invasion assays (Figure 4C, D). These data further provide evidence that MT1G functions as a potential tumor suppressor gene in vitro.
Knockdown of MT1G promotes cell colony formation, invasion and migration. A) Knockdown efficiency of MT1G with short hairpin RNA in MHCC97H and MHCC97L cells was measured by western blot. B) Knockdown of MT1G increased the number of colonies in the colony formation assay. C) Knockdown of MT1G markedly promoted the cell invasion ability. D) Knockdown of MT1G elevated the migration ability of cancer cells.
MT1G affects xenograft tumor growth in nude mice
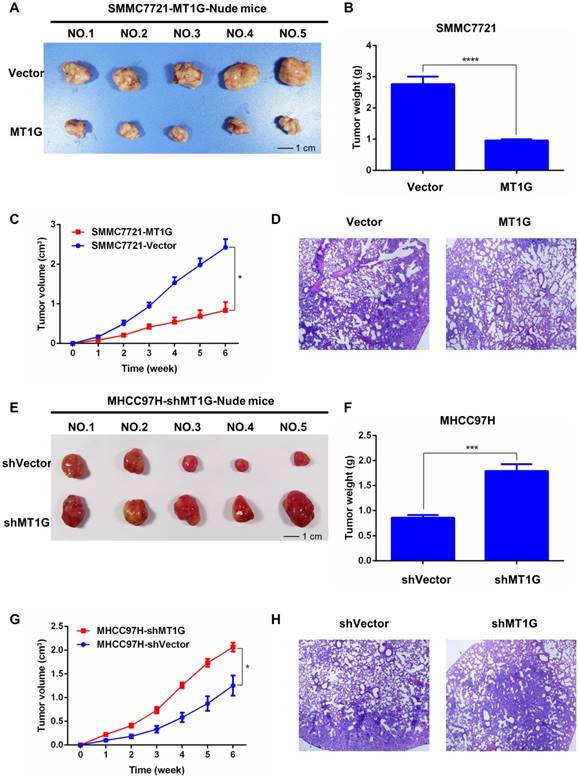
Based on the antitumor effect of MT1G in vitro, we further tested whether MT1G functions as a tumor suppressor in vivo. We generated xenograft tumors in nude mice with SMMC7721 cells stably transfected with MT1G or vector-transfected control cells injected subcutaneously into the dorsal right flank. After the mice were sacrificed and the tumors were collected (Figure 5A), the results revealed that MT1G expression significantly reduced xenograft tumor weight and volume (Figure 5B, C). In addition, hematoxylin-eosin (HE) staining of tumor tissues showed no apparent morphological alteration between the MT1G-overexpression and control groups (Figure 5D). Moreover, to further confirm our hypothesis, we also generated a xenograft model with MHCC97H cells stably expressing MT1G shRNA (Figure 5E). These MT1G-knockdown tumors displayed an enhanced growth rate as measured by tumor weight and volume compared with the control tumors (Figure 5F, G). In addition, HE staining showed no significant change in morphology (Figure 5H). These data demonstrate that MT1G functions as a tumor suppressor in a xenograft tumor mouse model.
MT1G affects xenograft tumor growth in nude mice. A) MT1G overexpression inhibited xenograft tumor growth in nude mice. Xenograft tumors were collected at the end of the study from nude mice that received subcutaneous injection of SMMC7721 cells overexpressing MT1G or carrying a control vector. B) The tumor weights of those injected with MT1G-overexpressing or control cells were compared by student's t-test. C) MT1G overexpression suppressed tumor growth in vivo. D) H&E staining of MT1G-overexpressing and control tumors. E) Knockdown of MT1G in MHCC97H cells promoted xenograft tumor growth in nude mice. F) The tumor weights of shMT1G and shVector groups were compared by student's t-test. G) Knockdown of MT1G accelerated the growth of the tumor in vivo. H) H&E staining of shMT1G and shVector tumors.
MT1G mainly modulates the epithelial-mesenchymal transition (EMT) process
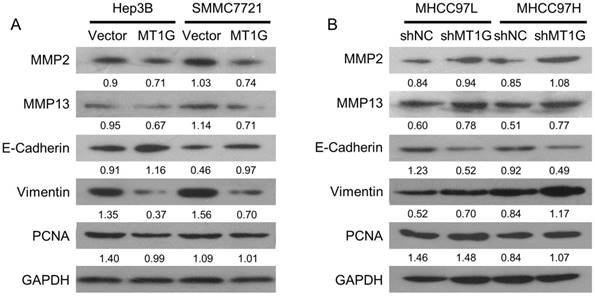
To elucidate the molecular mechanism of MT1G in HCC, we examined the molecules involved in proliferation and EMT, including PCNA, MMP2, MMP13, E-Cadherin, and Vimentin. The cell proliferation marker, PCNA, was downregulated in MT1G-transfected cell lines compared to its expression in control cells (Figure 6A, B). In addition, MT1G overexpression moderately downregulated MMP expression and markedly reversed the expression of EMT-related proteins (Figure 6A, B). In contrast, knockdown of MT1G regulated MMP and EMT marker expression in an opposing fashion (Figure 6A, B). These data demonstrate that MT1G mainly modulates the EMT process to suppress the tumor cell malignant phenotype.
MT1G mainly modulates the EMT process. The effects of A) ectopic expression or B) knockdown of MT1G on MMP2, MMP13, E-cadherin, Vimentin and PCNA expression were analyzed by western blot assay.
The evaluation of MT1G as a biomarker in patients with HCC
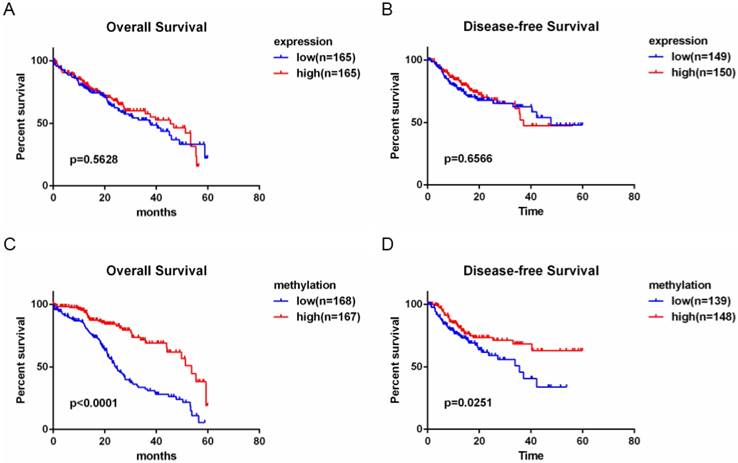
The above data show that MT1G is silenced by promoter hypermethylation in HCC and functions as a tumor suppressor in hepatocarcinogenesis. To evaluate the clinical potential of MT1G expression and methylation status, we performed survival analysis using clinical features combined with expression and methylation data of HCC patients from TCGA. As shown in the Kaplan-Meier survival curve (Figure 7A, B), HCC patients with high MT1G mRNA expression (median overall survival time (OS) = 45.53 months, median disease-free survival time (DFS) = 37.12 months) showed no apparent survival benefits compared to patients with lower MT1G expression (median OS = 37.75 months, median DFS = 47.73 months). Surprisingly, however, overall survival and disease-free survival of HCC samples with relatively high methylation status (median OS = 53.65 months, median DFS > 5 years) were significantly better than those with lower methylation (median OS of 24.44 months, median DFS = 35.58 months, Figure 7C, D). These survival analysis data indicate that hypermethylation of MT1G is a predictor of good survival in HCC patients.
Hypermethylation of MT1G is associated with good prognosis of HCC patients. A) Overall survival time (OS) and disease-free survival time (DFS) with low or high MT1G expression in HCC samples obtained from TCGA were analyzed by Kaplan-Meier survival analysis. No different outcome was observed between the two groups. B) HCC Patients with a higher methylation level of MT1G had longer OS and DFS.
Discussion
In this study, we demonstrate that MT1G is frequently silenced in HCC primary tumor samples compared with its expression in adjacent normal tissues by using TCGA data and experimental methods. Downregulation of MT1G is largely related to aberrant promoter hypermethylation, which was confirmed by MSP and RT-PCR in both HCC cell lines and clinical samples. This result was further validated in MT1G-silenced cell lines treated with the demethylation reagent 5-aza-dC, which led to the partial restoration of MT1G expression. However, the HCC cell line Hep3B showed no restoration in the 5-aza-dC demethylation assay. This result indicates that other mechanisms also regulate MT1G expression, such as histone modification and metal-induced transcription factor regulation[22-24]. In general, these data reveal that MT1G silencing is mainly regulated by promoter methylation, which may contribute to the development of HCC.
The antioxidative role of MTs is a double-edged sword. As an antioxidant family, MTs can significantly resist oxidative damage, which is closely related to genomic instability, tumor progression and metastasis[25]. Meanwhile, the antioxidative attribute of MTs may assist cancer cells to remove reactive oxygen species (ROS) and attenuate the efficacy of chemotherapy and radiotherapy, which kill cancer cells based on their production of reactive oxygen species [26, 27]. Considering such a paradoxical effect of MTs, we overexpressed and knocked down MT1G expression in HCC cell lines and revealed that MT1G functions as a tumor suppressor gene in vivo and in vitro. Ectopic expression of MT1G in HCC cell lines significantly suppressed colony formation, cell invasion and migration. A xenograft mouse model also confirmed the tumor-suppressive function of MT1G. Based on our data, MT1G inhibits tumorigenicity in an ROS-free microenvironment.
Functional experiments showed that MT1G could markedly affect cell invasion and metastasis, which was characterized by the ability to degrade the extracellular matrix (ECM) and activate the epithelial-mesenchymal transition (EMT) process[28]. Our results showed that MT1G obviously reduced the expression of matrix metalloproteinases (MMPs), which promote cancer invasion due to their ability to degrade connective tissue[29]. In addition, MT1G significantly upregulated E-cadherin (epithelial marker) and downregulated vimentin (mesenchymal marker), indicating that MT1G strongly reverses the EMT process and suppresses the tumor migratory property[30].
Based on the tumor suppressor role of MT1G identified both in vitro and in vivo, we further estimated the clinical value of MT1G as a predictor in HCC prognosis. To our surprise, we found that hypermethylation of MT1G but not low methylation of MT1G predicted good survival of HCC patients. One of the explanation of this phenomenon is that MT1G may play a distinct role in different tumor stage. For example, MT1G is capable of scavenging excess ROS in precancerous tissue to slow the tumorigenesis. While in advanced stage of tumor development, MT1G may promote tumor progression through hampering the efficacy of therapies, which is based on their production of oxidative damage. We also noticed that the recent studies have reported that MT1G is able to induce sorafenib resistance via inhibiting ferroptosis [31] and acts as a biomarker for altered redox status in HCC cells subjected to sorafenib [32]. All these data combined our results indicated that the role of MT1G in tumor progression and its potential as a biomarker require further evaluation.
In conclusion, we demonstrate that MT1G is epigenetically silenced in HCC and acts as a tumor suppressor in HCC development. The high methylation level of MT1G is associated with good survival in HCC patients.
Acknowledgements
Supported by the National Natural Science Foundation of China (Nos: 81730081, 81772635, 81572440, 81460378 and 81401999), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholar (No: 2015A030306047), Pearl River S&T Nova Program of Guangzhou (201506010050) and the Research Fund of State Key Laboratory of Oncology in South China. Key raw data were verified and uploaded onto the Research Data Deposit platform (www.researchdata.org.cn, NO. RDDB2018000361).
Abbreviations
TCGA: The Cancer Genome Atlas; MTs: metallothioneins; HCC: hepatocellular carcinoma; MMP: matrix metalloproteinase; EMT: epithelial-mesenchymal transition; HBV: hepatitis B virus; HCV: hepatitis C virus; TACE: transarterial chemoembolization; HE: hematoxylin-eosin; shRNA: short hairpin RNA; PCNA: proliferating cell nuclear antigen; OS: overall survival; DFS: disease-free survival; ROS: reactive oxygen species.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer. 2010;127:2893-917
2. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. The Lancet. 2012;379:1245-55
3. Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Seminars in liver disease. 2005;25:181-200
4. Menahem B, Lubrano J, Duvoux C. et al. Liver transplantation versus liver resection for hepatocellular carcinoma in intention to treat: An attempt to perform an ideal meta-analysis. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2017;23:836-44
5. Cho YK, Kim JK, Kim MY. et al. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology. 2009;49:453-9
6. Lammer J, Malagari K, Vogl T. et al. Prospective randomized study of doxorubicin-eluting-bead embolization in the treatment of hepatocellular carcinoma: results of the PRECISION V study. Cardiovascular and interventional radiology. 2010;33:41-52
7. Lencioni R. Loco-regional treatment of hepatocellular carcinoma. Hepatology. 2010;52:762-73
8. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359:378-90
9. El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011;365:1118-27
10. Schulze K, Nault JC, Villanueva A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. Journal of hepatology. 2016;65:1031-42
11. Krizkova S, Fabrik I, Adam V. et al. Metallothionein-a promising tool for cancer diagnostics. Bratislavske lekarske listy. 2009;110:93-7
12. Simpkins CO. Metallothionein in human disease. Cellular and molecular biology. 2000;46:465-88
13. Thirumoorthy N, Manisenthil Kumar KT, Shyam Sundar A. et al. Metallothionein: an overview. World journal of gastroenterology. 2007;13:993-6
14. Zalewska M, Trefon J, Milnerowicz H. The role of metallothionein interactions with other proteins. Proteomics. 2014;14:1343-56
15. Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: hints from the EASL Basic School of Hepatology. Journal of hepatology. 2013;58:365-74
16. Maret W. Redox biochemistry of mammalian metallothioneins. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2011;16:1079-86
17. Ji XF, Fan YC, Gao S. et al. MT1M and MT1G promoter methylation as biomarkers for hepatocellular carcinoma. World journal of gastroenterology. 2014;20:4723-9
18. Kanda M, Nomoto S, Okamura Y. et al. Detection of metallothionein 1G as a methylated tumor suppressor gene in human hepatocellular carcinoma using a novel method of double combination array analysis. International journal of oncology. 2009;35:477-83
19. Heger Z, Rodrigo MA, Krizkova S. et al. Metallothionein as a Scavenger of Free Radicals - New Cardioprotective Therapeutic Agent or Initiator of Tumor Chemoresistance? Current drug targets. 2016;17:1438-51
20. Kwee JK. A paradoxical chemoresistance and tumor suppressive role of antioxidant in solid cancer cells: a strange case of Dr. Jekyll and Mr. Hyde. BioMed research international. 2014;2014:209845
21. Lazo JS, Kuo SM, Woo ES. et al. The protein thiol metallothionein as an antioxidant and protectant against antineoplastic drugs. Chemico-biological interactions. 1998;111-112:255-62
22. Egli D, Selvaraj A, Yepiskoposyan H. et al. Knockout of 'metal-responsive transcription factor' MTF-1 in Drosophila by homologous recombination reveals its central role in heavy metal homeostasis. The EMBO journal. 2003;22:100-8
23. Radtke F, Heuchel R, Georgiev O. et al. Cloned transcription factor MTF-1 activates the mouse metallothionein I promoter. The EMBO journal. 1993;12:1355-62
24. Shimazu T, Hirschey MD, Newman J. et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211-4
25. Fuchs-Tarlovsky V. Role of antioxidants in cancer therapy. Nutrition. 2013;29:15-21
26. Block KI, Koch AC, Mead MN. et al. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. International journal of cancer. 2008;123:1227-39
27. Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox biology. 2013;1:244-57
28. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178-96
29. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nature reviews Drug discovery. 2014;13:904-27
30. Santamaria PG, Moreno-Bueno G, Portillo F. et al. EMT: Present and future in clinical oncology. Molecular oncology. 2017;11:718-38
31. Sun X, Niu X, Chen R. et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488-500
32. Houessinon A, Francois C, Sauzay C. et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Molecular cancer. 2016;15:38
Author contact
![]() Corresponding authors: Xiao-xing Li: Lixiaoxorg.cn; Tel: +86-20-8734-2283, Steven Xiao-feng Zheng: zhengstrutgers.edu; Tel: 732-235-6879, Hui-yun Wang: wanghyorg.cn; Tel: +86-20-8734-3308
Corresponding authors: Xiao-xing Li: Lixiaoxorg.cn; Tel: +86-20-8734-2283, Steven Xiao-feng Zheng: zhengstrutgers.edu; Tel: 732-235-6879, Hui-yun Wang: wanghyorg.cn; Tel: +86-20-8734-3308